
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical-mediated vicinal addition of alkoxysulfonyl/fluorosulfonyl and trifluoromethyl groups to aryl alkyl alkynes†
Xinrui
Dong‡
 a,
Wenhua
Jiang‡
a,
Dexiang
Hua
a,
Xiaohui
Wang
a,
Liang
Xu
*b and
Xiaoxing
Wu
*a
a,
Wenhua
Jiang‡
a,
Dexiang
Hua
a,
Xiaohui
Wang
a,
Liang
Xu
*b and
Xiaoxing
Wu
*a
aState Key Laboratory of Natural Medicines, Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Nanjing 211198, China. E-mail: wu_xiaoxing@cpu.edu.cn
bSchool of Chemistry and Chemical Engineering, Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan, Shihezi University, Shihezi 832003, China. E-mail: xuliang4423@shzu.edu.cn
First published on 3rd August 2021
Abstract
The addition of sulfonyl radicals to alkenes and alkynes is a valuable method for constructing useful highly functionalized sulfonyl compounds. The underexplored alkoxy- and fluorosulfonyl radicals are easily accessed by CF3 radical addition to readily available allylsulfonic acid derivatives and then β-fragmentation. These substituted sulfonyl radicals add to aryl alkyl alkynes to give vinyl radicals that are trapped by trifluoromethyl transfer to provide tetra-substituted alkenes bearing the privileged alkoxy- or fluorosulfonyl group on one carbon and a trifluoromethyl group on the other. This process exhibits broad functional group compatibility and allows for the late-stage functionalization of drug molecules, demonstrating its potential in drug discovery and chemical biology.
Introduction
Sulfonic acid derivatives are valuable pharmaceutical compounds, biochemical probes, novel materials, and synthetic intermediates (Scheme 1a).1 Moreover, sulfur(VI)-fluoride exchange (SuFEx) has recently gained intense interest as one of the most powerful reactions in click chemistry.2 This stems from the incredible stability and unique reactivity of the fluorosulfonyl (SO2F) motif and provides an additional need for the development of efficient fluorosulfonylation methods. Sulfonate esters3 and sulfonyl fluorides4 are usually obtained from the corresponding sulfonyl chlorides. However, these sulfonyl chloride precursors are generally prepared under harsh conditions with highly corrosive reagents or by the oxidative chlorination of organosulfur compounds.5 Consequently, these methods are not compatible with sensitive functional groups and often suffer from tedious purification procedures or limited sources of the organosulfur starting materials. Recent methods for introduction of a sulfonate moiety focus on arylsulfonates,6 while the diversity-oriented synthesis of alkenylsulfonate esters by Wittig reactions is limited by the strongly basic conditions employed.1d,7 Alkenylsulfonyl fluorides are of special interest due to the electrophilic character of both the sulfur center and the β-carbon, representing a unique warhead for covalent binders.1j To access alkenylsulfonyl fluorides, the groups of Sharpless, Arvidsson, Qin, Yu, Huestis, Willis, and others reported transition-metal catalyzed cross-coupling reactions with ethenesulfonyl fluoride (ESF),8 1-bromoethenesulfonyl fluoride (BESF),9 or 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) (DABSO)/N-fluorobenzenesulfonimide (NFSI).10 These reactions work well, but don't provide access to tetra-substituted alkenyl sulfonyl fluorides that should also be valuable for SuFEx click chemistry.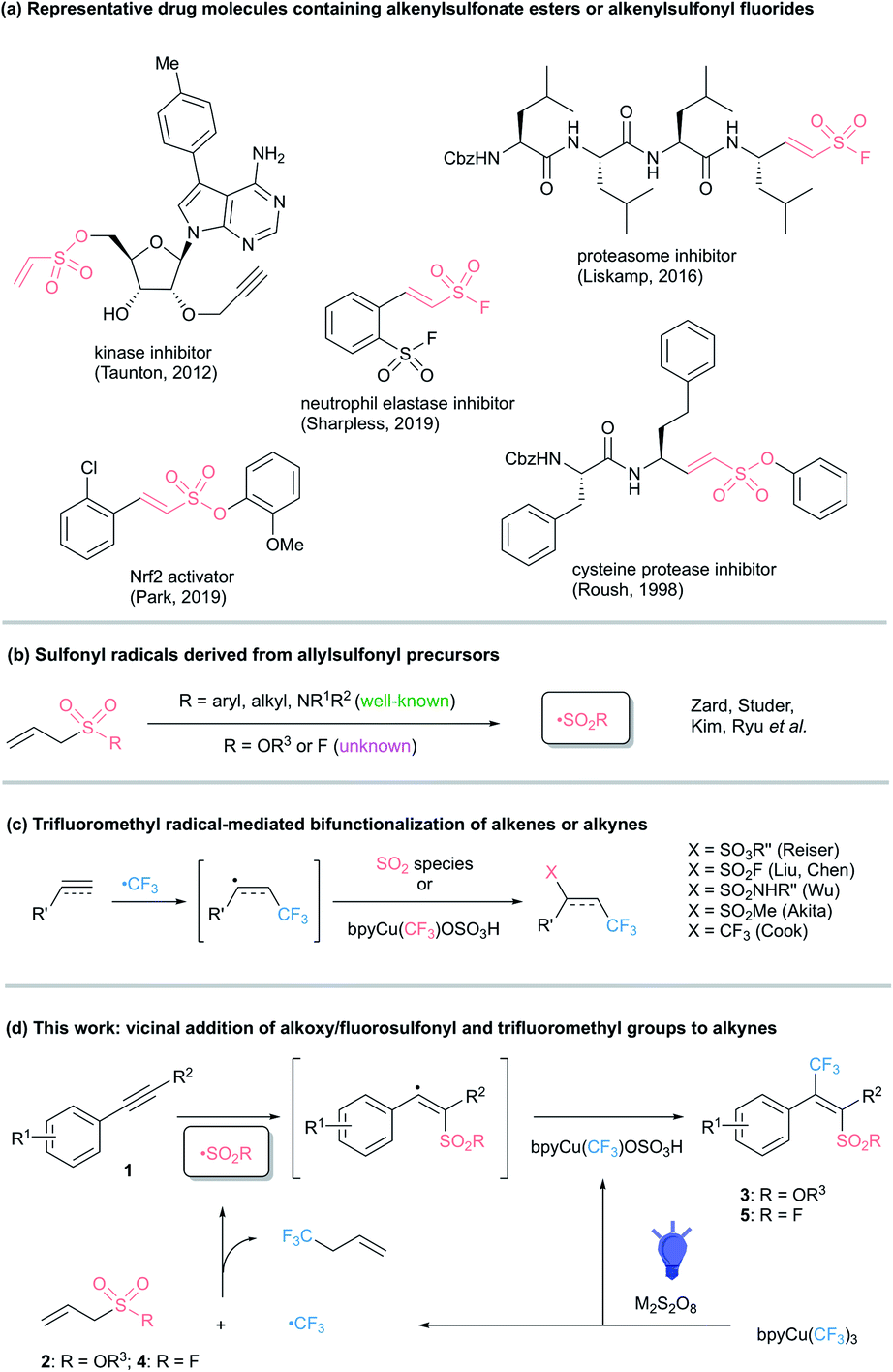 | ||
| Scheme 1 Alkenyl SO3R- or SO2F-containing drug molecules and sulfonyl radical-involved reactions. | ||
Recently, sulfonyl radical-mediated processes have been developed that provide direct methods to construct valuable carbon–SO2 bonds.11 In this context, alkoxysulfonyl radicals (ROSO2˙)12 were found to add to alkenes in hot benzene.13 Later, other groups reported the cyclization of alkoxysulfonyl radicals tethered to alkenes and alkynes.14,15 Unfortunately, these methods either suffer from narrow substrate scope or restriction to intramolecular processes, limiting their applicability in the context of late-stage functionalization of complex molecules. The use of the fluorosulfonyl radical (FSO2˙) in organic synthesis has rarely been reported, presumably due to its instability and limited synthetic access.16 In 2021, the Liao group disclosed a radical fluorosulfonylation of alkenes using sulfuryl chlorofluoride (FSO2Cl) as the fluorosulfonyl radical source.17 Thus, the discovery of new SO2-containing reagents and the development of efficient synthetic methods to access alkoxy- or fluorosulfonyl compounds are of great significance. Allylsulfonyl compounds have been recognized as facile and practical precursors for generating such radical species through an addition–fragmentation process (Scheme 1b).18 However, only aryl, alkyl, and amide groups have been used as substituents on the allylsulfonyl reagents. The generation of alkoxy- or fluorosulfonyl radicals by this process has not been reported. This prompted us to explore the use of alkyl allylsulfonates and allylsulfonyl fluorides to form useful carbon–SO2 bonds by radical processes.
Trifluoromethyl compounds have been prepared by direct trifluoromethylation with the CF3 radical.19 Thus, a few groups have independently reported the addition of the CF3 radical to alkenes or alkynes followed by trapping to form sulfonic acid derivatives (Scheme 1c).20 In our continuing efforts to discover new trifluoromethylation methods,21 we envisioned that the addition of an alkoxysulfonyl or fluorosulfonyl radical to an alkyne or alkene followed by trapping with a trifluoromethyl moiety would provide an alternative route to vicinal alkoxy- or fluorosulfonyltrifluoromethylation (Scheme 1d). This would give 3 and 5 with the opposite regiochemistry to the reactions shown in Scheme 1c, opening up access to new scaffolds for drug discovery and chemical biology. To realize this conception, we sought to use readily-prepared allylsulfonyl reagents as the sulfonyl radical sources, in combination with commercially-available bpyCu(CF3)3.22 This was inspired by the recent work described by Cook, in which photo-induced bistrifluoromethylation of terminal alkynes was efficiently realized in the presence of bpyCu(CF3)3 and persulfate (Scheme 1c).23 As shown in Scheme 1d, the CF3 radical could react preferentially with the unhindered double bond of alkyl allylsulfonate 2 or allylsulfonyl fluoride 4, rather than the triple bond in alkyne 1 that would initiate the bistrifluoromethylation reported by Cook. Subsequent β-fragmentation would lead to RSO2˙ and volatile 4,4,4-trifluoro-1-butene.24 The addition of RSO2˙ across the triple bond of 1 followed by carbon–CF3 bond formation with bpyCu(CF3)OSO3H would produce trifluoromethylalkenylsulfonate esters 3 and trifluoromethylalkenylsulfonyl fluorides 5, respectively. These tetra-substituted alkenes are challenging to access through known cross-coupling methods.8–10
Results and discussion
Reaction condition optimization
The optimized reaction conditions are outlined in Table 1 (see ESI† for details). Treatment of alkyne 1a25 with methyl allylsulfonate 2a (3 equiv.), bpyCu(CF3)3 (1.2 equiv.), and (NH4)2S2O8 (3 equiv.) under blue LED irradiation (λmax = 460 nm) affords 77% (19F NMR yield) of the desired CF3-containing alkenylsulfonate 3a (Table 1, entry 1). The yield is lower with sodium or potassium persulfate, and only 4% of 3a is obtained in the absence of (NH4)2S2O8 (Table 1, entries 2–4). The yield is slightly lower in the absence of water (Table 1, entry 5). The reaction does not proceed in the dark and the yield is 13% lower under irradiation with 365 nm light (Table 1, entries 6 and 7). Under most conditions, an approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 3a and the isomer Z-3a is obtained (see ESI† for details). The methyl protons of 3a absorb at δ 2.44 in the 1H NMR spectra, whereas those of the minor isomer Z-3a absorb at δ 2.01 due to shielding by the aryl group. The selectivity likely arises from the greater steric bulk of the methoxysulfonyl moiety as compared to the methyl group. To our delight, bistrifluoromethylated product bis-3a was produced in no more than 15% 19F NMR yield under the standard conditions. This allowed a good isolated yield (73%) of desired product 3a, whose structure was unambiguously determined by single-crystal X-ray diffraction.
:1 mixture of 3a and the isomer Z-3a is obtained (see ESI† for details). The methyl protons of 3a absorb at δ 2.44 in the 1H NMR spectra, whereas those of the minor isomer Z-3a absorb at δ 2.01 due to shielding by the aryl group. The selectivity likely arises from the greater steric bulk of the methoxysulfonyl moiety as compared to the methyl group. To our delight, bistrifluoromethylated product bis-3a was produced in no more than 15% 19F NMR yield under the standard conditions. This allowed a good isolated yield (73%) of desired product 3a, whose structure was unambiguously determined by single-crystal X-ray diffraction.
|
|
||
|---|---|---|
| Entry | Reaction conditions | Yield of 3a/Z-3ab |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), bpyCu(CF3)3 (0.24 mmol), (NH4)2S2O8 (0.6 mmol) under blue LED irradiation for 4 h at room temperature. b Yields were determined by 19F NMR spectroscopy with trifluoromethoxybenzene as an internal standard. c Isolated yield. | ||
| 1 | Standard conditions | 77% (73%)c/6% |
| 2 | Na2S2O8 instead of (NH4)2S2O8 | 52%/5% |
| 3 | K2S2O8 instead of (NH4)2S2O8 | 46%/4% |
| 4 | Without (NH4)2S2O8 | 4%/0% |
| 5 | Without H2O | 64%/8% |
| 6 | 365 nm instead of blue LEDs | 64%/5% |
| 7 | Performed in dark | 0%/0% |
| 8 | 2 equiv. of 2a used instead | 66%/7% |
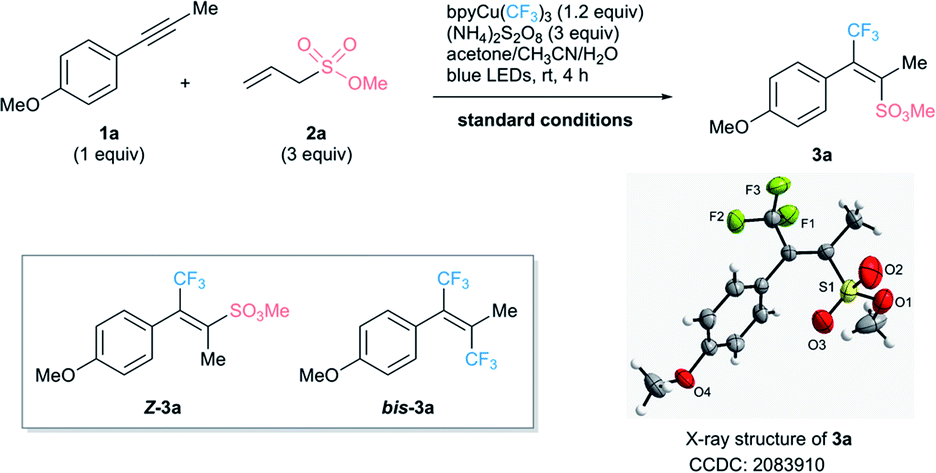
The use of only 2 equiv. of 2a leads to a decreased yield (66%) of 3a and the formation of more bis-3a (26%) as expected (Table 1, entry 8).
Substrate scope
With the optimized protocol in hand, we explored the generality of this transformation on a range of alkynes with several alkyl allylsulfonates (Scheme 2). Various functional groups, such as halides, ethers, aldehydes, esters, amides, sulfonamides, nitriles, and hydroxyl groups are well tolerated, giving products 3a–3aq in moderate to good yields. Meta-substitution on the aryl ring has little effect on the reaction efficiency (3w–3y). With ortho-substitution on the aryl ring, the yield of 3z is higher than that of 3aa, presumably because a hydroxy group is smaller than a methoxy group. As exemplified by 3v, aryl alkyl alkynes are much more reactive than bisalkyl alkynes. We have established that a variety of heteroarenes, such as pyrazole, (benzo)thiophenes, and furans, are suitable substrates for this transformation as shown in 3ac–3ag. The stereochemistry of the major products was assigned by analogy to that of 3a and by the chemical shift of the methyl group at δ 2.51–2.41.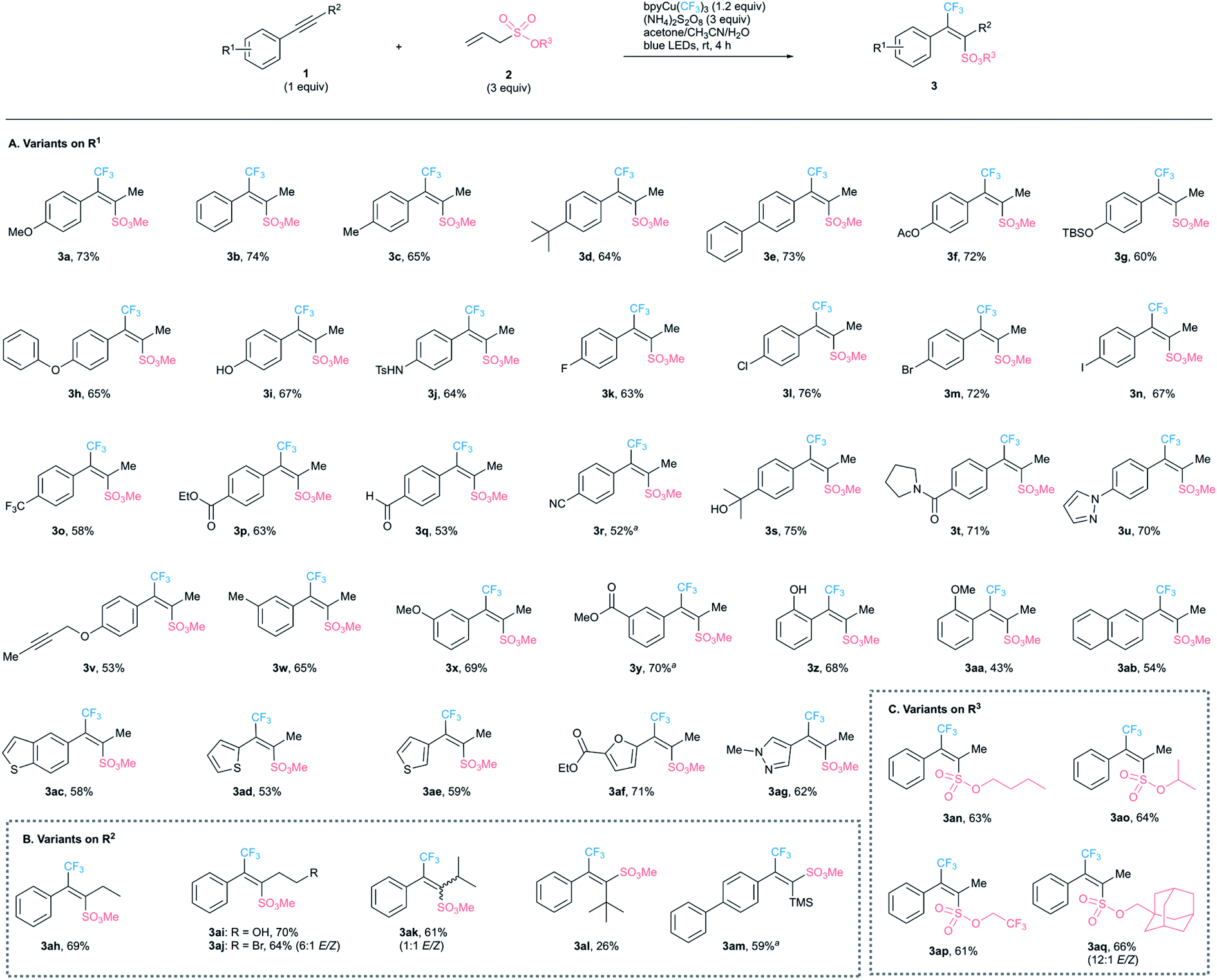 | ||
| Scheme 2 Substrate scope for alkoxysulfonyltrilfuoromethylation of alkynes. Unless otherwise noted, all the reactions were run on a 0.2 mmol scale under standard conditions. a 1.5 equiv. of bpyCu(CF3)3 was used for 8 h. | ||
This reaction is compatible with primary, secondary, and tertiary alkyl groups and trimethylsilyl group as the R2 substituent on the alkyne (3ah–3am). Intriguingly, the configuration in products shifts from anti- to syn-addition with the increased bulkiness of the R2 group. For instance, a nearly 1:1 E/Z mixture is obtained for 3ak with an isopropyl group, while exclusively syn-addition occurs to give 3al with a tert-butyl group and 3am with a TMS substituent. The structures of 3al and 3am were determined by single-crystal X-ray analysis (see ESI† for details). These results indicate that the methoxysulfonyl group is larger than a methyl group, smaller than a tert-butyl or TMS group, and about the same size as an isopropyl group. Other alkyl allylsulfonates smoothly undergo this transformation to afford 3an–3aq in good yields.
Considering the high value of SO2F-containing compounds in SuFEx click chemistry, we next examined the reaction of alkynes 1 with allylsulfonyl fluoride 4 (Scheme 3). To our delight, a variety of alkynes undergo this transformation smoothly to afford the desired products 5a–5o using 365 nm irradiation. In contrast to the reaction with methyl allylsulfonate 2 to give 3, which worked slightly better with blue LED than 365 nm irradiation, the yields of 5 are 10–20% lower with blue LED irradiation. The optimal wavelength appears to be a balance between its efficiency in generating trifluoromethyl radicals and photo-induced side reactions.
 | ||
| Scheme 3 Substrate scope for fluorosulfonyltrifluoromethylation of alkynes. Unless otherwise noted, all the reactions were run on a 0.2 mmol scale under standard conditions. a 4.0 equiv. of 4 was used for 8 h. | ||
A variety of electron-donating or electron-withdrawing aryl substituents and heteroarenes (5h, 5l, and 5m) are compatible with these reaction conditions. Anti-addition occurs predominately for alkynes with primary alkyl R2 substituents (5a–5n), and syn-addition to give 5o occurs exclusively with the bulky tert-butyl group. The structure of 5o was established by single-crystal X-ray analysis (see ESI† for details). In comparison to the alkoxysulfonyl trifluoromethylation discussed above, slightly increased amounts of undesired Z-isomers and bistrifluoromethylated products were detected. This might arise from the subtle differences in size, stability, and reactivity between fluorosulfonyl and alkoxysulfonyl radicals.12a,16b,c
Synthetic application
To demonstrate the potential of this transformation, we applied this methodology to the late-stage functionalization of both drug-derived alkynes and allylsulfonates. As shown in Scheme 4, alkynes derived from estrone (3ar and 5p), ezetimibe (3as), 4-methylumbelliferone (3at and 5q), and fenofibrate (3au and 5r) react smoothly to afford the expected products in moderate to good yields. In addition, the etofylline-derived allylsulfonate (3av) undergo alkoxysulfonyl trifluoromethylation as well. | ||
| Scheme 4 Late-stage functionalization of complex drug molecules. | ||
To further demonstrate the synthetic utility of the products, sulfonate ester 3a was efficiently converted to sulfonamide 6 in 68% yield (Scheme 5). Nucleophilic substitution on sulfonyl fluoride 5f was achieved by reaction with imidazole to afford sulfonamide 7 in 48% yield, with TMSN3 to afford sulfonyl azide 8 in 71% yield, and with phenol to afford sulfonate ester 9 in 83% yield. This suggests that this method is applicable to construct molecular complexity and diversity, which is beneficial for drug discovery and chemical biology.
 | ||
| Scheme 5 Derivatization on alkenyl sulfonate esters or alkenyl sulfonyl fluorides. | ||
Computational studies
We then performed DFT calculations to provide insights on the reaction mechanism and selectivity of this process (Fig. 1, see ESI† for details). On the basis of Cook's work22c,23 and our TEMPO-trapping studies (see ESI† for details), the alkyne 1b, methyl allylsulfonate 2a, and the initial products upon light irradiation, the CF3 radical and bpyCu(CF3)OSO3H, were chosen as the starting point. The energy of TS1, the transition state for the addition of the CF3 radical to methyl allylsulfonate 2a to give INT1, was calculated to be 9.2 kcal mol−1. This is 0.8 kcal mol−1 lower than the energy of TSA, the transition state for the addition of the CF3 radical to the alkyne to give INTA. The newly-formed intermediates INT1 and INTA are more stable than the corresponding transition states TS1 and TSA by >30 kcal mol−1, rendering the radical addition processes irreversible. The steps from INT1 to final product 3 proceed readily because the activation barriers in the subsequent transition states are all less than 13 kcal mol−1. The calculated energy for TS1 that is 0.8 kcal mol−1 lower than that of TSA is consistent with the observed preferential formation of methoxysulfonyltrifluoromethylation to form 3/Z-3 rather than bistrifluoromethylation to form bis-3. Moreover, the transition state TS4E leading to the formation of anti-addition product 3 is 1.7 kcal mol−1 more stable than transition state TS4Z leading to syn-addition product Z-3, in good agreement with the approximately 10:1 ratio of E- and Z-isomers observed.
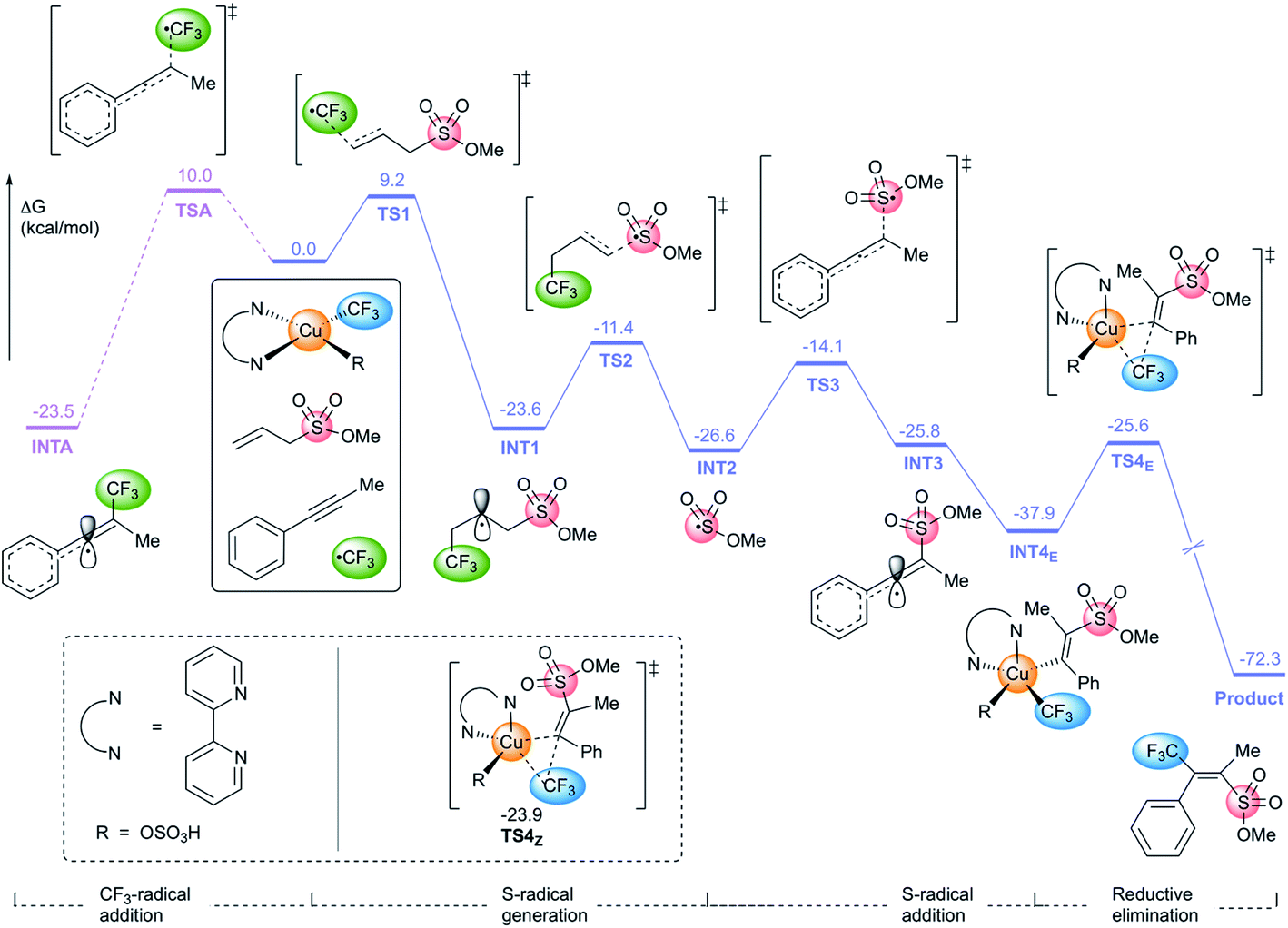 | ||
| Fig. 1 Computational energy profiles in alkoxysulfonyl trifluoromethylation of alkynes. | ||
Conclusion
In conclusion, a method for alkoxy- and fluorosulfonyltrifluoromethylation of alkynes has been developed. This process uses readily-prepared alkyl allylsulfonates or allylsulfonyl fluoride as sulfonyl radical donors and good selectivity is achieved. Numerous functional groups and heteroarenes are tolerated, allowing access to a broad range of tetra-substituted alkenes that are valuable for drug discovery, chemical biology and other fields. In addition, this methodology further expands the application of allylsulfonyl reagents in radical coupling reactions.Data availability
The datasets (experimental procedures, characterization, copies of NMR spectra for all new compounds, mechanistic studies, and computational studies) supporting this article have been uploaded as part of the ESI.† Crystallographic data for 3a, 3al, 3am, and 5o has been deposited at the CCDC under 2083910–2083913 and can be obtained from https://www.ccdc.cam.ac.uk/structures.Author contributions
X. D., W. J., D. H., and X. W. planned, conducted, and analysed the experiments. L. X. conducted the computational studies. Prof. X. W. conceived this concept, directed the project, and prepared this manuscript. All authors contributed to discussions and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was received from the National Natural Science Foundation of China (grant 21877128 and 21963010), and the Double First-Class University Project (CPU2018GY03).Notes and references
- (a) A. Haddow and W. C. J. Ross, Nature, 1956, 177, 995–996 CrossRef CAS; (b) S. O. Ciurea and B. S. Andersson, Biol. Blood Marrow Transplant., 2009, 15, 523–536 CrossRef CAS; (c) J. W. Choi, S. J. Shin, H. J. Kim, J.-H. Park, H. J. Kim, E. H. Lee, A. N. Pae, Y. S. Bahn and K. D. Park, ACS Med. Chem. Lett., 2019, 10, 1061–1067 CrossRef CAS; (d) W. R. Roush, S. L. Gwaltney II, J. Cheng, K. A. Scheidt, J. H. McKerrow and E. Hansell, J. Am. Chem. Soc., 1998, 120, 10994–10995 CrossRef CAS; (e) S. M. Pauff and S. C. Miller, Org. Lett., 2011, 13, 6196–6199 CrossRef CAS PubMed; (f) K. A. Pardeshi, G. Ravikumar and H. Chakrapani, Org. Lett., 2018, 20, 4–7 CrossRef CAS PubMed; (g) C.-B. Kim, H. Jo, B.-K. Ahn, C. K. Kim and K. Park, J. Org. Chem., 2009, 74, 9566–9569 CrossRef CAS PubMed; (h) Q. Zheng, J. L. Woehl, S. Kitamura, D. Santos-Martins, C. J. Smedley, G. Li, S. Forli, J. E. Moses, D. W. Wolan and K. B. Sharpless, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18808–18814 CrossRef CAS; (i) N. N. Gushwa, S. Kang, J. Chen and J. Taunton, J. Am. Chem. Soc., 2012, 134, 20214–20217 CrossRef CAS; (j) A. J. Brouwer, N. H. Álvarez, A. Ciaffoni, H. van de Langemheen and R. M. J. Liskamp, Bioorg. Med. Chem., 2016, 24, 3429–3435 CrossRef CAS PubMed; (k) J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Oakdale and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 9466–9470 CrossRef CAS.
- (a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS; (b) A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC; (c) L. Xu and J. Dong, Chin. J. Chem., 2020, 38, 414–419 CrossRef CAS; (d) Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919–2925 CrossRef CAS PubMed; (e) D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS; (f) Q. Li, Q. Chen, P. C. Klauser, M. Li, F. Zheng, N. Wang, X. Li, Q. Zhang, X. Fu, Q. Wang, Y. Xu and L. Wang, Cell, 2020, 182, 85–97 CrossRef CAS; (g) Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS; (h) G. J. Brighty, R. C. Botham, S. Li, L. Nelson, D. E. Mortenson, G. Li, C. Morisseau, H. Wang, B. D. Hammock, K. B. Sharpless and J. W. Kelly, Nat. Chem., 2020, 12, 906–913 CrossRef; (i) P. Martín-Gago and C. A. Olsen, Angew. Chem., Int. Ed., 2019, 58, 957–966 CrossRef; (j) S. Kitamura, Q. Zheng, J. L. Woehl, A. Solania, E. Chen, N. Dillon, M. V. Hull, M. Kotaniguchi, J. R. Cappiello, S. Kitamura, V. Nizet, K. B. Sharpless and D. W. Wolan, J. Am. Chem. Soc., 2020, 142, 10899–10904 CrossRef CAS PubMed; (k) B. Gao, L. Zhang, Q. Zheng, F. Zhou, L. M. Klivansky, J. Lu, Y. Liu, J. Dong, P. Wu and K. B. Sharpless, Nat. Chem., 2017, 9, 1083–1088 CrossRef CAS.
- M. Gagné-Boulet, C. Bouzriba, A. C. C. Alvarez and S. Fortin, Eur. J. Med. Chem., 2021, 213, 113136–113151 CrossRef.
- A. Talko and M. Barbasiewicz, ACS Sustainable Chem. Eng., 2018, 6, 6693–6701 CrossRef CAS.
- (a) L. Zhang, C. Guo, X. Zheng, R. Zhu, X. Yang and C. Tang, Chin. J. Synth. Chem., 2009, 17, 133–139 CAS; (b) S. Madabhushi, R. Jillella, V. Sriramoju and R. Singh, Green Chem., 2014, 16, 3125–3131 RSC; (c) G. K. S. Prakash, T. Mathew, C. Panja and G. A. Olah, J. Org. Chem., 2007, 72, 5847–5850 CrossRef CAS PubMed.
- (a) S. P. Blum, D. Schollmeyer, M. Turks and S. R. Waldvogel, Chem.–Eur. J., 2020, 26, 8358–8362 CrossRef CAS PubMed; (b) X. Hong, Q. Tan, B. Liu and B. Xu, Angew. Chem., Int. Ed., 2017, 56, 3961–3965 CrossRef CAS.
- C. Gennari, B. Salom, D. Potenza and A. Williams, Angew. Chem., Int. Ed., 1994, 33, 2067–2069 CrossRef.
- (a) H.-L. Qin, Q. Zheng, G. A. L. Bare, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2016, 55, 14155–14158 CrossRef CAS; (b) G.-F. Zha, Q. Zheng, J. Leng, P. Wu, H.-L. Qin and K. B. Sharpless, Angew. Chem., Int. Ed., 2017, 56, 4849–4852 CrossRef CAS; (c) P. K. Chinthakindi, K. B. Govender, A. S. Kumar, H. G. Kruger, T. Govender, T. Naicker and P. I. Arvidsson, Org. Lett., 2017, 19, 480–483 CrossRef CAS; (d) C. Li, S.-M. Wang and H.-L. Qin, Org. Lett., 2018, 20, 4699–4703 CrossRef CAS PubMed; (e) X.-Y. Chen, Y. Wu, J. Zhou, P. Wang and J.-Q. Yu, Org. Lett., 2019, 21, 1426–1429 CrossRef CAS; (f) G. Ncube and M. P. Huestis, Organometallics, 2019, 38, 76–80 CrossRef CAS.
- J. Leng, N. S. Alharbi and H.-L. Qin, Eur. J. Org. Chem., 2019, 6101–6105 CrossRef CAS.
- T. S.-B. Lou, S. W. Bagley and M. C. Willis, Angew. Chem., Int. Ed., 2019, 58, 18859–18863 CrossRef CAS.
- (a) S. M. Hell, C. F. Meyer, G. Laudadio, A. Misale, M. C. Willis, T. Noël, A. A. Trabanco and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 720–725 CrossRef; (b) S. Yang, X. Wu, S. Wu and C. Zhu, Org. Lett., 2019, 21, 4837–4841 CrossRef CAS; (c) Y. Ning, Q. Ji, P. Liao, E. A. Anderson and X. Bi, Angew. Chem., Int. Ed., 2017, 56, 13805–13808 CrossRef CAS; (d) A. García-Domínguez, S. Müller and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 9949–9952 CrossRef; (e) X.-J. Tang and W. R. Dolbier Jr, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS; (f) Q. Lu, J. Zhang, G. Zhao, Y. Qi, H. Wang and A. Lei, J. Am. Chem. Soc., 2013, 135, 11481–11484 CrossRef CAS; (g) K. Liu and A. Studer, J. Am. Chem. Soc., 2021, 143, 4903–4909 CrossRef CAS PubMed.
- (a) C. Chatgilialoglu, D. Griller and S. Rossini, J. Org. Chem., 1989, 54, 2734–2737 CrossRef CAS; (b) J. J. Douglas, H. Albright, M. J. Sevrin, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2015, 54, 14898–14902 CrossRef CAS PubMed; (c) M. Bossart, R. Fässler, J. Schoenberger and A. Studer, Eur. J. Org. Chem., 2002, 2742–2757 CrossRef CAS.
- M. S. Heller, D. P. Lorah and C. G. Cox, J. Chem. Eng. Data, 1983, 28, 134–137 CrossRef CAS.
- E. Bonfand, W. B. Motherwell, A. M. K. Pennell, M. K. Uddin and F. Ujjainwalla, Heterocycles, 1997, 46, 523–534 CrossRef CAS.
- L. Cala, O. García-Pedrero, R. Rubio-Presa, F. J. Fañanás and F. Rodríguez, Chem. Commun., 2020, 56, 13425–13428 RSC.
- (a) X. Zeng, H. Beckers and H. Willner, J. Am. Chem. Soc., 2013, 135, 2096–2099 CrossRef CAS PubMed; (b) C. A. McDowell, F. G. Herring and J. C. Tait, J. Chem. Phys., 1975, 63, 3278–3283 CrossRef CAS; (c) Y. R. Sekhar, H. Bill and D. Lovy, Chem. Phys. Lett., 1987, 136, 57–61 CrossRef CAS.
- X. Nie, T. Xu, J. Song, A. Devaraj, B. Zhang, Y. Chen and S. Liao, Angew. Chem., Int. Ed., 2021, 60, 3956–3960 CrossRef CAS.
- (a) B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 1996, 118, 1209–1210 CrossRef CAS; (b) S. Kim and C. J. Lim, Angew. Chem., Int. Ed., 2002, 41, 3265–3267 CrossRef CAS; (c) S. Kim, S. Kim, N. Otsuka and I. Ryu, Angew. Chem., Int. Ed., 2005, 44, 6183–6186 CrossRef CAS; (d) Y. Xia and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9836–9840 CrossRef CAS PubMed; (e) C. Moutrille and S. Z. Zard, Chem. Commun., 2004, 1848–1849 RSC; (f) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940–12944 CrossRef CAS; (g) L. Wang, Y. Xia, K. Bergander and A. Studer, Org. Lett., 2018, 20, 5817–5820 CrossRef CAS.
- (a) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (b) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294–8308 CrossRef CAS; (c) P. Gao, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Chem.–Eur. J., 2015, 21, 7648–7661 CrossRef CAS; (d) S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2020, 26, 11065–11084 CrossRef CAS PubMed.
- (a) Y. Li, Y. Xiang, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 1493–1497 RSC; (b) T. Rawner, M. Knorn, E. Lutsker, A. Hossain and O. Reiser, J. Org. Chem., 2016, 81, 7139–7147 CrossRef PubMed; (c) Y. Liu, H. Wu, Y. Guo, J.-C. Xiao, Q.-Y. Chen and C. Liu, Angew. Chem., Int. Ed., 2017, 56, 15432–15435 CrossRef CAS PubMed; (d) S. Tanaka, Y. Nakayama, Y. Konishi, T. Koike and M. Akita, Org. Lett., 2020, 22, 2801–2805 CrossRef CAS PubMed.
- (a) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962–6966 CrossRef CAS; (b) J. Lei, X. Liu, S. Zhang, S. Jiang, M. Huang, X. Wu and Q. Zhu, Chem.–Eur. J., 2015, 21, 6700–6703 CrossRef CAS PubMed; (c) J. Lei, X. Wu and Q. Zhu, Org. Lett., 2015, 17, 2322–2325 CrossRef CAS PubMed.
- (a) X. Tan, Z. Liu, H. Shen, P. Zhang, Z. Zhang and C. Li, J. Am. Chem. Soc., 2017, 139, 12430–12433 CrossRef CAS; (b) M. Paeth, W. Carson, J.-H. Luo, D. Tierney, Z. Cao, M.-J. Cheng and W. Liu, Chem.–Eur. J., 2018, 24, 11559–11563 CrossRef CAS; (c) S. Guo, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2018, 140, 12378–12382 CrossRef CAS PubMed; (d) H. Shen, Z. Liu, P. Zhang, X. Tan, Z. Zhang and C. Li, J. Am. Chem. Soc., 2017, 139, 9843–9846 CrossRef CAS PubMed; (e) G. Choi, G. S. Lee, B. Park, D. Kim and S. H. Hong, Angew. Chem., Int. Ed., 2021, 60, 5467–5474 CrossRef CAS PubMed; (f) A. M. Romine, N. Nebra, A. I. Konovalov, E. Martin, J. Benet-Buchholz and V. V. Grushin, Angew. Chem., Int. Ed., 2015, 54, 2745–2749 CrossRef CAS PubMed.
- S. Guo, D. I. AbuSalim and S. P. Cook, Angew. Chem., Int. Ed., 2019, 58, 11704–11708 CrossRef CAS.
- The boiling point of 4,4,4-trifluoro-1-butene is 10 °C: R. N. Haszeldine, K. Leedham and B. R. Steele, J. Chem. Soc., 1954, 2040–2042 RSC.
- Reaction with a terminal alkyne under the standard conditions formed an approximately 1:1 mixture of the desired E-product and the bistrifluoromethylated one, indicating that the alkyl substituent on alkyne 1 is necessary to reduce the reactivity of the alkyne so that the trifluoromethyl radical adds mainly to the allylsulfonate 2 to generate the alkoxysulfonyl radical rather than directly to the alkyne (see ESI† for details).
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2083910–2083913. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03315h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |