
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereodivergent synthesis of β-iodoenol carbamates with CO2via photocatalysis†
Lu
Wang
a,
Fuxing
Shi
b,
Chaorong
Qi
 *a,
Wenjie
Xu
a,
Wenfang
Xiong
a,
Bangxiong
Kang
a and
Huanfeng
Jiang
*a
*a,
Wenjie
Xu
a,
Wenfang
Xiong
a,
Bangxiong
Kang
a and
Huanfeng
Jiang
*a
aKey Laboratory of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: crqi@scut.edu.cn; jianghf@scut.edu.cn
bState Key Laboratory of Chemical Resource Engineering, Institute of Computational Chemistry, College of Chemistry, Beijing University of Chemical Technology, Beijing 100029, P. R. China
First published on 3rd August 2021
Abstract
Photocatalytic conversion of carbon dioxide (CO2) into value-added chemicals is of great significance from the viewpoint of green chemistry and sustainable development. Here, we report a stereodivergent synthesis of β-iodoenol carbamates through a photocatalytic three-component coupling of ethynylbenziodoxolones, CO2 and amines. By choosing appropriate photocatalysts, both Z- and E-isomers of β-iodoenol carbamates, which are difficult to prepare using existing methods, can be obtained stereoselectively. This transformation featured mild conditions, excellent functional group compatibility and broad substrate scope. The potential synthetic utility of this protocol was demonstrated by late-stage modification of bioactive molecules and pharmaceuticals as well as by elaborating the products to access a wide range of valuable compounds. More importantly, this strategy could provide a general and practical method for stereodivergent construction of trisubstituted alkenes such as triarylalkenes, which represents a fascinating challenge in the field of organic chemistry research. A series of mechanism investigations revealed that the transformation might proceed through a charge-transfer complex which might be formed through a halogen bond.
Introduction
The development of new and efficient methodologies for the conversion of carbon dioxide (CO2) into valuable chemicals is of great significance, because CO2 is considered as an ideal C1 feedstock in organic synthesis.1 Recently, visible-light photocatalysis has become a powerful strategy for CO2 transformation and attracted ever-increasing attention from chemists.2 Many elegant methods that cannot be achieved through conventional thermal catalysis have been successfully developed to produce various valued-added chemicals and fuels from CO2, such as CO,3 CH3OH,4 CH4 (ref. 5) and carboxylic acids.6 Organic carbamates constitute an important class of compounds, which are widely used in agrochemicals,7 pharmaceuticals8 and as intermediates in organic synthesis.9 Traditional methods for the preparation of carbamates are mainly based on the use of toxic phosgene and its derivatives, which are environmentally unfriendly.10 In the past decades, many phosgene-free protocols have been explored for the synthesis of carbamates.11 Among them, the methods using CO2 are appealing because CO2 is non-toxic, cheap, and readily available.12 In general, these methods rely on the in situ formation of nucleophilic CO2-amine adduct followed by the interaction with electrophiles. However, some of the methods still suffered from one or more drawbacks such as the need for harsh reaction conditions, poor functional group tolerance and limited substrate scope. Moreover, the stereoselective addition of the CO2-amine adduct to carbon–carbon triple bond remain very rare, as the reversibility in the interaction between CO2 and amine is usually a major issue.On the other hand, 1,2-difunctionalized alkenes, especially those containing vinyl halide moiety, are versatile and valuable building blocks in organic synthesis, because they can be further elaborated to access a variety of synthetically useful multisubstituted alkenes and functional molecules through transition metal-catalyzed cross-coupling reactions in a step-wise manner.13 Although many methods based on halofunctionalization of alkynes or hydrofunctionalization of haloalkynes have been established for producing these compounds, their stereodivergent synthesis and stereospecific transformation still remain a great challenge (Scheme 1a).14
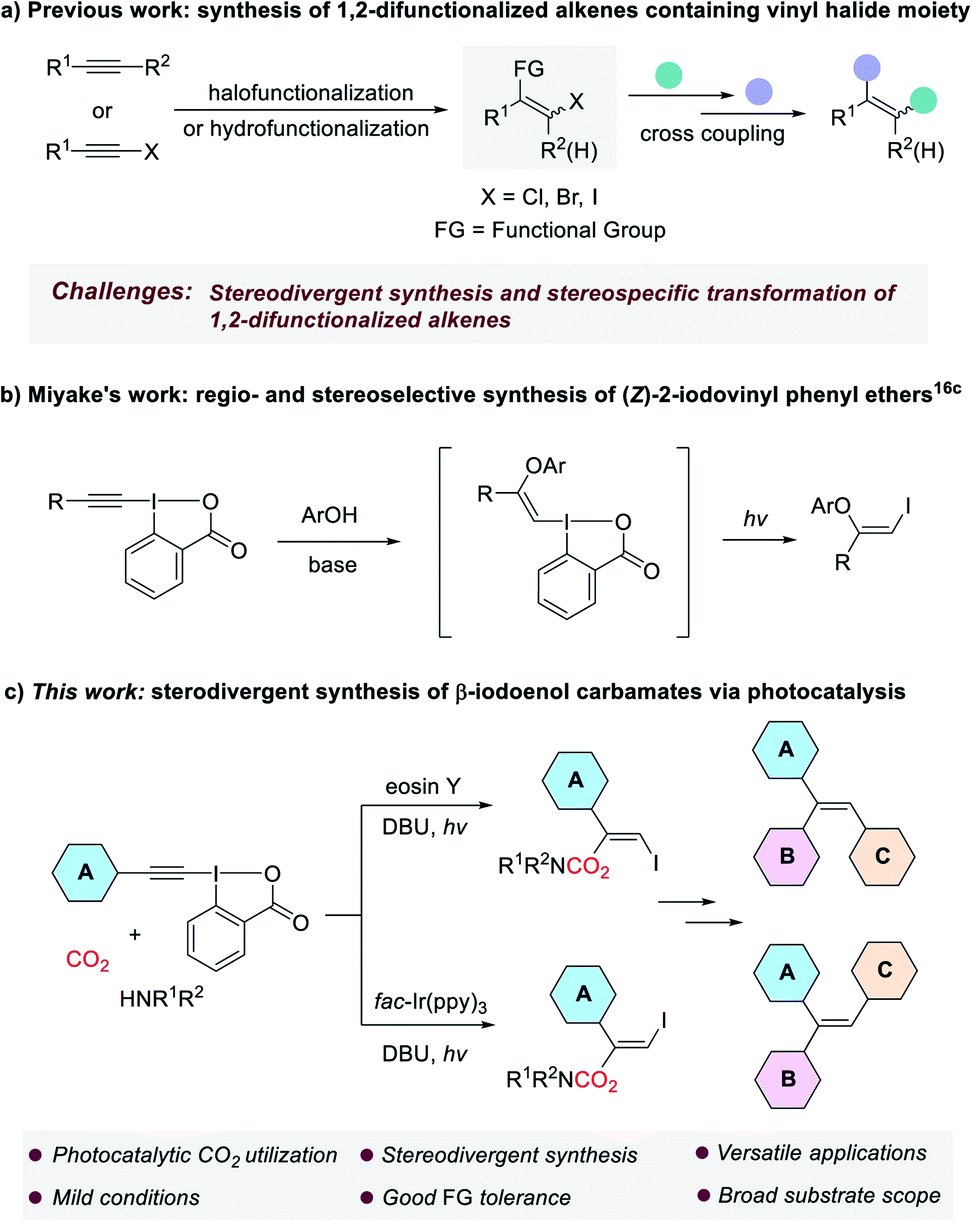 | ||
| Scheme 1 Synthesis and transformation of 1,2-difunctionalized alkenes containing vinyl halide moiety. | ||
Ethynylbenziodoxolones (EBXs), one of the most important classes of hypervalent iodine reagents, are versatile synthetic intermediates in organic chemistry.15 Especially, in the past six years the addition of nucleophiles to EBXs to construct functionalized vinylbenziodoxolones (VBXs) have been extensively studied by the groups of Yoshikai, Waser and Miyake.16 Interestingly, in 2018, Miyake and co-workers reported the regio- and stereoselective synthesis of (Z)-2-iodovinyl phenyl ethers via a visible light-driven reaction of EBXs and phenols, which was proposed to proceed through the addition of phenols to EBXs to form vinylbenziodoxolone intermediates, followed by light-mediated fragmentation via phenolate electron donor–acceptor (EDA) complexes (Scheme 1b).16c Although great achievement has been made in this field, to our knowledge, the addition of amine-CO2 adduct to the highly electrophilic EBXs is unprecedented.
Inspired by the excellent works mentioned above, and as part of our continuous interest in the synthesis of organic carbamates with CO2 and amines,17 herein, we wish to report a visible light-mediated stereodivergent synthesis of β-iodoenol carbamates from ethynylbenziodoxolones, CO2 and amines under very mild reaction conditions (Scheme 1c). The transformation was proposed to proceed through a charge-transfer complex (CTC),18 which might be formed through a halogen bond.19 The obtained products that encompass the different reactivity of enol carbamates and vinyl iodides have proven to be versatile alkenyl bis-electrophiles to access a range of valuable compounds. More importantly, the strategy could provide a general and practical method for stereodivergent construction of trisubstituted alkenes such as triarylalkenes, which represents a great challenge in synthetic organic chemistry.
Results and discussion
We began our study by evaluating the three-component reaction of phenyl-EBX (1a), diethylamine (2a) and CO2 (1 atm) under different conditions. To our delight, when the reaction was conducted in ethyl acetate with fac-Ir(ppy)3 as the photocatalyst and 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU) as the base, the desired product 3aa was obtained in 70% yield with a Z/E ratio of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :82 (Table 1, entry 1). Other catalysts including Ir(ppy)2(dtbbpy)PF6, Ir[dF(CF3)ppy]2(dtbbpy)PF6, Ru(bpy)3Cl2, 4CzIPN and eosin Y were then examined for the reaction. It was shown that the catalyst employed has a great impact on the stereoselectivity of the product and a reverse Z/E ratio could be achieved (entries 2–6). Thus, by employing eosin Y as the photocatalyst, which possesses lower triplet excited state energies, 3aa could be obtained in 70% yield with a Z/E ratio more than 99:1 (entry 6). Considering the important role of the base in CO2 conversion,20 we also investigated other organic and inorganic bases. 1,5-Diazabicyclo[4.3.0]non-5-ene (DBN) could also give the desired product in 65% yield with a Z/E ratio of 94:6 (entry 7), while the use of triethylamine, Cs2CO3 or tBuOK led to low yields due to the formation of large amounts of N,N-diethyl-2-iodobenzamide as the byproduct (entries 8–10). Subsequent optimization revealed that the use of dimethyl sulfoxide as the solvent led to higher yield (entries 11–13). To our delight, decreasing the amount of the base to 1.0 equivalent relative to 1a and even shortening the reaction time to 0.5 h did not affect the reaction efficiency and selectivity (entry 14). Finally, control experiments demonstrated that no desired product was formed in the absence of visible light (entry 15), while low yield was observed in the absence of photocatalyst or base (entries 16 and 17). Moreover, when the reaction was performed under blue LEDs irradiation conditions, low stereoselectivity was observed albeit high yield was obtained (entry 18). We speculated that the product could undergo light-induced isomerization due to the considerable UV-vis spectral overlap of (Z)-3aa and (E)-3aa with the blue LEDs spectrum (for the UV-vis spectrum of 3aa see Fig. S1†).
:82 (Table 1, entry 1). Other catalysts including Ir(ppy)2(dtbbpy)PF6, Ir[dF(CF3)ppy]2(dtbbpy)PF6, Ru(bpy)3Cl2, 4CzIPN and eosin Y were then examined for the reaction. It was shown that the catalyst employed has a great impact on the stereoselectivity of the product and a reverse Z/E ratio could be achieved (entries 2–6). Thus, by employing eosin Y as the photocatalyst, which possesses lower triplet excited state energies, 3aa could be obtained in 70% yield with a Z/E ratio more than 99:1 (entry 6). Considering the important role of the base in CO2 conversion,20 we also investigated other organic and inorganic bases. 1,5-Diazabicyclo[4.3.0]non-5-ene (DBN) could also give the desired product in 65% yield with a Z/E ratio of 94:6 (entry 7), while the use of triethylamine, Cs2CO3 or tBuOK led to low yields due to the formation of large amounts of N,N-diethyl-2-iodobenzamide as the byproduct (entries 8–10). Subsequent optimization revealed that the use of dimethyl sulfoxide as the solvent led to higher yield (entries 11–13). To our delight, decreasing the amount of the base to 1.0 equivalent relative to 1a and even shortening the reaction time to 0.5 h did not affect the reaction efficiency and selectivity (entry 14). Finally, control experiments demonstrated that no desired product was formed in the absence of visible light (entry 15), while low yield was observed in the absence of photocatalyst or base (entries 16 and 17). Moreover, when the reaction was performed under blue LEDs irradiation conditions, low stereoselectivity was observed albeit high yield was obtained (entry 18). We speculated that the product could undergo light-induced isomerization due to the considerable UV-vis spectral overlap of (Z)-3aa and (E)-3aa with the blue LEDs spectrum (for the UV-vis spectrum of 3aa see Fig. S1†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Yieldb (%) | Z/E |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.3 mmol), CO2 (1 atm), photocatalyst (1 mol%), base (0.2 mmol), solvent (1 mL), green LEDs irradiation for 14 h, room temperature. b Yields and Z/E ratios were determined by 1H NMR analysis with CH2Br2 as internal standard. c Base (0.1 mmol), 0.5 h. d The reaction was performed in the dark. e n.d. = not detected. f The reaction was conducted under blue LEDs irradiation. | |||||
| 1 | fac-Ir(ppy)3 | DBU | EtOAc | 70 | 18:82 |
| 2 | Ir(ppy)2(dtbbpy)PF6 | DBU | EtOAc | 66 | 41:59 |
| 3 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | DBU | EtOAc | 67 | 89:11 |
| 4 | Ru(bpy)3Cl2 | DBU | EtOAc | 44 | >99:1 |
| 5 | 4CzIPN | DBU | EtOAc | 66 | 98:2 |
| 6 | Eosin Y | DBU | EtOAc | 70 | >99:1 |
| 7 | Eosin Y | DBN | EtOAc | 65 | 94:6 |
| 8 | Eosin Y | NEt3 | EtOAc | 12 | >99:1 |
| 9 | Eosin Y | Cs2CO3 | EtOAc | 18 | >99:1 |
| 10 | Eosin Y | tBuOK | EtOAc | 8 | >99:1 |
| 11 | Eosin Y | DBU | CH3CN | 67 | >99:1 |
| 12 | Eosin Y | DBU | DMF | 56 | 88:13 |
| 13 | Eosin Y | DBU | DMSO | 74 | >99:1 |
| 14c | Eosin Y | DBU | DMSO | 73 | >99:1 |
| 15d | Eosin Y | DBU | DMSO | n.d.e | — |
| 16 | — | DBU | DMSO | 12 | >99:1 |
| 17 | Eosin Y | — | DMSO | 11 | >99:1 |
| 18f | Eosin Y | DBU | DMSO | 71 | 46:54 |

With the optimized reaction conditions in hand, we first examined the generality and robustness of the reaction with respect to the EBXs using eosin Y as the photocatalyst. As showed in Scheme 2, a wide range of aryl-EBXs could undergo the reaction efficiently and gave the corresponding products (Z)-3aa–(Z)-3na in moderate to good yields with excellent stereoselectivity. The Z-configuration of the carbamate products was confirmed by X-ray crystallographic analysis of (Z)-3fa.21 A variety of electron-donating and -withdrawing substituents at the para-, meta-, and ortho-positions of the benzene ring, including methyl, methoxyl, halogen (F, Cl and Br), cyano, nitro, trifluoromethyl and ester groups were well tolerated. The electronic and steric properties of the substituents have no significant effects on the reaction. Notably, EBXs derived from alkylacetylenes, alkynols and enynes were also suitable substrates for the reaction, providing the desired products (Z)-3oa–(Z)-3ra in moderate to high yields. Noted that the reaction of 1a on 1 mmol scale proceeded smoothly to give the desired product (Z)-3aa in 62% yield, demonstrating the scalability of the reaction.
 | ||
| Scheme 2 Synthesis of (Z)-β-iodoenol carbamates. Reaction conditions: 1 (0.1 mmol), 2a (0.3 mmol), eosin Y (1 mol%), DBU (0.1 mmol), DMSO (1 mL), CO2 (1 atm), green LEDs, rt, 0.5 h. Yields of the isolated products are reported. All of the Z/E values are greater than 99:1 as determined by 1H NMR analysis of the crude reaction mixtures prior to purification. aThe reaction was conducted on 1 mmol scale. | ||
Encouraged by these results, we continued to investigate the substrate scope with respect to amines under the standard conditions (Scheme 3). A series of structurally diverse acyclic dialkylamines, including symmetric and unsymmetric ones, could be applied to the transformation with good to high efficiency regardless of their alkyl chain length and steric hinderance ((Z)-3ab–(Z)-3an). Moreover, the visible light-promoted three-component reaction could be extended to various cyclic amines, affording the corresponding carbamates (Z)-3ao–(Z)-3ax in good yields. The substrate bearing a free hydroxyl group was also accommodated, providing the desired product (Z)-3aw in 65% yield.
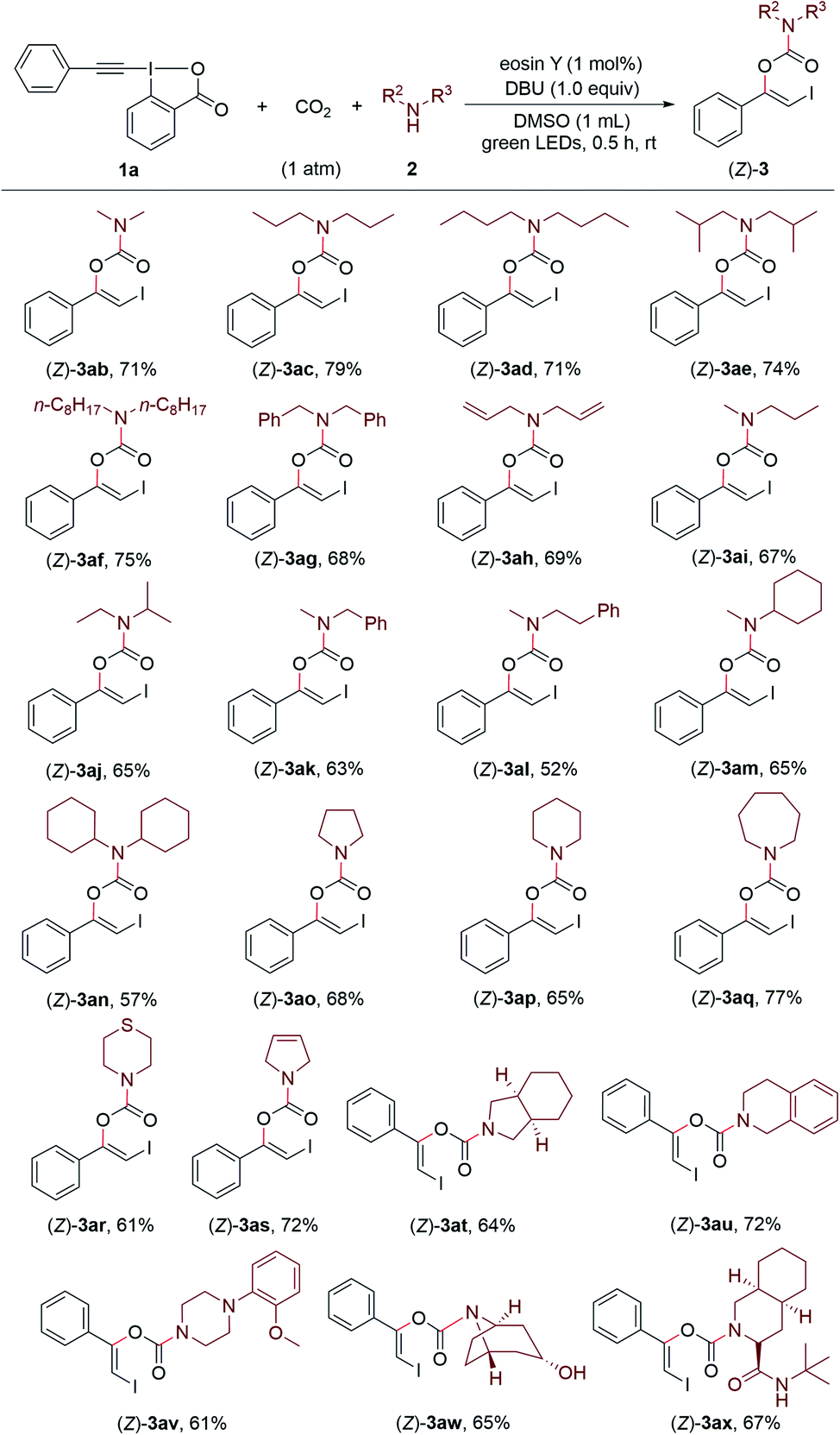 | ||
| Scheme 3 Synthesis of (Z)-β-iodoenol carbamates. Reaction conditions: 1a (0.1 mmol), 2 (0.3 mmol), eosin Y (1 mol%), DBU (0.1 mmol), DMSO (1 mL), CO2 (1 atm), green LEDs, rt, 0.5 h. Yields of the isolated products are reported. All of the Z/E values are greater than 99:1 as determined by 1H NMR analysis of the crude reaction mixtures prior to purification. | ||
Remarkably, this method provides an efficient and convenient approach for late-stage modification of bioactive molecules and pharmaceuticals containing free secondary amino group (Scheme 4). For example, three simple amino acid esters derived from glycine, L-valine and sarcosine, respectively, could readily undergo the reaction with 1a and CO2 under standard conditions and gave the carbamate products (Z)-3ay–(Z)-3aaa. Both antidepressant drugs Fluoxetine22a (marketed as Prozac) and Amoxapine22b were suitable coupling partners to give the corresponding products (Z)-3aba and (Z)-3aca in moderate yields. The late-stage modifications of Sitagliptin22c and Cytisine,22d which are used for treating diabetes and smoking cessation, respectively, were successful to afford the products (Z)-3ada and (Z)-3aea. Furthermore, the antihistamine drug Desloratadine,22e the potent bruton tyrosine kinase (BTK) inhibitor Ibrutinib N-1,22f and the antimigraine drug Lomerizine22g could undergo the modification process without difficulty ((Z)-3afa–(Z)-3aha).
 | ||
| Scheme 4 Synthesis of (Z)-β-iodoenol carbamates. Reaction conditions: 1a (0.1 mmol), 2 (0.3 mmol), eosin Y (1 mol%), DBU (0.1 mmol), DMSO (1 mL), CO2 (1 atm), green LEDs, rt, 0.5 h. Yields of the isolated products are reported. All of the Z/E values are greater than 99:1 as determined by 1H NMR analysis of the crude reaction mixtures prior to purification. | ||
Subsequently, the substrate scope of the three-component reaction catalyzed by fac-Ir(ppy)3 was explored to assemble (E)-β-iodoenol carbamates (Scheme 5). Pleasingly, a variety of aryl-EBXs bearing electron-withdrawing or electron-donating groups on the benzene rings could enter into the reaction and gave the anticipated products (E)-3aa–(E)-3fa, (E)-3ia, (E)-3ka and (E)-3sa in moderate to good yields. Although the catalyst fac-Ir(ppy)3 gave the adducts as a mixture of stereoisomers in these cases, the minor (Z)- isomers were easily separated from the major (E)-isomers by column chromatography. Moreover, the reaction of 1a could be performed on a 1 mmol scale to give the desired product in moderate yield. The electronic effect of substituents on the aromatic rings has an obvious influence on the stereoselectivity. The EBX reagents bearing a strong electron-withdrawing group on the aryl ring delivered the products with lower stereoselectivities than those with weak electron-withdrawing or electron-donating groups. However, alkyl-substituted EBXs could not give the desired (E)-β-iodoenol carbamate products. For example, the reaction of EBX 1p under the same conditions only afforded (Z)-3pa in 45% yield (not shown), indicating that (Z)-3pa could not undergo photocatalytic Z to E isomerization under these conditions, which might be due to the absence of a conjugated π system in such a molecule.23
 | ||
| Scheme 5 Synthesis of (E)-β-iodoenol carbamates. Reaction conditions: 1 (0.1 mmol), 2a (0.3 mmol), fac-Ir(ppy)3 (1 mol%), DBU (0.2 mmol), EtOAc (1 mL), CO2 (1 atm), green LEDs, rt, 14 h. Yields of the isolated products are reported. Compounds isolated as pure stereoisomers contained <1% of the alternative geometrical isomer. The E/Z values reported in parentheses were determined by 1H NMR analysis of the crude reaction mixtures prior to purification. aThe reaction was conducted on 1 mmol scale. | ||
A series of secondary amines could be converted to the corresponding products (E)-3ab–(E)-3af, (E)-3ah, (E)-3ak, (E)-3ao–(E)-3ar in 48–73% yield with good stereoselectivities (Scheme 6). Moreover, the Ir-catalyzed three-component reaction could be applied to amino acid esters and drugs, as exemplified by glycine ester and Amoxapine, which gave the desired products (E)-3ay and (E)-3aca in 61% and 53% yields, respectively. The geometry of the product (E)-3aca was also confirmed by the X-ray diffraction analysis.24
 | ||
| Scheme 6 Synthesis of (E)-β-iodoenol carbamates. Reaction conditions: 1a (0.1 mmol), 2 (0.3 mmol), fac-Ir(ppy)3 (1 mol%), DBU (0.2 mmol), EtOAc (1 mL), CO2 (1 atm), green LEDs, rt, 14 h. Yields of the isolated products are reported. Compounds isolated as pure stereoisomers contained <1% of the alternative geometrical isomer. The E/Z values reported in parentheses were determined by 1H NMR analysis of the crude reaction mixtures prior to purification. | ||
To unravel the underlying synthetic value of the protocol, a series of subsequent transformations of the newly formed product (Z)-3aa was performed. As can be seen from Scheme 7A, (Z)-3aa could take part in a range of Cu- or Pd-catalyzed cross-coupling reactions via C(sp2)–I bond cleavage, including thiolization (4), methoxycarbonylation (5), Sonogashira (6) and Suzuki cross-coupling reactions (7 and 8). In addition, the palladium-catalyzed [2 + 2 + 1] cycloaddition reaction of (Z)-3aa with diphenylacetylene could also proceed smoothly, affording a structurally intriguing pentasubstituted fulvene derivative 9 in 62% yield.25 The structure of compound 9 was confirmed by X-ray crystallographic analysis.26
 | ||
| Scheme 7 Versatile transformations of the β-iodoenol carbamates. | ||
Stereodivergent synthesis of triarylalkenes represents a long-standing challenge because of the small energy difference between E and Z isomers of triarylalkenes. With the visible light-mediated stereodivergent synthesis of β-iodoenol carbamates in hand, we hypothesized whether this transformation would provide a general method for stereodivergent synthesis of triarylalkenes, since both iodine atom and carbamate group might serve as convenient handles for cross-coupling to form C–C bonds. With this goal in mind, we attempted to synthesize 1-fluoro-4-(2-(4-methoxyphenyl)-1 phenylvinyl)benzene (11), an alkene with three different aryl groups. As can be seen in Scheme 7B, both (Z)-3aa and (E)-3aa could undergo the Pd-catalyzed Suzuki cross-coupling with (4-methoxyphenyl)boronic acid to give the desired products (Z)-10 and (E)-10 in 72% and 84% yields, respectively. Subsequent Pd-catalyzed Kumada–Corriu cross-coupling with Grignard reagent (4-fluorophenyl)magnesium bromide would afforded the desired products (Z)-11 and (E)-11 in high yields with excellent stereoselectivities. Notably, these types of complementary pairs of alkenes would otherwise be difficult to prepare through classic Wittig olefination reaction.
To gain some insight into the mechanism, we conducted a series of control experiments and spectroscopic investigations. Firstly, it was found that 1a could react with CO2 and 2a in the absence of visible light and photosensitizer to form a stable vinylbenziodoxolone product 12 in excellent yield, and irradiation of 12 with green LEDs in the presence of eosin Y and DBU led to the formation of (Z)-3aa, indicating compound 12 was the key intermediate for the transformation. Control experiments showed that light, eosin Y and DBU are all essential for the formation of (Z)-3aa from 12 (Scheme 8A).
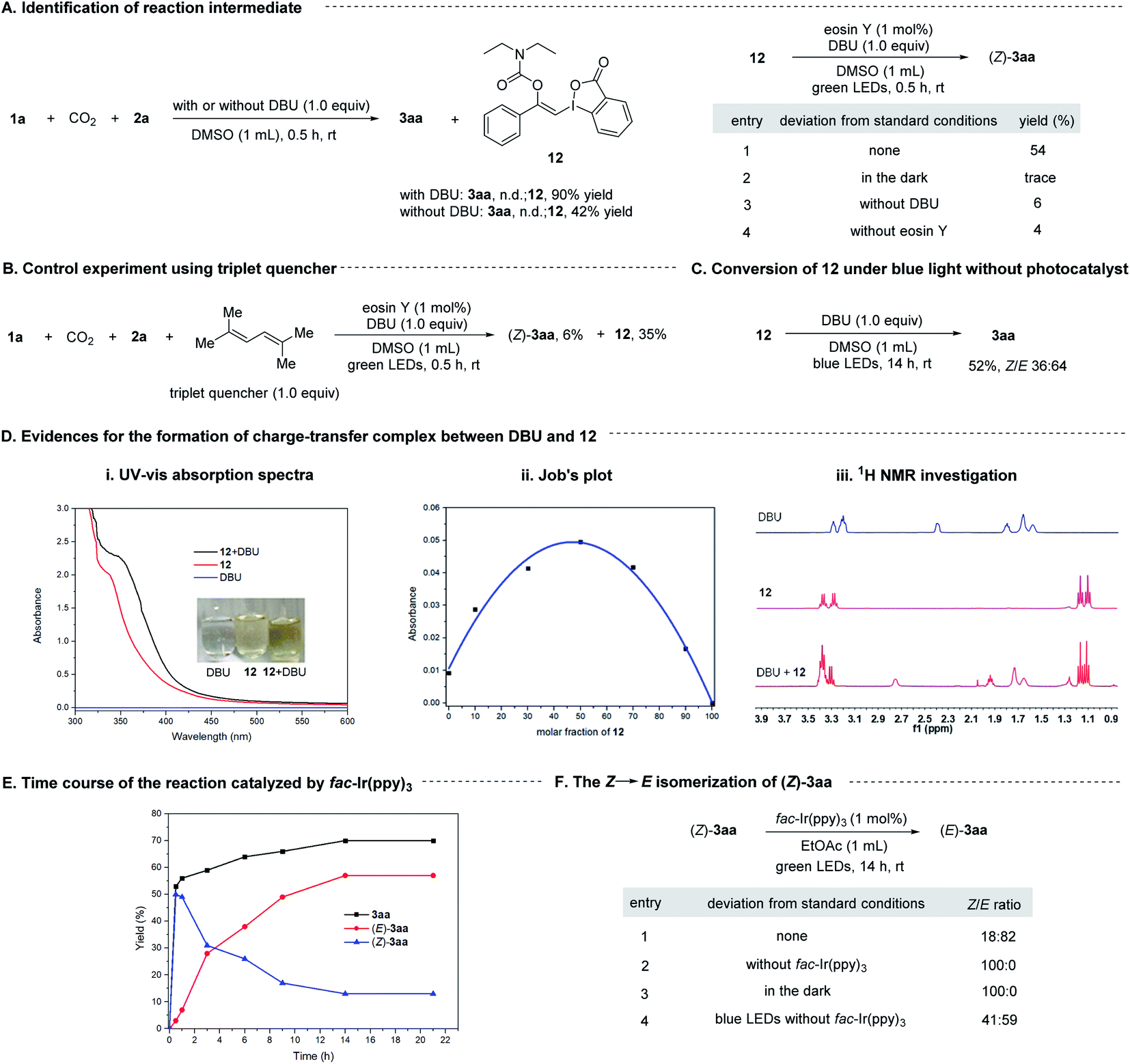 | ||
| Scheme 8 Mechanistic studies. | ||
Next, we tried to explore the role of the photocatalyst eosin Y in this transformation. Stern–Volmer quenching studies showed that the excited eosin Y was effectively quenched by the addition of a mixture of 12 and DBU (1:1 molar ratio) rather than by 12 alone (Fig. S2†), indicating that DBU might be capable of interacting with 12 to accelerate the quenching of eosin Y*. The redox potential of 12 (Eoxp/2 = +1.05 V vs. SCE, Eredp/2 = −1.22 V vs. SCE; Fig. S4 and S5†) were also measured by cyclic voltammetry experiments, which demonstrated that the redox potential of 12 is outside the redox range of eosin Y (Eox1/2 = +0.83 V vs. SCE, Ered1/2 = −1.11 V vs. SCE).27 Moreover, the reaction was significantly inhibited by addition of 1.0 equivalent of 2,5-dimethylhexa-2,4-diene, which is a well-known triplet quencher.28 In this case, only little amount of (Z)-3aa was formed along with intermediate 12 in 35% yield (Scheme 8B). The above results suggested that electron transfer between 12 and eosin Y might not be involved in the reaction, precluding a photoinduced redox process. In support of the energy transfer catalysis, we observed that irradiation of a mixture of 12 and DBU (1:1, molar ratio) in DMSO with blue LEDs in the absence of any photocatalysts would give 3aa in 52% yield with a Z/E ratio of 36:64 (Scheme 8C). Furthermore, the density functional theory (DFT) calculations showed that the triplet excited state energies of 12 and complex 12-DBU were computed to be endergonic by 89.7 and 41.6 kcal mol−1, respectively (see ESI† for more details). These results suggested that a thermodynamical activation of 12-DBU complex by eosin Y (ET = 44.0 kcal mol−1) via energy transfer might be feasible, while the thermodynamical activation of 12 by eosin Y via energy transfer is rather difficult.27
To further confirm the role of DBU, the UV-vis absorption spectra tests were conducted on 12 and DBU in DMSO at 0.1 M concentration for each species. A significant bathochromic shift was observed when mixing 12 and DBU in a 1:1 ratio (Scheme 8D(i)). The Job's plot further confirmed a 1:1 binding stoichiometry for 12 and DBU (Scheme 8D(ii)). 1H NMR analysis showed that all the proton signals of DBU in CDCl3 were significantly shifted to lower field when 1 equivalent of 12 was added, also indicating that DBU would interact with 12 directly (Scheme 8D(iii)). Moreover, the DFT studies revealed that the calculated N⋯I distance of the 1:1 complex of DBU and 12 is 3.15 Å in singlet ground state, which is significantly smaller than the sum of the van der Waals radii of N and I atoms (3.7 Å), and the atomic polar tensors (APT) charges29 on the N and I atom are −0.43 and +0.88 e−, respectively. However, in the excited triplet state the N⋯I distance will be shorten to 2.83 Å, and the APT charges on the N and I atom are changed to −0.39 and +0.57 e−, respectively (Scheme 9, and see ESI† for more details). All these results suggested that 12 and DBU might form a charge-transfer complex (CTC) through an attractive non-covalent halogen bond.
 | ||
| Scheme 9 Proposed mechanism. | ||
Then, the time course of the reaction catalyzed by fac-Ir(ppy)3 under green LEDs irradiation was studied. It was shown that the yield of (Z)-3aa increased rapidly at the early stage of the reaction and reached a maximal value of about 50% after 0.5 h, and then decreased gradually while the yield of its E-isomer increased (Scheme 8E), indicating (Z)-3aa was first generated and then isomerized to (E)-3aa under the visible light irradiation conditions. This could be confirmed by irradiation of pure (Z)-3aa with green LEDs in the presence of fac-Ir(ppy)3, which gave (E)-3aa with high efficiency. It is also found that (Z)-3aa could also undergo the isomerization under blue LEDs irradiation in the absence of any photocatalysts albeit with low efficiency (Scheme 8F), revealing the importance of photocatalyst in controlling the stereoselectivity of the transformation.
Finally, the source of the vinylic hydrogens in the final product 3aa was investigated. When diethylamine-N-D1 was used for the reaction under standard reaction conditions, products (Z)- and (E)-3aa were obtained with 83% and 71% of D-form, respectively, indicating that amines are the major proton source for the carbamate products (see ESI† for more details).
Although the mechanism of the visible light-mediated three-component reaction has not yet been fully elucidated, on the basis of the above-described observations and previous literature, a plausible mechanism is proposed in Scheme 9. Initially, CO2 reacts with ethylamine (2a) in the presence of DBU to give carbamate salt I,17 which will nucleophilically attack the C–C triple bond of EBX 1a to form a vinyl carbanion intermediate II. Subsequent protonation of intermediate II will lead to the formation of vinylbenziodoxolone 12.16a,c–e Then, interaction between 12 and DBU via a halogen bond forms a charge-transfer complex III. Under the irradiation of green LEDs, a triplet triplet energy transfer (TTEnT) between complex III and excited state PC* occurs to generate an excited charge-transfer complex III*, which will undergo a rapid intermolecular single-electron transfer (SET) process to give the product (Z)-3aa, along with the formation of aryl radical IV and radical cation V. Subsequent hydrogen atom transfer (HAT) from V to IV will generate benzoate VI and VII. In the case of eosin Y as the photocatalyst, the resulting (Z)-3aa cannot further undergo photoinduced isomeriation to yield its isomer (E)-3aa due to the low ET of eosin Y. However, when photocatalysts with higher ET, such as fac-Ir(ppy)3 is employed,30 a contra-thermodynamic, photocatalytic Z to E isomerization of (Z)-3aa will proceed via an energy transfer process,31 furnishing the (E)-3aa product. Additionally, in this case, since photoexcited fac-Ir(ppy)3 30 might be responsible for the single-electron reduction of 12 on the basis of the Stern–Volmer quenching results (Fig. S3†), an alternative reaction pathway for the formation of (Z)-3aa that involves direct electron transfer between 12 and fac-Ir(ppy)3 cannot be excluded at present (Fig. S9†).
30 might be responsible for the single-electron reduction of 12 on the basis of the Stern–Volmer quenching results (Fig. S3†), an alternative reaction pathway for the formation of (Z)-3aa that involves direct electron transfer between 12 and fac-Ir(ppy)3 cannot be excluded at present (Fig. S9†).
Conclusions
We have reported a visible light-mediated stereodivergent synthesis of β-iodoenol carbamates from ethynylbenziodoxolones, CO2 and amines for the first time. By choosing appropriate photocatalysts, both Z- and E-isomers of β-iodoenol carbamates, which are difficult to prepare using existing methods, can be obtained stereoselectively. The mild reaction conditions allowed excellent functional group compatibility and broad substrate scope. The potential utility of this protocol was demonstrated by late-stage modification of second amino group-containing bioactive molecules and drugs as well as by elaborating the products to access a wide range of synthetically valuable compounds. More importantly, this strategy could provide a general and practical method for stereodivergent synthesis of trisubstituted alkenes such as triarylalkenes, which represents a fascinating challenge in the field of organic chemistry research. A series of mechanism investigations revealed that the transformation might proceed through a charge-transfer complex which might be formed through a halogen bond.Data availability
All data supporting the findings of this study are available within the paper and its ESI,† include experimental details, the results of the DFT calculations, characterization data, and 1H and 13C NMR spectra of all new compounds. Crystallographic data for the crystal structures has been deposited at the CCDC under CCDC 2041438 for (Z)-3fa, 2041439 for (E)-3aca and 2041440 for 9.Author contributions
L. W., H. J. and C. Q. designed the research and experiments; L. W., W. X. (Wenjie Xu), W. X. (Wenfang Xiong) and B. K. contributed to the experiment work and F. S. contributed to the DFT calculations; L. W. and C. Q. wrote the manuscript; all authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Ministry of Science and Technology of the People’s Republic of China (2016YFA0602900), the National Natural Science Foundation of China (21971073 and 22001075) and the Natural Science Foundation of Guangdong Province (2019A1515011468). We also thank Professor Jiarong Chen (Central China Normal University) for his helpful discussion.Notes and references
- For selected reviews, see: (a) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS; (b) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (c) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948 CrossRef CAS PubMed; (d) V. A. Peshkov, O. P. Pereshivko, A. A. Nechaev, A. A. Peshkov and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3861 RSC; (e) X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962 CrossRef CAS; (f) S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382 RSC; (g) Y. Yang and J.-W. Lee, Chem. Sci., 2019, 10, 3905 RSC; (h) L. Song, Y.-X. Jiang, Z. Zhang, Y.-Y. Gui, X.-Y. Zhou and D.-G. Yu, Chem. Commun., 2020, 56, 8355 RSC; (i) H.-R. Li and L.-N. He, Organometallics, 2020, 39, 1461 CrossRef CAS.
- For selected reviews, see: (a) Y.-Y. Gui, W.-J. Zhou, J.-H. Ye and D.-G. Yu, ChemSusChem, 2017, 10, 1337 CrossRef CAS PubMed; (b) Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644 CrossRef CAS; (c) J. Hou, J.-S. Li and J. Wu, Asian J. Org. Chem., 2018, 7, 1439 CrossRef CAS; (d) C. S. Yeung, Angew. Chem., Int. Ed., 2019, 58, 5492 CrossRef CAS; (e) X. He, L.-Q. Qiu, W.-J. Wang, K.-H. Chen and L.-N. He, Green Chem., 2020, 22, 7301 RSC; (f) C. Xi, Z. Fan and Z. Zhang, ChemSusChem, 2020, 13, 6201 Search PubMed; (g) Z. Zhang, J.-H. Ye, T. Ju, L.-L. Liao, H. Huang, Y.-Y. Gui, W.-J. Zhou and D.-G. Yu, ACS Catal., 2020, 10, 10871 CrossRef; (h) B. Cai, H. W. Cheo, T. Liu and J. Wu, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202010710; (i) G. Zhang, Y. Cheng, M. Beller and F. Chen, Adv. Synth. Catal., 2021, 363, 1583 CrossRef CAS.
- (a) Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao and P. K. Wong, Adv. Mater., 2018, 30, 1706 Search PubMed; (b) Z. Jiang, X. Xu, Y. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Peng, L. Zan and H. Deng, Nature, 2020, 586, 549 CrossRef CAS PubMed; (c) D. Saito, Y. Yamazaki, Y. Tamaki and O. Ishitani, J. Am. Chem. Soc., 2020, 142, 19249 CrossRef CAS; (d) H. Zhang, Y. Wang, S. Zuo, W. Zhou, J. Zhang and X. W. D. Lou, J. Am. Chem. Soc., 2021, 143, 2173 CrossRef CAS.
- (a) R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-j. Kong and J.-O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728 CrossRef CAS; (b) Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C. J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957 CrossRef CAS.
- (a) G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292 CrossRef CAS PubMed; (b) H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74 CrossRef CAS; (c) L.-Y. Wu, Y.-F. Mu, X.-X. Guo, W. Zhang, Z.-M. Zhang, M. Zhang and T.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491 CrossRef CAS.
- (a) S. Wang, B.-Y. Cheng, M. Sršen and B. König, J. Am. Chem. Soc., 2020, 142, 7524 CrossRef CAS PubMed; (b) H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122 CrossRef CAS; (c) H. Huang, J.-H. Ye, L. Zhu, C.-K. Ran, M. Miao, W. Wang, H. Chen, W.-J. Zhou, Y. Lan, B. Yu and D.-G. Yu, CCS Chem., 2020, 2, 1746 Search PubMed; (d) D. Kong, M. Munch, Q. Qiqige, C. J. C. Cooze, B. H. Rotstein and R. J. Lundgren, J. Am. Chem. Soc., 2021, 143, 2200 CrossRef CAS PubMed; (e) L.-L. Liao, G.-M. Cao, Y.-X. Jiang, X.-H. Jin, X.-L. Hu, J. J. Chruma, G.-Q. Sun, Y.-Y. Gui and D.-G. Yu, J. Am. Chem. Soc., 2021, 143, 2812 CrossRef CAS PubMed; (f) Y. Gao, H. Wang, Z. Chi, L. Yang, C. Zhou and G. Li, CCS Chem., 2021, 3, 1848 CrossRef.
- (a) Y. Zhou, Z. Ke, H. Ye, M. Hong, Y. Xu, M. Zhang, W. Jiang and Q. Hong, J. Agric. Food Chem., 2020, 68, 14739 CrossRef CAS; (b) V. Mariyappan, M. Keerthi and S.-M. Chen, J. Agric. Food Chem., 2021, 69, 2679 CrossRef CAS.
- (a) J. Skácel, B. S. Slusher and T. Tsukamoto, J. Med. Chem., 2021, 64, 279 CrossRef; (b) P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339 CrossRef CAS PubMed; (c) Y. Wu, B. Wang, H. Lu, H. Zhao, B. Yang, L. Li, Y. Lu, D. Zhang, N. Sun and H. Huang, J. Med. Chem., 2021, 64, 3234 CrossRef CAS PubMed.
- (a) M. R. Harris, L. E. Hanna, M. A. Greene, C. E. Moore and E. R. Jarvo, J. Am. Chem. Soc., 2013, 135, 3303 CrossRef CAS PubMed; (b) D. Holt and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 7857 CrossRef CAS; (c) A. C. P. Rivera, R. Still and D. E. Frantz, Angew. Chem., Int. Ed., 2016, 55, 6689 CrossRef CAS; (d) A. Nishizawa, T. Takahira, K. Yasui, H. Fujimoto, T. Iwai, M. Sawamura, N. Chatani and M. Tobisu, J. Am. Chem. Soc., 2019, 141, 7261 CrossRef CAS PubMed; (e) M. Wilkovitsch, M. Haider, B. Sohr, B. Herrmann, J. Klubnick, R. Weissleder, J. C. T. Carlson and H. Mikula, J. Am. Chem. Soc., 2020, 142, 19132 CrossRef CAS PubMed; (f) C. Wata and T. Hashimoto, J. Am. Chem. Soc., 2021, 143, 1745 CrossRef CAS PubMed.
- (a) S. Ozaki, Chem. Rev., 1972, 72, 457 CrossRef; (b) H. Babad and A. G. Zeiler, Chem. Rev., 1973, 73, 75 CrossRef CAS.
- (a) A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035 CrossRef CAS; (b) D. Chaturvedi, Tetrahedron, 2012, 68, 15 CrossRef CAS; (c) G. S. Kumar, C. U. Maheswari, R. A. Kumar, M. L. Kantam and K. R. Reddy, Angew. Chem., Int. Ed., 2011, 50, 11748 CrossRef CAS.
- For selected reviews, see: (a) Z. Zhang, J.-H. Ye, D.-S. Wu, Y.-Q. Zhou and D.-G. Yu, Chem.–Asian J., 2018, 13, 2292 CrossRef CAS; (b) W. Schilling and S. Das, ChemSusChem, 2020, 13, 6246 CAS.
- (a) N. T. Barczak, D. A. Rooke, Z. A. Menard and E. M. Ferreira, Angew. Chem., Int. Ed., 2013, 52, 7579 CrossRef CAS PubMed; (b) J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518 CrossRef CAS; (c) M. J. Koh, T. T. Nguyen, H. Zhang, R. R. Schrock and A. H. Hoveyda, Nature, 2016, 531, 459 CrossRef CAS PubMed; (d) T. T. Nguyen, M. J. Koh, T. J. Mann, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 552, 347 CrossRef CAS PubMed.
- (a) T. Xu, C. W. Cheung and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 4910 CrossRef CAS PubMed; (b) L. Wang, J. M. Lear, S. M. Rafferty, S. C. Fosu and D. A. Nagib, Science, 2018, 362, 225 CrossRef CAS; (c) X. Zeng, S. Liu, G. B. Hammond and B. Xu, ACS Catal., 2018, 8, 904 CrossRef CAS PubMed; (d) X. Zeng, S. Liu, Y. Yang, Y. Yang, G. B. Hammond and B. Xu, Chem, 2020, 6, 1018 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. P. Brand, D. F. González, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102 RSC; (b) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed; (c) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436 CrossRef CAS; (d) D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212 CrossRef CAS PubMed.
- For selected recent examples, see: (a) J. Wu, X. Deng, H. Hirao and N. Yoshikai, J. Am. Chem. Soc., 2016, 138, 9105 CrossRef CAS; (b) J. Wu, K. Xu, H. Hirao and N. Yoshikai, Chem.–Eur. J., 2017, 23, 1521 CrossRef CAS; (c) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 12829 CrossRef CAS; (d) P. Caramenti, N. Declas, R. Tessier, M. D. Wodrich and J. Waser, Chem. Sci., 2019, 10, 3223 RSC; (e) R. Tessier, J. Ceballos, N. Guidotti, R. Simonet-Davin, B. Fierz and J. Waser, Chem, 2019, 5, 2243 CrossRef CAS; (f) W. Ding, J. Chai, C. Wang, J. Wu and N. Yoshikai, J. Am. Chem. Soc., 2020, 142, 8619 CrossRef CAS PubMed; (g) B. Liu, J. V. Alegre-Requena, R. S. Paton and G. M. Miyake, Chem.–Eur. J., 2020, 26, 2386 CrossRef CAS.
- (a) W. Xiong, C. Qi, H. He, L. Ouyang, M. Zhang and H. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3084 CrossRef CAS; (b) Y. Peng, J. Liu, C. Qi, G. Yuan, J. Li and H. Jiang, Chem. Commun., 2017, 53, 2665 RSC; (c) W. Xiong, C. Qi, R. Cheng, H. Zhang, L. Wang, D. Yan and H. Jiang, Chem. Commun., 2018, 54, 5835 RSC; (d) L. Wang, C. Qi, R. Cheng, H. Liu, W. Xiong and H. Jiang, Org. Lett., 2019, 21, 7386 CrossRef CAS PubMed; (e) W. Xiong, R. Cheng, B. Wu, W. Wu, C. Qi and H. Jiang, Sci. China: Chem., 2020, 63, 331 CrossRef CAS; (f) R. Cheng, C. Qi, L. Wang, W. Xiong, H. Liu and H. Jiang, Green Chem., 2020, 22, 4890 RSC; (g) L. Wang, P. Wang, T. Guo, W. Xiong, B. Kang, C. Qi, G. Luo, Y. Luo and H. Jiang, Org. Chem. Front., 2021, 8, 1851 RSC.
- (a) C. G. S. Lima, T. d. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS; (b) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef CAS; (c) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed; (d) C. Wang, R. Qi, H. Xue, Y. Shen, M. Chang, Y. Chen, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2020, 59, 7461 CrossRef CAS PubMed; (e) B. Sun, X. Shi, X. Zhuang, P. Huang, R. Shi, R. Zhu and C. Jin, Org. Lett., 2021, 23, 1862 CrossRef CAS.
- For selected reviews, see: (a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS; (b) G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772 RSC; (c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS.
- M. Hulla and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 1002 CrossRef CAS PubMed.
- CCDC 2041438 ((Z)-3fa) contains the supplementary crystallographic data for this paper.†.
- (a) C. J. Wenthur, M. R. Bennett and C. W. Lindsley, ACS Chem. Neurosci., 2014, 5, 14 CrossRef CAS; (b) B. M. Cohen, P. Q. Harris, R. I. Altesman and J. O. Cole, Am. J. Psychiatry, 1982, 139, 1165 CrossRef CAS PubMed; (c) K. B. Hansen, Y. Hsiao, F. Xu, N. Rivera, A. Clausen, M. Kubryk, S. Krska, T. Rosner, B. Simmons, J. Balsells, N. Ikemoto, Y. Sun, F. Spindler, C. Malan, E. J. J. Grabowski and J. D. Armstrong, J. Am. Chem. Soc., 2009, 131, 8798 CrossRef CAS PubMed; (d) A. E. M. Blom, H. R. Campello, H. A. Lester, T. Gallagher and D. A. Dougherty, J. Am. Chem. Soc., 2019, 141, 15840 CrossRef CAS PubMed; (e) E. A. Stone, K. J. Cutrona and S. J. Miller, J. Am. Chem. Soc., 2020, 142, 12690 CrossRef CAS PubMed; (f) S. v. Hoppe, J. J. M. Rood, L. Buil, E. Wagenaar, R. W. Sparidans, J. H. Beijnen and A. H. Schinkel, Mol. Pharmaceutics, 2018, 15, 5124 CrossRef; (g) M. Thevis, K. Walpurgis and A. Thomas, Anal. Chem., 2020, 92, 506 CrossRef CAS.
- (a) J. L. Magee, W. Shand Jr and H. Eyring, J. Am. Chem. Soc., 1941, 63, 677 CrossRef CAS; (b) A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796 RSC; (c) J. J. Molloy, T. Morack and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 13654 CrossRef CAS PubMed; (d) J. J. Molloy, M. Schäfer, M. Wienhold, T. Morack, C. G. Daniliuc and R. Gilmour, Science, 2020, 368, 302 CrossRef.
- CCDC 2041439 ((E)-3aca) contains the supplementary crystallographic data for this paper.†.
- (a) A. D. Finke, O. Dumele, M. Zalibera, D. Confortin, P. Cias, G. Jayamurugan, J.-P. Gisselbrecht, C. Boudon, W. B. Schweizer, G. Gescheidt and F. Diederich, J. Am. Chem. Soc., 2012, 134, 18139 CrossRef CAS PubMed; (b) A. D. Finke, S. Haberland, W. B. Schweizer, P. Chen and F. Diederich, Angew. Chem., Int. Ed., 2013, 52, 9827 CrossRef CAS; (c) P. Preethalayam, K. S. Krishnan, S. Thulasi, S. S. Chand, J. Joseph, V. Nair, F. Jaroschik and K. V. Radhakrishnan, Chem. Rev., 2017, 117, 3930 CrossRef CAS PubMed.
- CCDC 2041440 (9) contains the supplementary crystallographic data for this paper.†.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC.
- (a) M. Zhu, C. Zheng, X. Zhang and S.-L. You, J. Am. Chem. Soc., 2019, 141, 2636 CrossRef CAS; (b) Z. Zhang, D. Yi, M. Zhang, J. Wei, J. Lu, L. Yang, J. Wang, N. Hao, X. Pan, S. Zhang, S. Wei and Q. Fu, ACS Catal., 2020, 10, 10149 CrossRef CAS.
- J. Cioslowsk, J. Am. Chem. Soc., 1989, 111, 8336 CrossRef.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156 CrossRef CAS PubMed.
- (a) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2015, 137, 11254 CrossRef CAS; (b) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040 CrossRef CAS; (c) C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, Nat. Catal., 2019, 2, 678 CrossRef CAS; (d) C. Onneken, K. Bussmann and R. Gilmour, Angew. Chem., Int. Ed., 2020, 59, 330 CrossRef CAS PubMed; (e) J. J. Molloy, M. Schäfer, M. Wienhold, T. Morack, C. G. Daniliuc and R. Gilmour, Science, 2020, 368, 302 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2041438, 2041439 and 2041440. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03366b |
| This journal is © The Royal Society of Chemistry 2021 |