
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoprocesses of the tyrosine kinase inhibitor gefitinib: from femtoseconds to microseconds and from solution to cells†
Lorena
Tamarit
 ab,
Meryem
El Ouardi
ab,
Inmaculada
Andreu
ab,
Ignacio
Vayá
*ab and
Miguel A.
Miranda
*ab
ab,
Meryem
El Ouardi
ab,
Inmaculada
Andreu
ab,
Ignacio
Vayá
*ab and
Miguel A.
Miranda
*ab
aDepartamento de Química/Instituto de Tecnología Química UPV-CSIC, Universitat Politècnica de València, Camino de Vera s/n, 46022 València, Spain. E-mail: mmiranda@qim.upv.es; igvapre@qim.upv.es
bUnidad Mixta de Investigación, Universitat Politècnica de València, Instituto de Investigación Sanitaria (IIS) La Fe, Hospital Universitari i Politècnic La Fe, Avenida de Fernando Abril Martorell 106, 46026, València, Spain
First published on 13th July 2021
Abstract
Gefitinib (GFT) is a tyrosine kinase inhibitor currently used for the treatment of metastatic non-small cell lung cancer. Although it has been suggested that GFT can be phototoxic, there are no systematic studies on this issue. Here, the photosensitizing potential of GFT has been assessed by means of NRU assays and protein photooxidation. In addition, a thorough photophysical study is presented based on ultrafast transient absorption spectroscopy, fluorescence and laser flash photolysis. Transient species generated after excitation of GFT have been characterized in solution and in biological environments (i.e. HSA and HaCaT cells) to gain insight into the mechanisms involved in photodamage. The photobehavior of GFT was strongly medium-dependent. Excitation of the drug resulted in the formation of locally excited (LE) singlet states (1GFT*), which were found to be the main emissive species in non-polar solvents and also within HSA and HaCaT cells. By contrast, in polar solvents, LE states rapidly evolved (∼1 ps) towards the formation of longer-lived intramolecular charge transfer (ICT) states. The triplet excited state of GFT (3GFT*) can be formed through intersystem crossing from 1GFT* in non-polar solvents and from ICT states in the polar ones, or in the particular case of ethanol, by photosensitization using 2-methoxyacetophenone as an energy donor. In the HSA environment, 3GFT* was hardly detected due to quenching of its LE 1GFT* precursor by Trp through an electron transfer process. Accordingly, HSA photooxidation by GFT was demonstrated using the protein carbonylation method. In summary, a good correlation is established between the photophysical behavior and the photobiological properties of GFT, which provides a mechanistic basis for the observed phototoxicity.
Introduction
The epidermal growth factor receptor (EGFR) family is composed of four members (HER1–4), which are transmembrane glycoproteins with tyrosine kinase activity. They are able to regulate a number of signaling pathways within cells including cell proliferation, migration, differentiation, tissue repair and wound healing.1,2 Mutations and overexpression of tyrosine kinase receptors, especially HER1 and HER2, may result in the appearance of different types of cancers and may promote solid tumor growth.3 Therefore, EGFRs are major targets for the design of anticancer agents. In this regard, tyrosine kinase inhibitors (TKIs) are of high interest due to their ability to block the kinase activity of these receptors.4–8Gefitinib (GFT) is an orally active first-generation TKI.9 It is clinically used for patients with locally advanced or metastatic non-small cell lung cancer; the mode of action involves specific binding of GFT to the ATP site of HER1 preventing autophosphorylation in tumor cells.10 Although the benefits of this drug are evident, it can also induce adverse effects, which are normally associated with rash, diarrhea, dry skin, nausea and vomiting.11
Many drugs are known to absorb solar radiation and can induce photosensitivity reactions, such as phototoxicity or photoallergy, but also photoaging, weakening of the immune system and skin cancer.12 These side effects can be associated with damage to biomolecules (lipids, proteins and DNA) caused by radicals or reactive oxygen species (ROS) arising from excited singlet or triplet states.13–15 Interestingly, drugs containing the quinazoline moiety are known to produce photodermatosis.16 In this regard, it has recently been reported that lapatinib (LAP), a TKI used for the treatment of breast and lung cancer, can induce protein photooxidation and phototoxicity.17 The excited states arising from irradiation of LAP with UV light have been investigated by means of spectroscopic techniques in solution and in the presence of human serum albumin (HSA).18,19 It has been shown that short-lived (ps scale) intramolecular charge transfer states (ICT) are formed in the bulk solution, while longer-lived locally excited (LE) states predominate in the protein-bound LAP; these states must be related to the photosensitizing potential of the drug.
In this context, preliminary in vitro studies suggest that GFT may be phototoxic,20 although there are no reports about the involved photochemical mechanisms. In the present work, the photobiological response of GFT is investigated; thus, its phototoxic potential is evaluated by means of the NRU assay, while its photooxidation activity is assessed towards HSA, the main transport protein in human serum.21 Besides, fluorescence and transient absorption spectroscopies from the femtosecond to the microsecond time-scale are used to investigate the photobehavior of GFT in solution and in the presence of HSA, in addition to human keratinocytes (HaCaT) cells. As a result, it has been observed that the excited state properties of the drug are strongly affected by the environment: LE states are mainly formed in organic non-polar solvents and within HSA or HaCaT cells, while ICT sates are predominant in organic polar solvents. The triplet excited state of gefitinib (3GFT*) has been identified and completely characterized for the first time, and its potential to generate ROS has been assessed. All these features are of key importance in connection with the photosensitizing potential of this drug.
Experimental
Chemicals and reagents
Gefitinib was purchased from Quimigen. Chlorpromazine hydrochloride (CPZ), sodium dodecyl sulphate (SDS), anthracene, human serum albumin (HSA) were purchased from Sigma-Aldrich. For cell culture experiments, HaCaT cells and Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), and penicillin–streptomycin (1.0 × 105 U mL−1, 1.0 × 105 μg mL−1) were supplied by invitrogen. Trypsin–EDTA (0.25–0.02%) and glutamine solutions were provided by Cultek. 2,4-Dinitrophenylhydrazine hydrochloride (DNPH) was purchased from Santa Cruz Biotechnology (Dallas, USA). PBS buffer was prepared by dissolving phosphate-buffered saline tablets (Sigma) using ultrapure water from a Millipore (Milli-Q Synthesis) system. Spectrophotometric HPLC solvents were obtained from Scharlab and used without further purification.Irradiation equipment
Irradiations were performed using a LCZ-4 photoreactor fitted with six top and eight side Hitachi lamps (λmax = 350 nm, Gaussian distribution; Luzchem, Canada). Irradiation for in vitro NRU assay was performed in 96-well transparent plates while 6-well transparent plates were used for photooxidation assay. All experiments were performed keeping the plates on ice inside the photoreactor to avoid overheating.Spectroscopic measurements
Steady-state absorption spectra were recorded in a JASCO V-760 spectrophotometer. Steady-state fluorescence spectra were obtained using a JASCO spectrofluorometer system provided with a monochromator in the wavelength range 200–900 nm, with an excitation wavelength of 340 nm at 25 °C. Measurements on drug@protein complexes were performed in aerated PBS of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio mixtures at 10 μM. The absorbance of the samples at the excitation wavelength was kept below 0.1. Phosphorescence measurements were performed in a Photon Technology International (PTI, TimeMaster TM-2/2003) spectrophotometer equipped with a pulsed Xe lamp, operating in a time-resolved mode with a delay time of 0.5 ms. The sample was dissolved in ethanol, introduced in a quartz tube of 5 mm of diameter and cooled with liquid N2 (77 K).
:1 molar ratio mixtures at 10 μM. The absorbance of the samples at the excitation wavelength was kept below 0.1. Phosphorescence measurements were performed in a Photon Technology International (PTI, TimeMaster TM-2/2003) spectrophotometer equipped with a pulsed Xe lamp, operating in a time-resolved mode with a delay time of 0.5 ms. The sample was dissolved in ethanol, introduced in a quartz tube of 5 mm of diameter and cooled with liquid N2 (77 K).
Time-resolved fluorescence measurements were performed using an EasyLife X system containing a sample compartment composed of an automated peltier cuvette holder to control the temperature, a pulsed LED excitation source and a lifetime detector. The employed LED excitation source was 340 nm, with an emission filter of WG370.
Laser Flash Photolysis (LFP) measurements were performed using a pulsed Nd:YAG L52137 V LOTIS TII at an excitation wavelength of 355 nm. The single pulses were ca. 10 ns duration, and the energy was ∼12 mJ per pulse. The laser flash photolysis system consisted of the pulsed laser, a 77250 Oriel monochromator and an oscilloscope DP04054 Tektronix. The output signal from the oscilloscope was transferred to a personal computer. The absorbances of all solutions were adjusted at ∼0.20 at 355 nm. All UV, fluorescence and LFP measurements were performed using 10 × 10 mm2 quartz cuvettes at room temperature under deaerated conditions (25 min N2 bubbling), or in the case of the protein complexes and/or singlet oxygen detection in an aerated atmosphere. Control experiments indicated that the degree of decomposition of the samples after photolysis was lower than 5%.
Femtosecond transient absorption experiments were performed using a typical pump-probe system. The femtosecond pulses were generated with a compact regenerative amplifier that produces pulses centered at 800 nm (τpulse 100 fs approx., 1 mJ per pulse). The output of the laser was split into two parts to generate the pump and the probe beams. Thus, tunable femtosecond pump pulses were obtained by directing the 800 nm light into an optical parametric amplifier. In the present case, the pump was set at 330 nm and passed through a chopper prior to focusing onto a rotating cell (1 mm optical path) containing the samples in organic or aqueous solution. The white light used as a probe was produced after part of the 800 nm light from the amplifier travelled through a computer controlled 8 ns variable optical delay line and impinged on a CaF2 rotating crystal. This white light was in turn split in two identical portions to generate reference and probe beams that then are focused on the rotating cell containing the sample. The pump and the probe were made to coincide to probe the sample. The power of the pump beam was set to 180 μW. Under these conditions, the degree of photodegradation of the samples was lower than 5%. A computer-controlled imaging spectrometer was placed after this path to measure the probe and the reference pulses to obtain the transient absorption decays/spectra. The experimental data were treated and compensated by the chirp using the ExciPro program.
Phototoxicity assay
A neutral Red Uptake phototoxicity test (NRU) was selected for the study of the cellular phototoxic properties of GFT. The assay was performed in accordance with the OECD Guideline 432 (OECD 2019) in HaCaT cells instead of the 3T3 cell line from BALB/c, the standard method, due to the similarity with human skin cells.22The positive and negative phototoxic controls were CPZ and SDS, respectively. Chlorpromazine is a commonly used anti-psychotic drug which has demonstrated relevant phototoxic properties.23 Briefly, two 96-well plates were seeded at a density of 2.0 × 104 cells per well, and cells were treated the next day with GFT at eight concentrations ranging from 2.5 to 500 μM. Additional plates were treated with CPZ (from 1.57 μM to 500 μM) and SDS (from 3.13 μM to 500 μM) as the references for this experiment. Then, one plate was irradiated with 5 J cm−2 UVA dose (UVA light) whereas the other plate was kept in a dark box (DARK). The next day, cells were incubated with neutral red solution (50 μg mL−1) and further dye extraction from lysosomes was accomplished with a mix buffer [distilled water 50% (v/v), ethanol 49.5% (v/v) and acetic acid 0.5% (v/v)]. Afterwards, the absorbance of the plates was read at 540 nm on a Synergy H1 microplate reader. Dose–response curves for GFT and controls were obtained to determine the concentration causing a reduction of 50% of the neutral red uptake (IC50) under dark and UVA light conditions. Lastly, the photoirritation factor (PIF) values were calculated using the following equation:

Conforming to OECD guideline 432, a substance is labelled as “non-phototoxic” when PIF is <2, “probably phototoxic” if PIF is between 2 and 5 and “phototoxic” if PIF is >5.
Protein carbonyl content assay
Protein carbonyl content assay was carried out following the protocol described elsewhere,24 but with minor modifications. Briefly, a solution of HSA (5 mg mL−1, 1 mg protein/sample) was prepared in PBS and irradiated alone or in the presence of 100 μM of GFT followed by the exposure to UVA light at doses of 5, 10 and 15 J cm−2. Then, the amount of HSA oxidation was monitored spectrophotometrically after incubation of the samples with 2,4-dinitrophenylhydrazine (DNPH) 10 mM in order to generate quantifiable DNPH adducts. Later, proteins were precipitated with 20% trichloroacetic acid solution and a sequence of two washes was performed with ethanol/ethyl acetate 1:1 (v/v) containing 20% trichloroacetic acid. Finally, dried protein pellets were resolubilized with guanidine buffer (6 M) and absorbance was registered at 375 nm using a Synergy H1 microplate reader. Conclusively, the potential of protein oxidation was measured based on the content of carbonyl generated (nmol of carbonyl per mg protein) in the presence of GFT.
Results and discussion
As mentioned in the introduction, TKIs can exhibit photosensitizing potential in combination with sunlight.17 Hence, it appeared interesting to assess the phototoxicity of GFT in vitro based on the established neutral red uptake (NRU) method. Thus, human keratinocytes were incubated with GFT and irradiated with a UVA light dose of 5 J cm−2. The cytotoxic profiles were determined by measurements of the irradiated samples in comparison with those kept in the dark, using neutral red as a vital dye. Thus, from the obtained dose–response curves (see Fig. S1 in the ESI†), IC50 was determined, and the photoirritation factor was calculated as the ratio of IC50 between dark and UVA conditions (see the Experimental section). The PIF value of GFT was found to be 13. Therefore, following the OECD 432 guide (OECD 2019), GFT can be considered a phototoxic drug (Fig. 1 and 2). | ||
| Fig. 1 Chemical structure of GFT. | ||
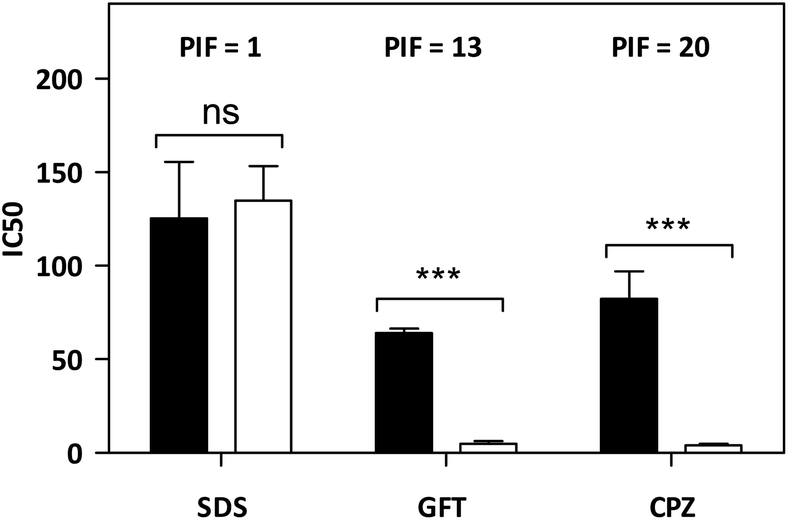 | ||
| Fig. 2 In vitro phototoxicity of GFT in the NRU assay. The concentration causing a reduction of 50% of the neutral red uptake (IC50) was calculated both in the dark (filled bars) and under UVA light conditions (empty bars). The represented data correspond to the mean ± SD from 4 independent dose–response curves. Chlorpromazine (CPZ) and sodium dodecyl sulfate (SDS) represent the selected positive and negative phototoxicity controls, respectively. The PIF value was determined from the ratio between IC50 dark and IC50 UVA for each compound. According to the OECD 432 guide (2019), PIF < 2 means “non-phototoxic”; 2 < PIF < 5 means “probable phototoxicity” and PIF > 5 means “phototoxicity”. Asterisks indicate significant differences by the t-Student test (ns: non-significant, ***p < 0.001). | ||
In view of the phototoxic potential of GFT, its photobehavior was studied by means of fluorescence and transient absorption spectroscopies. The photophysical properties of the drug were first investigated in organic solvents of different polarities. The UV absorption spectra barely varied from acetonitrile to cyclohexane (see Fig. S2 in the ESI†) while the fluorescence properties were strongly affected by the polarity. This is in agreement with previous observations in n-hexane, chloroform or in alcohol solutions, where no detailed explanation was given.25,26 The results show that the emission spectra of GFT were broad and unstructured in polar solvents (see Fig. 3A), displaying low quantum yield (ϕF) and peaking at long wavelengths (λmax > 450 nm). By contrast, ϕF values were much higher in non-polar solvents, and emission occurred at much shorter wavelengths with a lower fwhm (see Table 1). To give a comparison, the fluorescence quantum yield of GFT in acetonitrile was 0.05, with λmax ∼ 473 nm and a fwhm of about 118 nm, while in cyclohexane ϕF was 0.19, and the spectrum showed a fwhm of ca. 71 nm and λmax ∼ 378 nm. In addition, the fluorescence decay kinetics (see Fig. 3B) showed the longest lifetime for the emitting species in acetonitrile, while the shortest one was detected in cyclohexane. Due to the short fluorescence lifetimes (1–3 ns), the presence or absence of oxygen had only a marginal effect (see Fig. S3 in the ESI†).
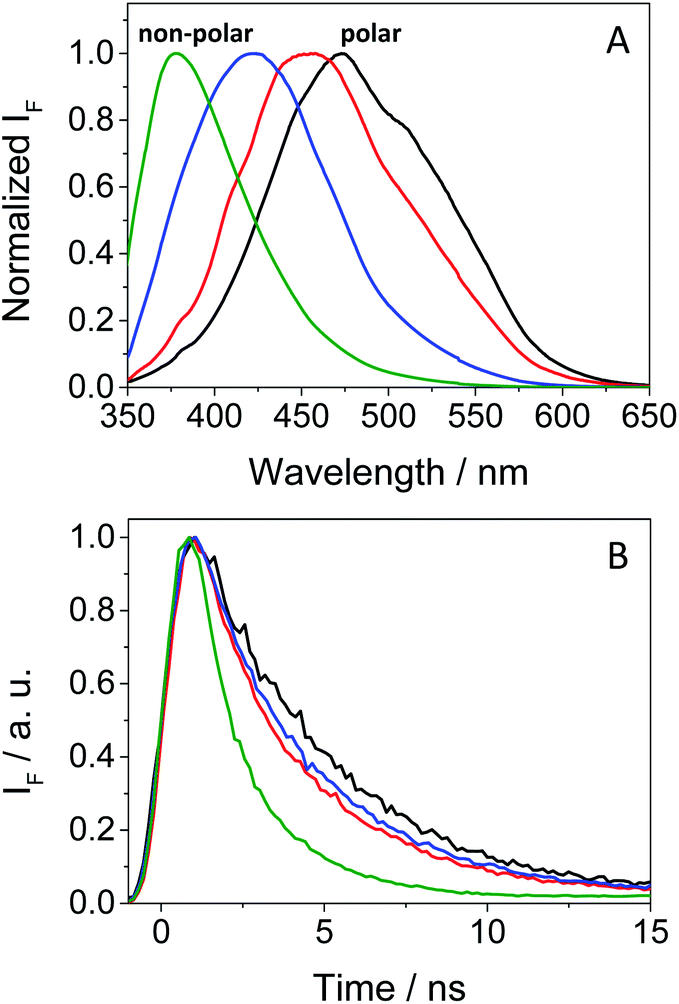 | ||
| Fig. 3 Normalized fluorescence spectra (A) and decays (B) under aerated conditions for GFT in acetonitrile (black), 1,4-dioxane (red), toluene (blue) and cyclohexane (green) after excitation at 340 nm. | ||
The combined results from both steady-state and time-resolved measurements can be interpreted as emission from locally excited (LE) singlet states in non-polar solvents or from intramolecular charge transfer (ICT) states in the polar ones. The energy of LE in cyclohexane, determined from the crossing point between the normalized excitation and emission spectra (see Fig. S4 in the ESI†), was ∼82 kcal mol−1, while that of ICT in MeCN was roughly estimated to be of about 64 kcal mol−1 from the bathochromic shift between the maxima spectra of cyclohexane and MeCN. At low temperatures, the fluorescence signal of GFT in a solid cyclohexane matrix at 77 K was very similar to that found in solution at 298 K. For acetonitrile, it was not possible to record the spectrum of a solid sample. Hence, low temperature measurements in a polar medium were performed in frozen ethanol, where two components were clearly distinguished in the 350–425 and 430–550 nm regions (see Fig. S5 in the ESI†), attributed to the LE and ICT states, respectively. From the wavelength corresponding to the first maximum of the ICT emission, a value of 65 kcal mol−1 was obtained for the energy of this state, which is compatible with the 64 kcal mol−1 estimated in MeCN from the bathochromic shift of the maximum (see above). In ethanol solution, at room temperature, the emission was very weak and no clear spectrum was recorded.
In order to get more insight into the formation of both LE and ICT states, femtosecond transient absorption measurements were performed on GFT in toluene, acetonitrile and ethanol (cyclohexane was not used due to solubility limitations). This is a highly sensitive technique which allows studying the formation of transient species in terms of spectral shape and kinetics resolution, and provides direct information on processes such as intersystem crossing (ISC), energy or electron transfer (ET) and charge separation.28–30 Thus, excitation of the drug at 330 nm in toluene gave rise to an absorption band peaking at λmax ∼ 460 nm. It evolved through two nearly isosbestic points (415 and 560 nm) towards the formation of a new band with two maxima ca. 605 and 410 nm (see Fig. 4A), which became clearly defined on the nanosecond scale. This band, mostly formed in about 10 ps (see Fig. S6 in the ESI†), can be tentatively ascribed to the triplet excited state of gefitinib (3GFT*). By contrast, in acetonitrile, the band at around 460 nm evolved in about 1 ps towards other species with a maximum ca. 430 nm, which persisted up to the nanosecond scale (see Fig. 4B). This behavior is comparable to that previously observed for LAP in MeCN, where LE was the precursor species of the ICT state, formed in about 1.5 ps and decayed in the nanosecond time-scale.18 Accordingly, a similar interpretation can be done for GFT in MeCN; thus, the band at 460 nm is associated with LE, while that at 430 nm to a ICT state. Interestingly, the transient absorption band at ∼605 nm was again visible on the ns scale, coexisting with that of ICT. A similar photobehavior was detected in ethanol; so, LE rapidly evolved (ca. 1.4 ps) towards the formation of ICT, which disappeared in about 700 ps (see Fig. 4C). Surprisingly, the long-lived band associated to 3GFT* with maxima around 410 and 605 nm was not detected.
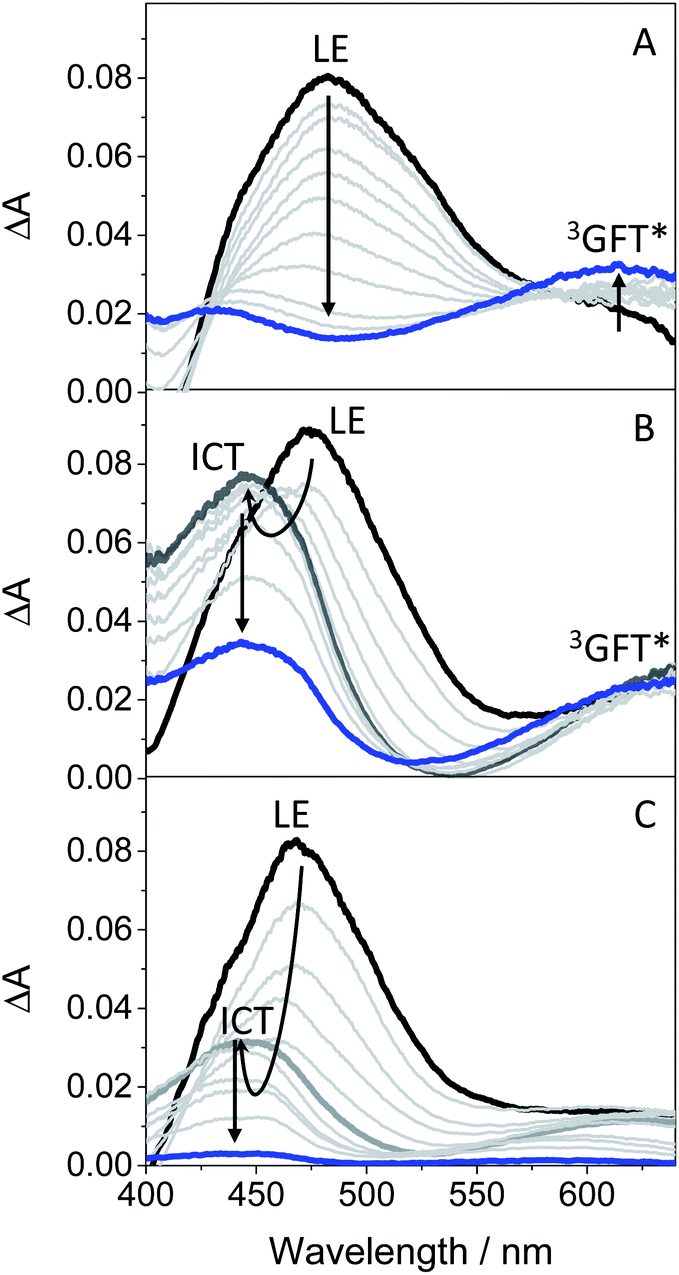 | ||
| Fig. 4 Femtosecond transient absorption spectra of GFT in (A) toluene, (B) acetonitrile and (C) ethanol, after excitation at 330 nm. The spectra were recorded from 0.5 ps (black line) to 2 ns (blue line) in (A) and (B), and from 0.5 ps (black line) to 0.7 ns (blue line) in (C). | ||
In order to further characterize the excited species of the drug at longer time scales, nanosecond LFP measurements were performed at λexc = 355 nm. The transient spectra obtained in toluene showed two maxima around 400 and 600 nm (see Fig. 5A), very similar to those detected in the ns window from ultrafast spectroscopy; the two bands decayed in a similar manner (ca. 2 μs), indicating that both are associated with the same species in the excited state. The photobehavior in acetonitrile (see Fig. 5B) was very similar to that observed in toluene, but with slightly lower efficiency and the excited species displaying shorter lifetimes (∼1.7 μs).
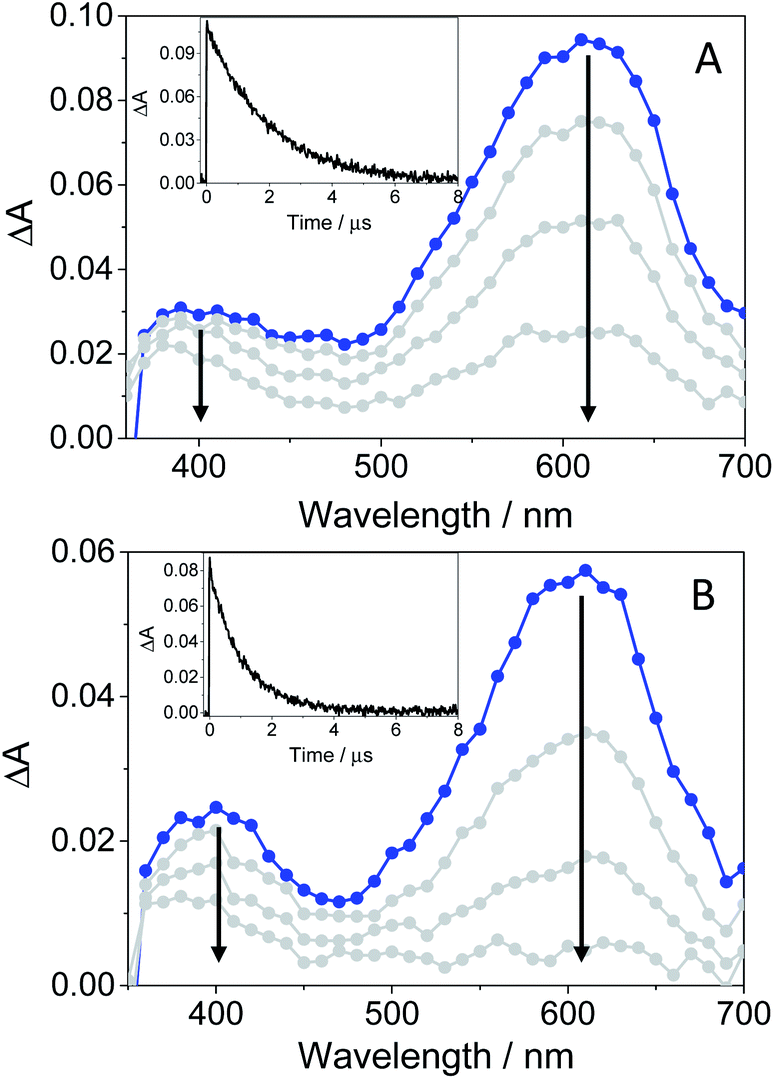 | ||
| Fig. 5 LFP spectra (from 0.2 to 3 μs) and decay traces at 600 nm of GFT in toluene (A) and MeCN (B), after excitation at 355 nm under deaerated conditions. | ||
The signal at 600 nm, assigned to 3GFT*, was strongly quenched by oxygen (kQ ∼ 5.6 × 109 M−1 s−1). In order to better characterize its triplet nature, photosensitization LFP measurements using naproxen (NPX) as an acceptor were performed. The energy of 3NPX* is 62 kcal mol−1,31 while that of 3GFT*, determined from the 4% rise of its phosphorescence spectrum in a solid matrix of ethanol at 77 K (see Fig. S7 in the ESI†), was of about 69 kcal mol−1. Selective excitation of GFT at 355 nm in the presence of NPX resulted in a strong quenching of the signal at 600 nm with a concomitant formation of 3NPX* (λmax ∼ 430 nm) through a triplet–triplet energy transfer process from GFT to NPX (see Fig. S8 in the ESI†). Therefore, the transient band peaking at ∼600 nm can be undoubtedly assigned to the first triplet excited state of the drug. It is worth noting that 3GFT* was much lower in ethanol (see Fig. S9 in the ESI†); all these results are in agreement with those obtained from ultrafast spectroscopy.
In this context, an interesting point of discussion is the formation of 3GFT* in both non-polar and polar solvents, as it can arise from different precursors (LE and/or ICT states, respectively). Interestingly, excitation of the drug in non-polar solvents such as toluene gives rise to LE states, which would evolve towards the formation of 3GFT* in ca. 10 ps through ISC. In contrast, ICT predominates in polar solvents such as acetonitrile; therefore, in this case, 3GFT* would be mainly formed from ICT rather than from LE states, as the latter disappear in a few ps. The minor formation of 3GFT* in ethanol might be due to an enhanced stabilization of the ICT species, whose energy becomes lower than that of 3GFT* (65 vs. 69 kcal mol−1), resulting in an endothermic ISC. However, markedly higher formation of triplet gefitinib was accomplished through triplet–triplet energy transfer from 2-methoxyacetophenone (MAP) as the photosensitizer.32 Thus, excitation of a mixture containing GFT and MAP in deaerated ethanol resulted in enhanced formation of 3GFT* (see the difference spectrum in the inset in Fig. 6A). The growth and decay of this species at its absorption maximum (ca. 600 nm) are clearly shown in Fig. 6B.
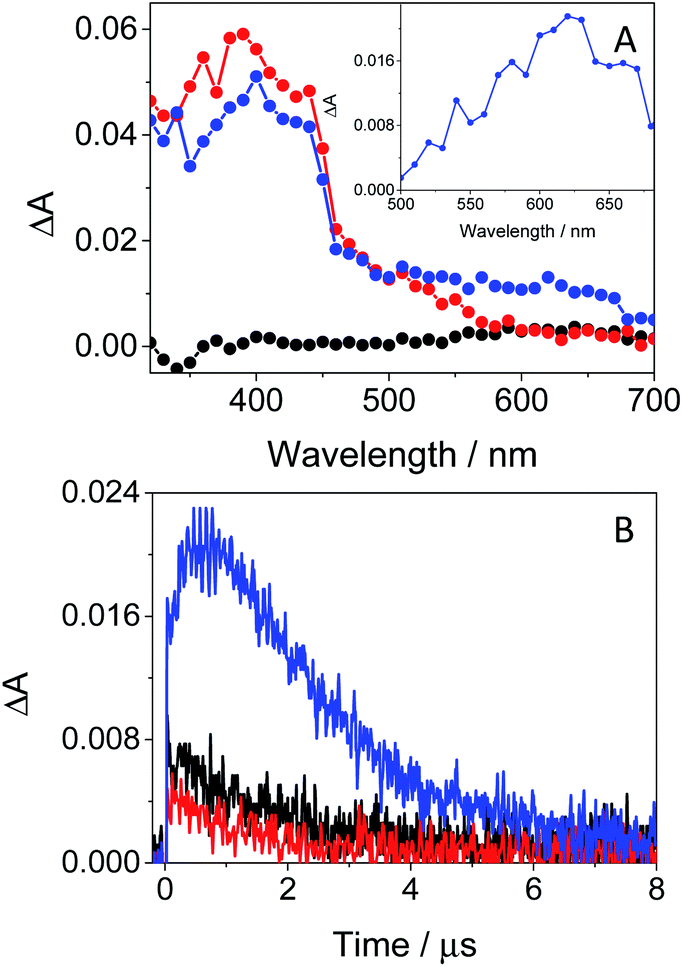 | ||
| Fig. 6 (A) Transient absorption spectra of GFT (black), MAP (red) and a mixture of GFT/MAP (blue). The inset shows the spectrum obtained from subtraction of the MAP spectrum to that of the GFT/MAP spectrum 0.6 μs after the laser pulse. (B) LFP decay traces at 610 nm for GFT (black), MAP (red) and a mixture of GFT/MAP. All measurements were performed in deaerated ethanol at λexc = 355 nm at concentrations of 40 μM for GFT and 30 mM for MAP. | ||
From a photobiological point of view, triplet excited species are key intermediates that can induce damage to proteins and other biological targets.33 This can involve radical pathways initiated by electron transfer or hydrogen abstraction (type I mechanism), and/or energy transfer from a photosensitizer to molecular oxygen, leading to singlet oxygen (1O2 type II mechanism).34,35 In this context, it has been observed by means of LFP (λexc = 355 nm) that GFT can induce formation of 1O2, which has been detected by time-resolved NIR emission at 1270 nm. In the case of GFT, the singlet oxygen quantum yields, determined using ketoprofen as a reference,36 were of about 0.17 and 0.1 in aerated toluene and MeCN, respectively; this agrees with the enhanced triplet formation in the former.
As GFT is phototoxic to cells (see above) and membrane proteins are major targets for photosensitized oxidation,37,38 the photobehavior of GFT was investigated in the presence of human serum albumin, a model protein which is the most abundant in plasma.21 It is known that GFT highly binds to HSA.39 Selective excitation of the protein-bound gefitinib at 340 nm (see Fig. S10 in the ESI†) evidenced a significant enhancement of its fluorescence compared with the drug free in aqueous solution, which is insignificant (see Fig. 7A). The stronger emission of complexed gefitinib may result from the higher restrictions in its degrees of freedom for conformational relaxation within HSA. Interestingly, the spectrum profile displays its maximum at ∼390 nm, and decays with a lifetime of about 1.3 ns, showing a very similar behavior to that observed in cyclohexane (see Table 1). Accordingly, the excited species detected for GFT@HSA can again be associated with LE singlet states. However, the lower fluorescence quantum yield of the drug within the protein compared with cyclohexane (0.02 vs. 0.19) is worth noting; this decrease can be the result of an electron transfer process to GFT in its excited state from appropriate donors, for instance the only tryptophan (Trp) residue of HSA.40 A similar process was previously observed for other drug@HSA systems.41,42 In order to check this possibility, application of the Weller equation,43 considering the singlet energy of GFT and the corresponding redox potentials,27 agrees with an exergonic electron transfer from Trp to the excited drug (ΔG = −18.5 kcal mol−1). As a matter of fact, the feasibility of this process was confirmed experimentally from fluorescence measurements. Thus, decay kinetics of 1GFT* was recorded in the non-polar solvent cyclohexane in the presence of increasing amounts of 3-methylindole, the chromophore present in the Trp residue (see Fig. S11 in the ESI†); a quenching rate constant of ∼4.3 × 1010 M−1 s−1 was determined. Therefore, the low ϕF observed for gefitinib within HSA can be explained as a result of an electron transfer from Trp to the LE 1GFT*.
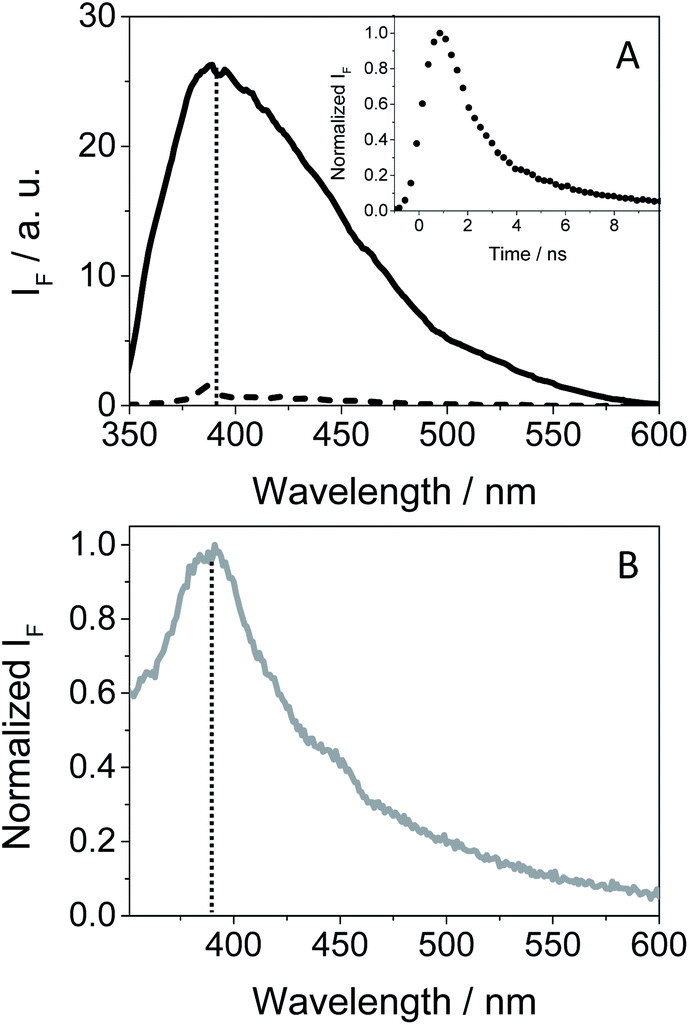 | ||
| Fig. 7 (A) Fluorescence spectra of isoabsorptive solutions at the excitation wavelength for GFT in the bulk aqueous solution (dashed line) and GFT@HSA at 1:1 molar ratio (solid line). The inset shows the normalized fluorescence decay for GFT@HSA at 1:1 molar ratio (10 μM) in PBS. (B) Normalized fluorescence spectra of GFT within HaCaT cells. All measurements were performed at λexc = 340 nm. The vertical dotted line marks the maximum emission at ca. 390 nm. | ||
An interesting point to highlight is the similarity of the emission spectra detected for GFT within HSA and HaCaT cells after selective excitation of the drug at 340 nm. As it can be observed, the emission of the drug in a cellular milieu (see Fig. 7B) was centered at the same position as in the protein (∼390 nm), showing a slightly higher quantum yield of about 0.05 (see Table 1). This may suggest that the photobehavior of GFT in the protein environment is similar to what could be expected in cells.
In order to obtain further information about the early processes occurring inside HSA, femtosecond transient absorption measurements were performed upon selective excitation of the protein-bound gefitinib at 330 nm. This resulted in the formation of a single transient band with a maximum at ∼460 nm (see Fig. 8A), assigned to LE 1GFT*. This species decays following a multi-exponential law (see Fig. 8B), which can be associated with parallel processes arising from the drug located in different binding sites of HSA. Thus, the shortest component, of ca. 5 ps, can be related to the binding of GFT in a site close to the Trp residue, where the electron transfer process from the amino acid to 1GFT* might take place. By contrast, the longest component, which persists up to the nanosecond time-scale, could correspond to the location of GFT in another site far from Trp, where the electron transfer process cannot occur.
 | ||
| Fig. 8 Femtosecond transient absorption spectra from 0.5 ps (black line) to 2 ns (blue line) (A), and decay trace at 460 nm (B) for GFT@HSA at 1:1 molar ratio in aerated PBS at λexc = 330 nm. | ||
It should be emphasized that 3GFT* was hardly detectable in the protein medium, where the band at ∼600 nm is marginal. In this regard, nanosecond LFP measurements on GFT@HSA at λexc = 355 nm indicated a very weak absorption around 600 nm compared with MeCN (see Fig. S12 in the ESI†). This may be the result of the LE 1GFT* quenching through electron transfer from the Trp residue of HSA. As stated above, this process might occur in about 5 ps, which is faster than ISC (∼10 ps); consequently, the yield of 3GFT* within HSA is greatly decreased. This has clear biological implications, since the photosensitization of GFT in biological media could involve the participation of LE singlet states rather than 3GFT*.
In this context, as the electron transfer process from Trp to 1GFT* has been detected not only in solution but also in the HSA-bound drug, it appeared interesting to evaluate the capability of GFT to induce protein photooxidation, since this process could be the origin of the above mentioned GFT-photosensitized damage occurring in HaCaT cells. To this end, the protein carbonylation method was used, which represents the most frequent irreversible oxidative modification affecting proteins. In this regard, PBS solutions containing HSA and GFT were irradiated at different UVA light doses (5, 10 and 15 J cm−2), and the carbonyl content, as an early biomarker of oxidative damage, was quantified using 2,4-dinitrophenylhydrazine (DNPH). The results shown in Fig. 9 revealed that GFT promotes a consistent photooxidative effect towards HSA, which agrees with the results obtained from the phototoxicity NRU assay.
 | ||
| Fig. 9 Protein photooxidation by GFT. Solutions of HSA (5 mg mL−1) in the presence or absence of 50 μM or 100 μM of GFT were irradiated at 5 J cm−2 UVA dose (dark gray bar), 10 J cm−2 (light gray bar) and 15 J cm−2 (empty bar) of UVA dose or kept under dark conditions (black bar). The carbonyl content was quantified spectrophotometrically after derivatization with 2,4-dinitrophenylhydrazine (DNPH). Data represent the mean ± SD of 4 independent experiments. Asterisks indicate significant differences relative to the carbonyl content in HSA in darkness by the t-Student test (*p < 0.05, **p < 0.01, ***p < 0.001, ns: non-significant). | ||
Scheme 1 summarizes the main species generated upon excitation of the drug in different media. In all cases, an instantaneous formation of LE 1GFT* is observed. In non-polar solvents, this species emits light at wavelengths around 380 nm within ca. 1 ns; in addition, LE 1GFT* undergoes ISC to the triplet excited state (∼10 ps), which displays a maximum at ca. 600 nm. By contrast, in polar solvents, LE rapidly evolves (∼1 ps) towards the formation of ICT states, which emit at longer wavelengths (∼470 nm) with much lower yields. In these media, 3GFT* is mainly populated from ICT states; surprisingly, it is formed in very low efficiency in ethanol solution, where conversion of ICT 1GFT* to 3GFT* is thermodynamically disfavored. Finally, in the biological environment, i.e. HSA and HaCaT cells, LE 1GFT* is the only detected species; its lifetime is significantly decreased through ET-quenching by electron donors.
 | ||
| Scheme 1 Schematic representation of the main species arising from the excited gefitinib in different environments: non-polar or polar organic solvents, and biological media such as protein or cells. | ||
Conclusions
The photophysical behavior of GFT has been investigated in solution and in biological environments, from the femtosecond to the microsecond time-scales, whereas the photosensitizing properties of the drug have been studied by means of the NRU and protein carbonylation methods. In vitro NRU assay using human keratinocytes (HaCaT) has proven the phototoxic potential of GFT. The main excited species arising from selective irradiation of the drug are the locally excited (LE) and intramolecular charge transfer (ICT) singlet states, as well as the triplet state. In general, the LE singlet is the only emitting species both in organic non-polar solution and in biological media (i.e. HSA and HaCaT cells). In the former, intersystem crossing to the triplet excited state of GFT occurs in the picosecond scale. By contrast, in organic polar solvents, LE states rapidly evolve towards the formation of ICT states. This species emits at longer wavelengths and shows higher lifetimes than LE states; they are also able to populate 3GFT* in acetonitrile. Surprisingly, ISC is not observed in ethanol, since ICT states are rapidly deactivated (in about 0.7 ns); however, 3GFT* is generated in this solvent by photosensitization with 2-methoxyacetophenone as an energy donor. In the HSA binding sites, formation of 3GFT* is hardly detected; instead, quenching of its LE singlet precursor by Trp through an electron transfer mechanism is observed. Accordingly, GFT photosensitized oxidation of HSA is demonstrated using the protein carbonylation method. In summary, a good correlation is established between the photophysical behavior and the photobiological properties of GFT, which provides a mechanistic basis for the observed phototoxicity.Author contributions
Research was conceived by all authors. Experiments were performed by L. T. and M. O., with the aid of I. A. and I. V. The research was supervised by I. A., I. V. and M. A. M. All authors contributed to the writing of the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Government (RYC-2015-17737, CTQ2017-89416-R, BEAGAL 18/00211, FPU19/00048 and PID2020-115010RB-I00; the Carlos III Institute (ISCIII) of Health, grants: PI16/01877, and CPII16/00052) and Consellería d’Educació, Cultura i Esport (PROMETEO/2017/075) is gratefully acknowledged.References
- S. R. Hubbard and J. H. Till, Annu. Rev. Biochem., 2000, 69, 373–398 CrossRef CAS PubMed.
- M. J. Wieduwilt and M. M. Moasser, Cell. Mol. Life Sci., 2008, 65, 1566–1584 CrossRef CAS PubMed.
- R. I. Nicholson, J. M. W. Gee and M. E. Harper, Eur. J. Cancer, 2001, 37, S9–S15 CrossRef CAS PubMed.
- L. Huang, S. Jiang and Y. Shi, J. Hematol. Oncol., 2020, 13, 143 CrossRef PubMed.
- J. Mendelsohn and J. Baselga, Oncogene, 2000, 19, 6550–6565 CrossRef CAS PubMed.
- C. Pottier, M. Fresnais, M. Gilon, G. Jerusalem, R. Longuespee and N. E. Sounni, Cancers, 2020, 12, 731 CrossRef CAS PubMed.
- T. Yamaoka, S. Kusumoto, K. Ando, M. Ohba and T. Ohmori, Int. J. Mol. Sci., 2018, 19, 3491 CrossRef PubMed.
- N. E. Hynes and H. A. Lane, Nat. Rev. Cancer, 2005, 5, 341–354 CrossRef CAS PubMed.
- M. H. Cohen, G. A. Williams, R. Sridhara, G. Chen and R. Pazdur, Oncologist, 2003, 8, 303–306 CrossRef CAS PubMed.
- A. Arora and E. M. Scholar, J. Pharmacol. Exp. Ther., 2005, 315, 971–979 CrossRef CAS PubMed.
- R. J. Cersosimo, Expert Opin. Drug Saf., 2006, 5, 469–479 CrossRef CAS PubMed.
- B. L. Diffey and I. E. Kochevar, in Photodermatology, ed. H. W. Lim, H. H. Hönigsmann and J. L. M. Hawk, Informa Healthcare, USA, 1st edn, 2007 Search PubMed.
- M. Gonçalo, in Contact Dermatitis, ed. J. D. Johansen, Springer-Verlag, Berlin, 2011, ch. 18, pp. 361–376 Search PubMed.
- S. Lembo, A. Raimondo, V. Conti and M. Venturini, Photodermatol., Photoimmunol. Photomed., 2020, 36, 172–178 CrossRef PubMed.
- K. R. Stein and N. S. Scheinfeld, Expert Opin. Drug Saf., 2007, 6, 431–443 CrossRef CAS PubMed.
- T. P. Selvam and P. V. Kumar, Res. Pharm., 2011, 1, 1–21 Search PubMed.
- G. García-Laínez, I. Vayá, M. P. Marín, M. A. Miranda and I. Andreu, Arch. Toxicol., 2021, 95, 169–178 CrossRef PubMed.
- I. Vayá, I. Andreu, E. Lence, C. González-Bello, M. C. Cuquerella, M. Navarrete-Miguel, D. Roca-Sanjuan and M. A. Miranda, Chem.–Eur. J., 2020, 26, 15922–15930 CrossRef PubMed.
- I. Andreu, E. Lence, C. Gonzalez-Bello, C. Mayorga, M. C. Cuquerella, I. Vayá and M. A. Miranda, Front. Pharmacol., 2020, 11, 576495 CrossRef CAS PubMed.
- https://ec.europa.eu/health/documents/community-register/2009/2009062459389/anx_59389_en.pdf .
- T. Peters, in All About Albumin - Biochemistry, Genetics, and Medical Applications, ed. Elsevier, Academic Press, San Diego, 1995, ch. 3, pp. 76–132 Search PubMed.
- A. R. Svobodova, J. Ulrichova and J. Vostalova, An. Bras. Dermatol., 2019, 94, 105–106 CrossRef PubMed.
- F. Palumbo, G. Garcia-Lainez, D. Limones-Herrero, M. D. Coloma, J. Escobar, M. C. Jimenez, M. A. Miranda and I. Andreu, Toxicol. Appl. Pharmacol., 2016, 313, 131–137 CrossRef CAS PubMed.
- G. Colombo, M. Clerici, M. E. Garavaglia, D. Giustarini, R. Rossi, A. Milzani and I. Dalle-Donne, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1019, 178–190 CrossRef CAS PubMed.
- B. J. Trummer, V. Iyer, S. V. Balu-Iyer, R. O'Connor and R. M. Straubinger, J. Pharm. Sci., 2012, 101, 2763–2776 CrossRef CAS PubMed.
- O. Domotor, K. Pelivan, A. Borics, B. K. Keppler, C. R. Kowol and E. A. Enyedy, J. Pharm. Biomed. Anal., 2018, 154, 321–331 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Taylor and Francis Group, Boca Raton, FL, 2006 Search PubMed.
- G. Cosa and J. C. Scaiano, Photochem. Photobiol., 2004, 80, 159–174 CrossRef CAS PubMed.
- C. Ruckebusch, M. Sliwa, P. Pernot, A. de Juan and R. Tauler, J. Photochem. Photobiol., C, 2012, 13, 1–27 CrossRef CAS.
- I. Vayá, V. Lhiaubet-Vallet, M. C. Jimenez and M. A. Miranda, Chem. Soc. Rev., 2014, 43, 4102–4122 RSC.
- L. J. Martínez and J. C. Scaiano, Photochem. Photobiol., 1998, 68, 646–651 CrossRef.
- O. R. Alzueta, M. C. Cuquerella and M. A. Miranda, Spectrochim. Acta, Part A, 2019, 218, 191–195 CrossRef CAS PubMed.
- M. J. Davies, Photochem. Photobiol. Sci., 2004, 3, 17–25 CrossRef CAS PubMed.
- M. S. Baptista, J. Cadet, P. Di Mascio, A. A. Ghogare, A. Greer, M. R. Hamblin, C. Lorente, S. C. Nunez, M. S. Ribeiro, A. H. Thomas, M. Vignoni and T. M. Yoshimura, Photochem. Photobiol., 2017, 93, 912–919 CrossRef CAS PubMed.
- C. S. Foote, Photochem. Photobiol., 1991, 54, 659 CrossRef CAS PubMed.
- D. de la Peña, C. Martí, S. Nonell, L. A. Martínez and M. A. Miranda, Photochem. Photobiol., 1997, 65, 828–832 CrossRef.
- M. J. Davies and R. J. W. Truscott, in Sun Protection in Man, ed. P. U. Giacomoni, Elsevier, 2001, ch. 12, pp. 251–275 Search PubMed.
- D. I. Pattison, A. S. Rahmanto and M. J. Davies, Photochem. Photobiol. Sci., 2012, 11, 38–53 CrossRef CAS PubMed.
- J. Li, J. Brahmer, W. Messersmith, M. Hidalgo and S. D. Baker, Invest. New Drugs, 2006, 24, 291–297 CrossRef CAS PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052–1061 CAS.
- I. Vayá, M. C. Jimenez and M. A. Miranda, J. Phys. Chem. B, 2007, 111, 9363–9371 CrossRef PubMed.
- I. Vayá, P. Bonancia, M. C. Jimenez, D. Markovitsi, T. Gustavsson and M. A. Miranda, Phys. Chem. Chem. Phys., 2013, 15, 4727–4734 RSC.
- A. Z. Weller, Phys. Chem., 1982, 133, 93–98 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03154f |
| This journal is © The Royal Society of Chemistry 2021 |