
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress and future challenges in the supramolecular polymerization of metal-containing monomers
Nils
Bäumer†
,
Jonas
Matern†
and
Gustavo
Fernández
 *
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 36, 48149 Münster, Germany. E-mail: fernandg@uni-muenster.de
First published on 8th September 2021
Abstract
The self-assembly of discrete molecular entities into functional nanomaterials has become a major research area in the past decades. The library of investigated compounds has diversified significantly, while the field as a whole has matured. The incorporation of metal ions in the molecular design of the (supra-)molecular building blocks greatly expands the potential applications, while also offering a promising approach to control molecular recognition and attractive and/or repulsive intermolecular binding events. Hence, supramolecular polymerization of metal-containing monomers has emerged as a major research focus in the field. In this perspective article, we highlight recent significant advances in supramolecular polymerization of metal-containing monomers and discuss their implications for future research. Additionally, we also outline some major challenges that metallosupramolecular chemists (will) have to face to produce metallosupramolecular polymers (MSPs) with advanced applications and functionalities.
 Nils Bäumer | Nils Bäumer received his B.Sc. and M.Sc. degrees from the WWU Münster in 2016 and 2018, respectively. He is currently working on his PhD degree under the supervision of Prof. Fernández at the WWU. His research focuses on controlling the self-assembly of discrete metal complexes, particularly through controlling coordination geometry. |
 Jonas Matern | Jonas Matern received his B.Sc. and M.Sc. degrees from the Ruprecht-Karls-Universität Heidelberg (2016) and Westfälische-Wilhelms Universität Münster (WWU, 2018), respectively. He is currently working on his PhD in the group of Prof. Fernández at the WWU. His research focuses on the systematic study of the factors governing supramolecular polymerization, particularly in metal-containing systems. |
 Gustavo Fernández | Gustavo Fernández has been Professor of Organic and Supramolecular Chemistry at the WWU Münster since 2015, where he heads a research group working on supramolecular functional materials. His research focuses on understanding the structure–property relationship in (stimuli-responsive) supramolecular assemblies of metal-containing π-systems and BODIPY dyes. |
1 Introduction
In the recent decades, the field of supramolecular chemistry has matured significantly, moving from discrete supramolecules, such as catenanes1 or rotaxanes,2 to dynamic self-assembled materials held together by non-covalent bonds.3 In this regard, molecular self-assembly has been established as a powerful tool to bridge the gap between the nanoscopic and mesoscopic scale by careful control over non-covalent interactions.4–6 More specifically, supramolecular polymers (SP) represent ideal model systems to investigate the diverse aspects of self-assembly of discrete molecular entities and to elaborate detailed structure–property relationships.7 These one- or two-dimensional self-assembled systems can shed relevant insights into more complex self-organization phenomena such as 3D crystallization,8–11 which are widespread both in natural and artificial functional systems.8,12–15 Since the first report more than three decades ago,16 SPs have been primarily constructed from purely organic building blocks,17,18 often incorporating additional functional groups for directional hydrogen bonding.The inclusion of metal centres in these dynamic systems intrigued academic researchers owing to new potential properties, such as semiconductivity or strong luminescence (Fig. 1),19–21 leading to the first reports of metallosupramolecular polymers (MSPs). These initial research efforts were focused on understanding the outcome of metal–ligand coordination in self-assembling di/polytopic ligands3,22,23 to create coordination polymers. However, a fundamental challenge of this approach lies in the uncontrolled formation of self-assembled structures as a result of random metal complexation in a 3D chemical space. Consequently, assessing the exact mechanism of self-assembly, the secondary interactions governing the process (apart from metal complexation events) and ultimately achieving precise control over the formed self-assembled structures, were still unsolved challenges at this stage.
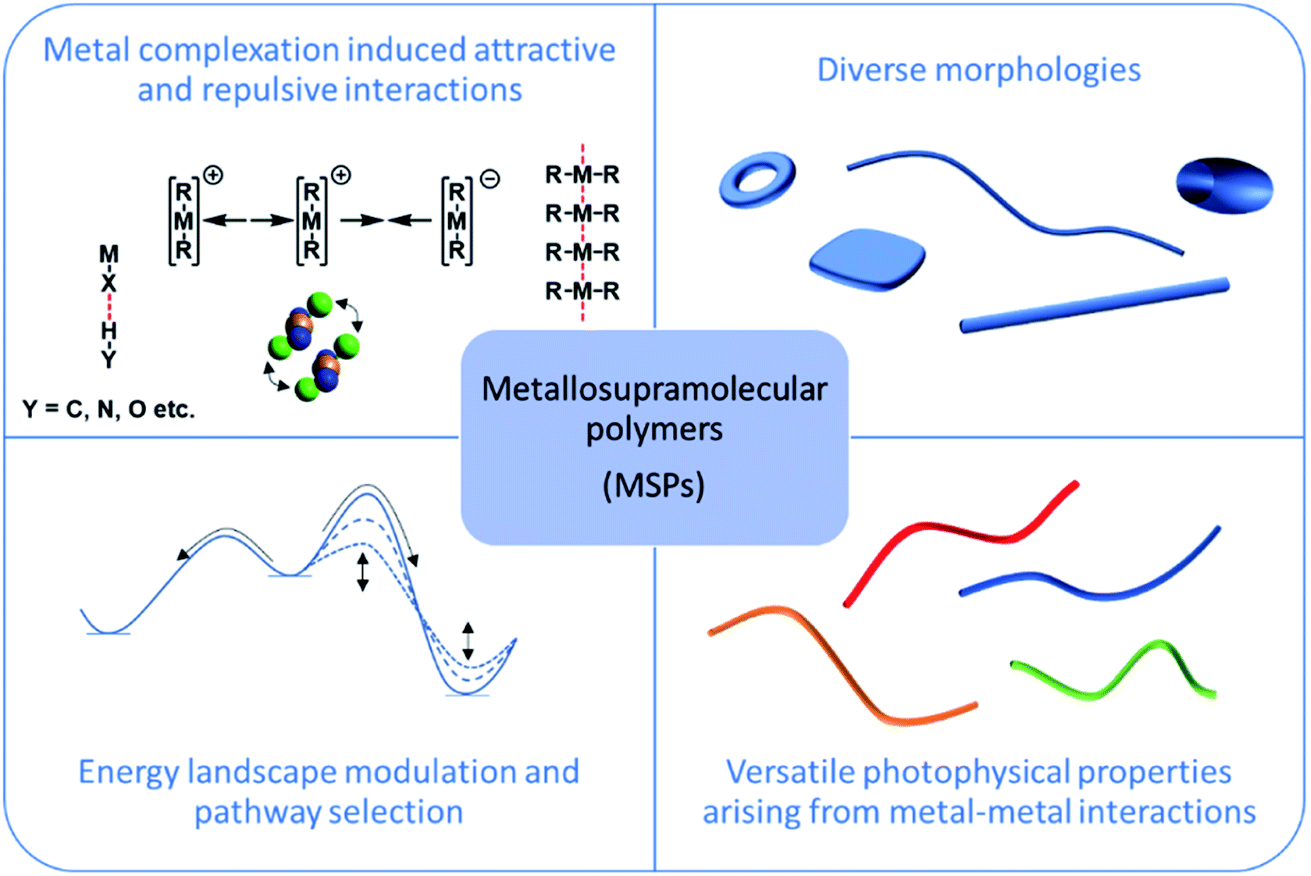 | ||
| Fig. 1 Schematic representation of commonly investigated aspects of MSPs. | ||
As a logical consequence, metallosupramolecular self-assembly had to be approached from an opposite direction; assembling previously synthesized discrete metal complexes into supramolecular architectures. Particularly, within the last decade, the research field has made significant advances beyond mere thermodynamic self-assembly. The investigation of kinetic effects on supramolecular polymerization has become a central aspect,24–26 which promoted the research field towards more biomimetic approaches, such as dissipative self-assembly.27–31
In the context of kinetically controlled SP, sample preparation methods and the precise adjustment of experimental conditions have been recognized as important tools to direct self-assembly. Rational choice of sample preparation protocols and molecular design has resulted in a vast variety of synthetic strategies and experimental handles available to supramolecular researchers today.
In this perspective, we aim at highlighting recent relevant advances in the field of MSPs, with special regard to the supramolecular polymerization of discrete metal complexes from a purely academic, as well as an application-oriented point of view. With increasingly complex systems being studied, more exhaustive investigations detailing multiple aspects of supramolecular polymers are being reported. Therefore, making a clear-cut classification of these examples is a tricky subject, as this unavoidably leads to some overlap between the sections. Consequently, we will focus the different sections on specific aspects of the presented examples to highlight key principles and properties of MSPs.
2 Scope and limitations
The term “self-assembly” has been used in different contexts in literature to describe multiple types of supramolecular systems.4,32,33 Thus, we will narrow the focus of this perspective, to reversible supramolecular polymerization, excluding those systems that are not self-assembled from discrete metal complexes, or where self-assembly is irreversible. Accordingly, coordination polymers,34 metal–organic frameworks35,36 as well as the self-assembly of discrete metallomacrocycles/-cages37–41 are out of the scope of this article. For the latter systems, recent relevant reports of metallomacrocycle/-cage-based SPs have been published,42,43 but an adequate discussion of these systems would require a very comprehensive introduction to the fundamentals of their self-assembly prior to supramolecular polymerization. We refer the reader to excellent articles published elsewhere, which document recent progress of the field.44,45 Further, we will focus our attention to self-assembly in solution, whereas purely solid-state research (e.g. crystal engineering)46–48 will not be reviewed. However, due to the fact that the self-assembly of metal-based building blocks has been greatly biased by “conventional” metal-free SPs, we will also highlight some relevant examples based on metal-free systems.Hence, we would like to point out that this article is not intended to be a thorough literature survey, but rather a condensed summary of selected relevant examples, with a particular focus on developments of the last five years. This is also evident from the fact that the great majority of the examples outlined in this perspective describes d8 metal complexes (namely PtII and PdII) besides a few highlights of complexes with a d10 metal configuration, as these metals cover the majority of literature examples on MSPs. Despite the beneficial properties (i.e. photophysical, or catalytic) of d8–d10 transition metals, the lack of comprehensive research on complexes of other transition metal ions remains a limitation of the field, hampering the discovery of new, unprecedented properties and self-assembly phenomena.
In many aspects, SPs and particularly MSPs, have shown exciting properties towards real-life applications, yet examples remain scarce. While showcasing recent progress, we will also discuss current roadblocks in the advancement of these compounds towards actual applications, ultimately giving our personal view on the potential scientific impact of metallosupramolecular polymerization.
3 Control over molecular packing and energy landscapes in supramolecular self-assembly
Manifestations of the potency of metal-containing systems
The potential of metal complexes as molecular building blocks in self-assembled systems with regard to different properties (photophysical, biological, electronic, catalytic, etc.) was recognized several decades ago.49–51 In addition to common molecular design strategies to induce supramolecular polymerization, such as the introduction of aromatic interactions or hydrogen bonding motifs, the inclusion of metal centres offers new intermolecular binding sites. For example, the polarized, metal-bound halogens can act as hydrogen bond acceptors, while the square planar coordination geometry of d8 complexes offers the possibility for short metal–metal contacts between adjacent molecules upon stacking. Moreover, the application of porphyrin moieties as binding sites for metal ions represents a common approach for biomimetic molecular designs. The growing interest in self-assembly and SPs in general also boosted MSP research and soon the first reports of metal-containing SPs of increased complexity were published. In this regard, in 2011, the group of Rybtchinski reported a remarkable example of a “pathway-dependent” self-assembly process of two PtII complexes in aqueous media.52 The complexes were shown to form multiple self-assembled structures with distinct properties. A similar phenomenon of diverse (competing) self-assembly pathways, termed as “pathway complexity”, was observed by Meijer and co-workers for an oligo(p-phenylenevinylene) derivative.24 In this seminal report, kinetic effects in complex supramolecular polymerization processes were elucidated both experimentally and using simulations.Whereas the early studies on metallosupramolecular polymerization primarily focused on the isolation of distinct aggregates using different experimental means, mechanistic studies and their link to the molecular structure were generally overlooked at that time. Meanwhile, after the introduction of the concept of “pathway complexity”, significant advances were made in the field of more conventional, purely organic SPs, where the focus had shifted towards understanding and controlling kinetic effects and their molecular origin. In light of these developments, the importance of correlating the molecular structure to kinetic effects and, in turn, complex self-assembly behaviour was soon recognized in the field of MSPs. To this end, a precise characterization of the secondary forces driving self-assembly, which is directly linked to the molecular design, was necessary. In addition to the supramolecular synthons known from organic SPs (e.g. π-surfaces, H-bonding sites, donor–acceptor moieties etc.), initial efforts were necessary to elucidate the role of new interaction motifs arising from the introduction of a metal fragment in MSPs.
In 2013, our group reported one of the first mechanistic investigations of the supramolecular polymerization of a metal-containing building block. The comparison of the self-assembly properties of an oligophenyleneethynylene (OPE)-based bispyridyldichlorido PdII complex 1 with a structurally analogous, metal-free reference compound 2 revealed intermolecular Pd⋯Cl interactions as efficient driving force for the supramolecular polymerization of 1 (Fig. 2).53 In addition to aromatic interactions, these orthogonal, secondary interactions induced a cooperative self-assembly mechanism, which was not observed for the reference compound lacking the metal fragment.
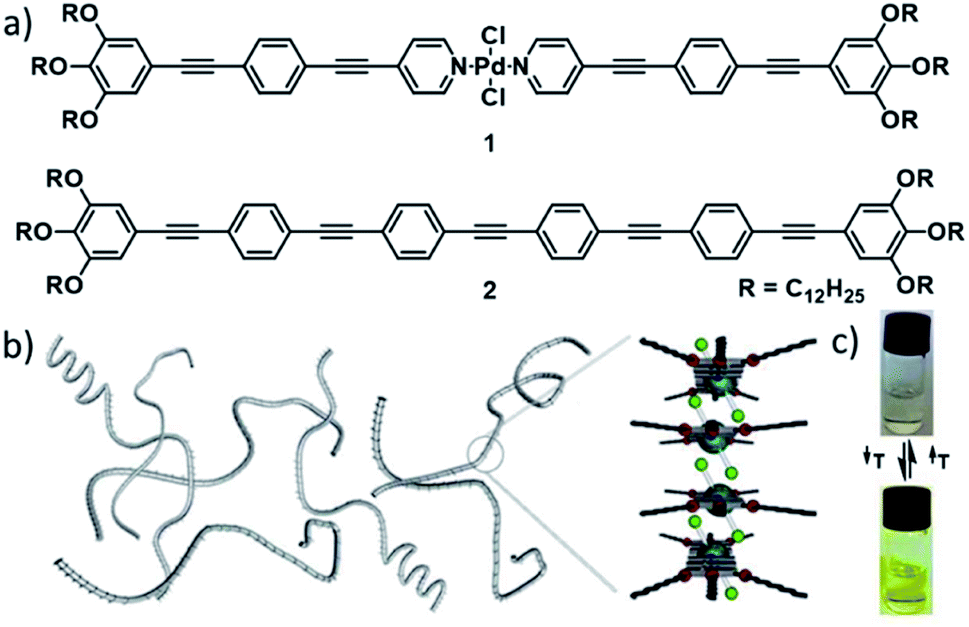 | ||
| Fig. 2 (a) Molecular structures of PdII complex 1 and reference OPE 2. (b) Schematic depiction of self-assembled fibres of 1 and the molecular packing around the metal centre. (c) Photographs of solutions of 1 showing the color change upon reversible self-assembly through temperature variation. Panels (b) and (c) adapted with permission from ref. 53. Copyright 2013 American Chemical Society. | ||
A plethora of studies investigating different molecular design aspects, such as the nature of the metal ion,54,55 coordination geometry,56,57 the influence of ancillary ligands,58 different linking units,59,60 chain length at the ligand backbone61 or the influence of counterions62 for charged complexes have brought about important insights on aspects of metallosupramolecular polymerization. From these diverse studies, a few generally applicable rules, which draw parallels to “conventional” SPs could be confirmed. For instance, it has become evident that orthogonal non-covalent interactions facilitate cooperative polymerization mechanisms, in analogy to SPs based on purely organic monomer units. However, in our view, a comprehensive understanding linking molecular design with self-assembly properties is in some aspects still lacking.
Pathway control and kinetic effects
Pathway complexity represents one of the most relevant research topics within the field of (metallo)supramolecular polymerization. Especially with regard to kinetic effects,24 more broadly applicable structure–property relationships remain to be established, particularly in MSPs. This can in part be attributed to the wide structural variety of metal ions and ligand designs, of which each combination can render individual, new and competing or cooperative secondary interactions. In this section, we will highlight some relevant examples that shed light onto the structure–property relationships regarding a certain family of molecules or a distinct aspect of the molecular design of metal complexes.As outlined in the previous section, the metal ion itself may have a significant impact on the supramolecular polymerization (e.g. through engagement in metal–metal interactions). Yet, the metal fragment can also be indirectly involved in the interactions governing the molecular packing and thus be responsible for the kinetic effects constituting diverse polymerization pathways. Among others, MII–X⋯Y interactions (X = halogen, Y = polarized hydrogen), or solvophobic effects play an important role.
In the recent past, our group has demonstrated the importance of N–H⋯Cl–MII interactions in enabling kinetic self-assembly pathways for different bispyridyldihalogen PtII/PdII complexes bearing amide linkers in their periphery (Fig. 3).56,63–66 These complexes can engage into either a kinetic or a thermodynamic self-assembly pathway, directed by cooperative aromatic and N–H⋯Cl–MII interactions or aromatic and N–H⋯O interactions, leading to a slipped or parallel molecular arrangement, respectively. In the case of PtII complex 3, the energy difference between the two self-assembled states is so small, that both packing modes can coexist concomitantly without interconversion, a phenomenon that bears close resemblance to polymorphism in crystals. For the PdII complex 4 on the other hand, the kinetic (N–H⋯Cl–Pd) pathway is “hidden” in nature due to its very low elongation temperature (i.e. the critical temperature below which the polymer growth sets in) compared to the thermodynamic (N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) pathway. Nevertheless, the appropriate aggregate preparation protocol by means of thermal or solvophobic quenching could enforce kinetic self-assembly processes, which once enabled, sequestered the monomers from the coupled polymerization equilibria in solution by a rapid hierarchical clustering process. The fact that related PtII/PdII complexes lacking the amide moiety self-assemble into a single SP driven dominantly by aromatic interactions67,68 nicely illustrates the important role of the metal fragment in regulating the energy landscape of SPs.
C) pathway. Nevertheless, the appropriate aggregate preparation protocol by means of thermal or solvophobic quenching could enforce kinetic self-assembly processes, which once enabled, sequestered the monomers from the coupled polymerization equilibria in solution by a rapid hierarchical clustering process. The fact that related PtII/PdII complexes lacking the amide moiety self-assemble into a single SP driven dominantly by aromatic interactions67,68 nicely illustrates the important role of the metal fragment in regulating the energy landscape of SPs.
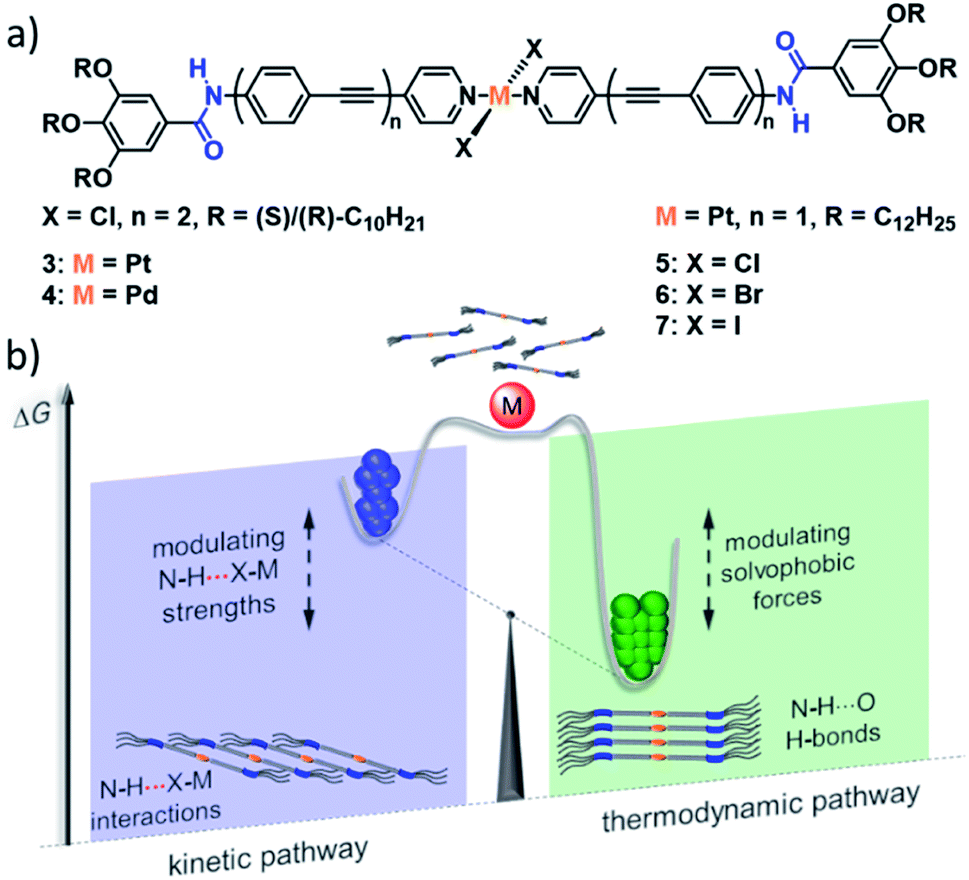 | ||
| Fig. 3 (a) PtII and PdII complexes 3–7 investigated by our group, unravelling N–H⋯X interactions of different strength leading to the competition of a slipped and a parallel molecular packing. (b) The relative magnitude of these interactions in comparison to other driving forces has a significant impact on the energy landscape of self-assembly, engendering hidden aggregates and supramolecular packing polymorphism. Panel (b) adapted with permission from ref. 58. Copyright 2021 American Chemical Society. | ||
Recently, we complemented our studies on the influence of N–H⋯halogen interactions by systematic variation of the Pt-bound halogen in complexes 5 (Cl), 6 (Br) and 7 (I).58 In accordance with a decreasing N–H⋯X binding strength, the chlorine-bearing molecule could form stable kinetic, slipped aggregates, whereas the Br-based derivative only exhibited a transient kinetic self-assembly and the iodine complex was not able to form a species stabilized by N–H⋯I interactions. Moreover, the study revealed that the steric demand imposed by the halogens is of minor importance, but instead, they exert increasing solvophobic effects stabilizing the parallel, thermodynamic packing. Thus, the metal centre can engage in a large variety of attractive and repulsive interactions, which can be exploited to modulate the energy landscape of SPs through a simple structural modification. This series of metal complexes also demonstrates the feasibility of theoretical methods to correlate supramolecular properties to molecular packing and pathway complexity. Semi-empirical PM6/7 methods were used to optimize dimers and tetramers of the respective molecular stack of 5–7. These optimizations not only confirmed the experimentally observed trend in stabilization energies, but also offered a reasonable explanation for the experimentally observed weakening of aromatic interactions, based on a distortion of the aromatic backbone to compensate the steric demand of the bulkier halogens in 6 and 7.
In addition to these fundamental research approaches, control over the aggregation pathways is also highly relevant to understand and eventually mimic complex biological self-assembly. It is widely known that biological functional systems operate under far-from-equilibrium conditions, where multiple metastable structures of the same monomer entity can exist. In this context, the group of Würthner has investigated the self-assembly of different ZnII chlorin derivatives as bioinspired semi-synthetic analogues of naturally occurring bacteriochlorophylls.69–72 The self-assembly of these compounds is conditioned by the formation of coordinative bonds between the hydroxyl group of one molecule and the metal centre of an adjacent molecule, leading to the formation of stable J-aggregates. For example, ZnII chlorine 8 can engage into two different J-type aggregation pathways, affording kinetically trapped dimers (J1-aggregate) or thermodynamically controlled, extended J2-aggregates (Fig. 4).73 The different molecular arrangements drastically affect the morphology (small nanoparticles for J1, 1D helical fibres for J2), UV/Vis absorption and chirality of the self-assembled structures. Detailed thermodynamic and kinetic analyses revealed that the two species are in competition, as the 1D structures, which are only 6.9 kJ mol−1 more stable than the J1-nanoparticles, can only be obtained directly from the monomer. Furthermore, the kinetic stability of the off-pathway J1-aggregates could be increased to the point that their transformation to J2 species was inhibited, simply by increasing the fraction of water in the water/MeOH solvent system. This behaviour bears close resemblance to out-of-equilibrium self-assembled structures in natural chlorosomes.
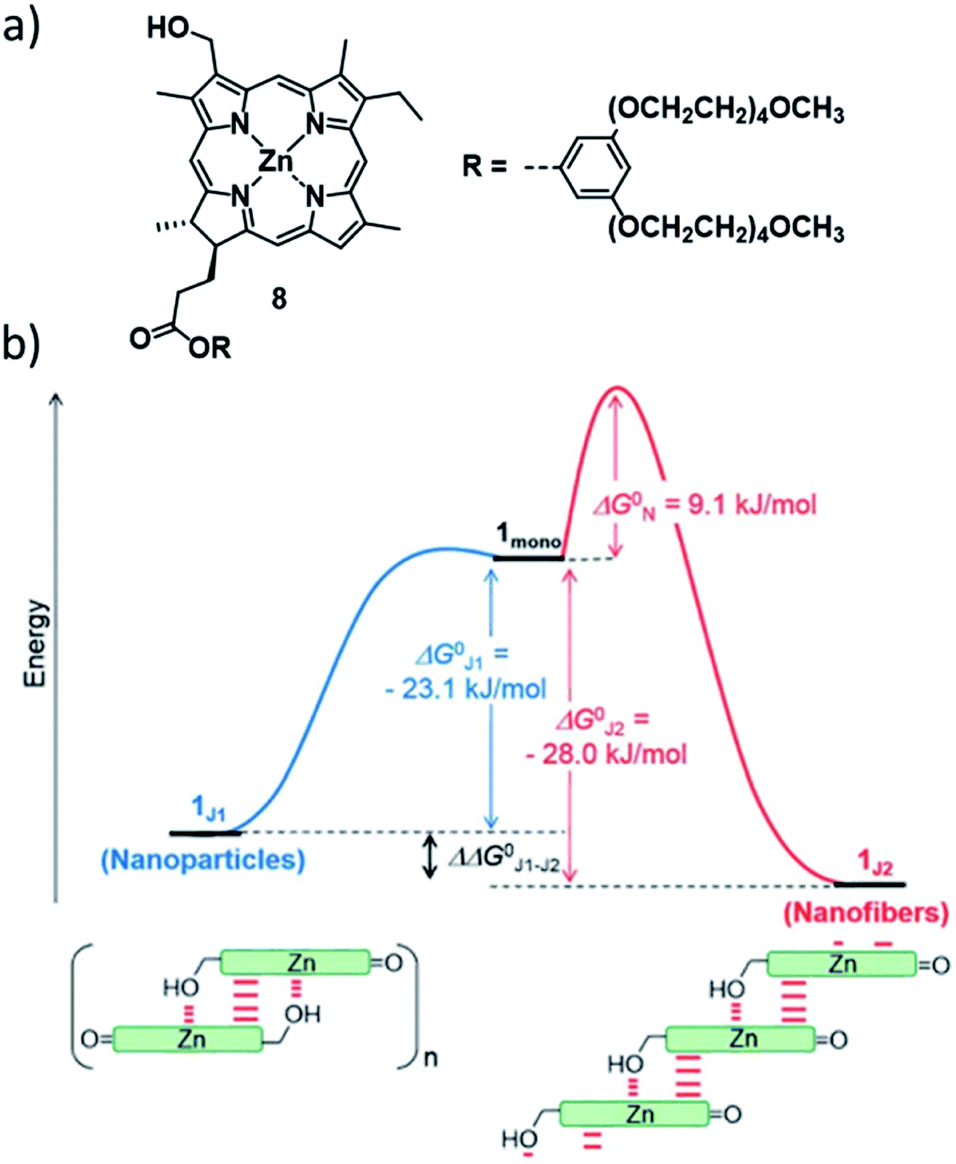 | ||
| Fig. 4 (a) Molecular structure of ZnII chlorine 8 investigated by the group of Würthner. (b) Energy landscape of the self-assembly of 8 illustrating the supramolecular polymerization into two different J-type aggregate species. Panel (b) adapted with permission from ref. 73. Copyright 2018 Royal Society of Chemistry. | ||
These representative examples of self-assembled metal complexes outline the significant impact of diverse interaction patterns involving either the metal ion itself or the (polarized) coordinated ligands attached to it. This underlines the capability of including metals into the molecular design of self-assembling monomers to regulate self-assembly pathways, which is relevant for the preparation of supramolecular materials with intriguing properties and also to understand more complex natural systems.
Supramolecular packing & photophysical properties
The outstanding inherent (photo-)physical properties of metal complexes, which can be further tuned by self-assembly, have attracted researchers' interest in this class of materials. Self-assembly-associated photophysical phenomena such as tuneable absorption wavelengths and Stokes shifts, aggregation-induced enhanced emission (AIEE) and/or increased lifetimes of excited states, among others, make discrete metal complexes interesting candidates for applications in the fields of e.g. optoelectronics or bioimaging.21,74–76 The photophysical properties of the complexes are strongly conditioned by their molecular packing and close chemical environment, which can be controlled through supramolecular polymerization along different pathways. Several groups have investigated a plethora of compounds, dominantly based on d8–d10 transition metals. In this section, we will highlight some key strategies to achieve desirable photophysical properties based on diverse research approaches.A strategy to enable metal–metal interactions in the aggregated state and, in turn, control the emission properties was recently reported by our group using a combination of molecular and coordination geometry changes. V-shaped, bipyridine-based complex 9, which displays a high degree of preorganization, self-assembles into H-type 1D SPs with short Pt–Pt contacts (Fig. 5).57 Linear, (bis)pyridine-based complex 10, in contrast, does not exhibit close metal–metal interactions as a result of steric effects leading to a short-slipped J-type packing mode in the aggregated state. Besides the effect on the luminescent properties (longer lifetimes, red MMLCT emission for 9 due to Pt–Pt contacts vs. shorter lifetimes, blue ligand centred emission for 10), the altered coordination geometry also promoted different energy landscapes of supramolecular polymerization: although both compounds self-assemble in a cooperative manner, 9 exhibits pathway complexity via an on-pathway kinetic species, whereas the aggregation mechanism of 10 is characterized by a single-step assembly behaviour.
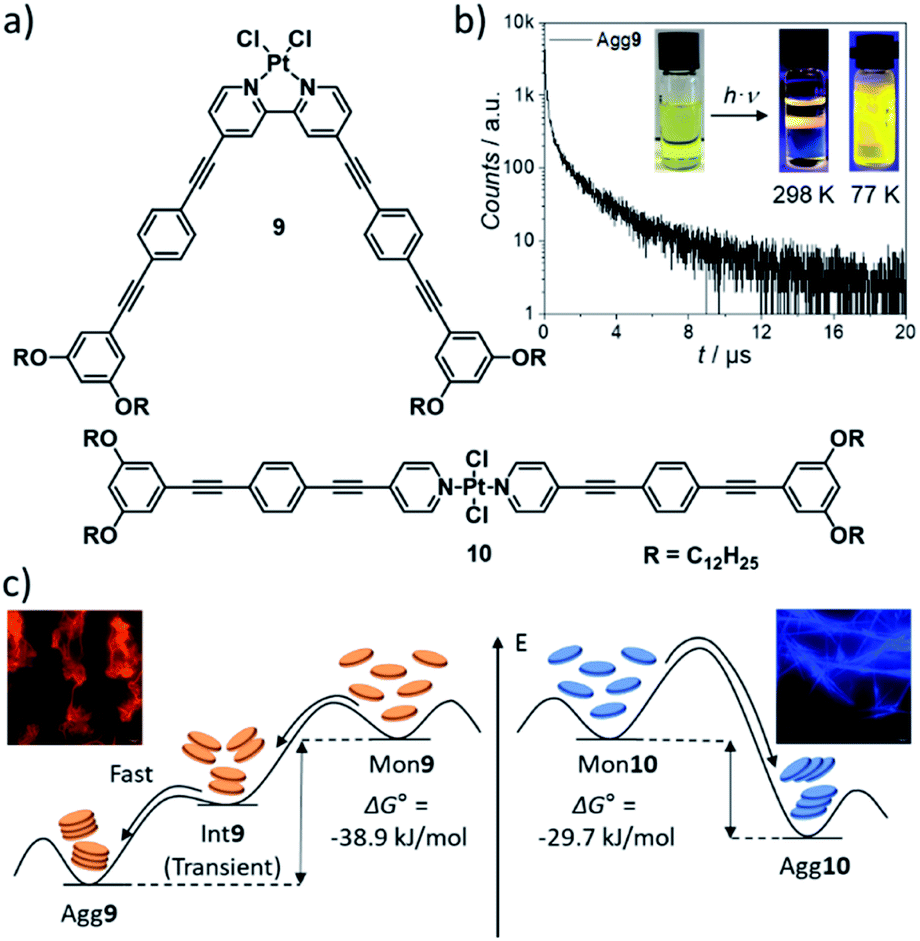 | ||
| Fig. 5 (a) Molecular structures of V-shaped complex 9 and linear complex 10. (b) Time-resolved photoluminescence decay of Agg9 at 298 K. (c) Schematic energy landscape illustrating the distinct supramolecular polymerization behaviour induced by a variation of geometry with luminescence micrographs of Agg9 and Agg10. Panels (b) and (c) adapted with permission from ref. 57. Copyright 2021 Royal Society of Chemistry. | ||
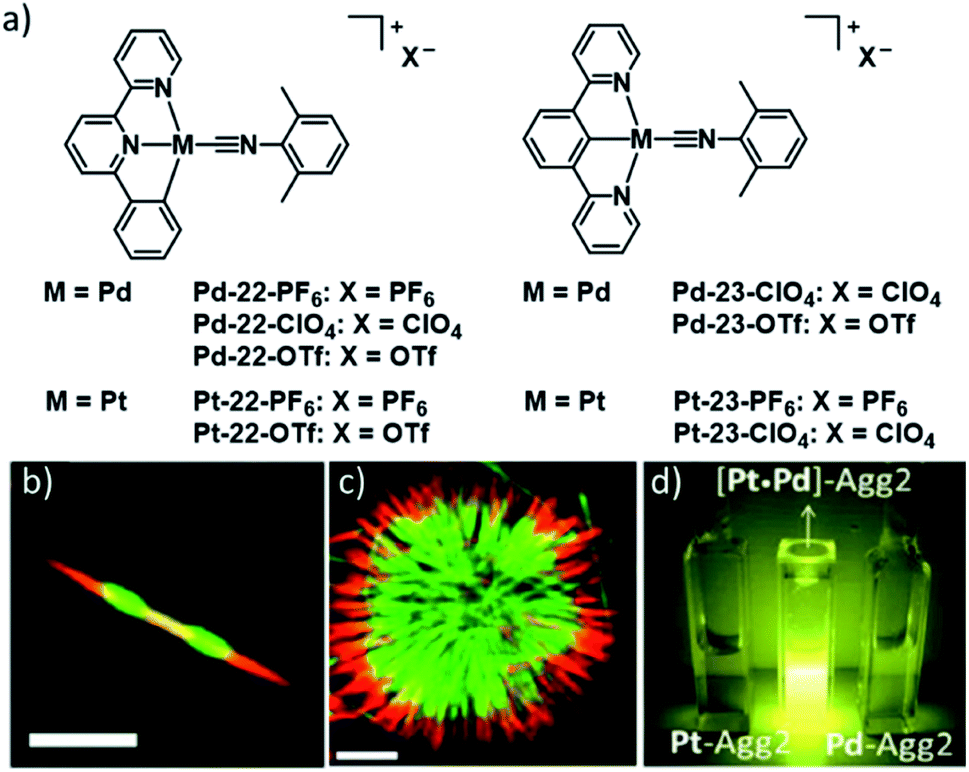
A more intricate example highlighting the potential to fine tune the extend of Pt–Pt interactions was recently demonstrated by the group of Che. In this report, a series of different (weakly) coordinating counterions was used to control intermolecular metallophilic interactions. The authors reported on the supramolecular polymerization of a series of luminescent pincer PdII isocyanide complexes 11–13 showing differing photophysical properties depending on the extent of anion–cation interactions (Fig. 6).62 Depending on the anion, the authors found two different aggregated species, a metastable state with “counterion-blocked” cation pairs (Agg1), and the thermodynamically favoured 1D SPs with close metal–metal contacts (Agg2). The metallophilic interactions in the 1D arrays gave rise to intense 1MMLCT absorption and 3MMLCT emission bands, which were not detected for Agg1. The competition between attractive cation–anion electrostatic interactions and attractive cation–cation dispersive interactions in Agg1 and Agg2, respectively, was identified to be the origin of the two pathways. Modulation of the anion's tendency to interact with a pyridyl ring of the tridentate ligand was exploited to tune the self-assembly: substitution of the PF6 ion (fastest formation of Agg2) by an anion exhibiting stronger anion-π interactions (ClO4−), led to a retarded formation of Agg2, or even a suppression of this pathway in the case of OTf− or CF3CO2−. The different excited states of the two aggregated species were investigated by means of DFT/TDDFT, corroborating the MMLCT character of absorption and emission due to a larger metal-centred character of the HOMO in Agg2, whereas the HOMO in Agg1 is ligand-centred, enabling only non-emissive IL-π–π* transitions.
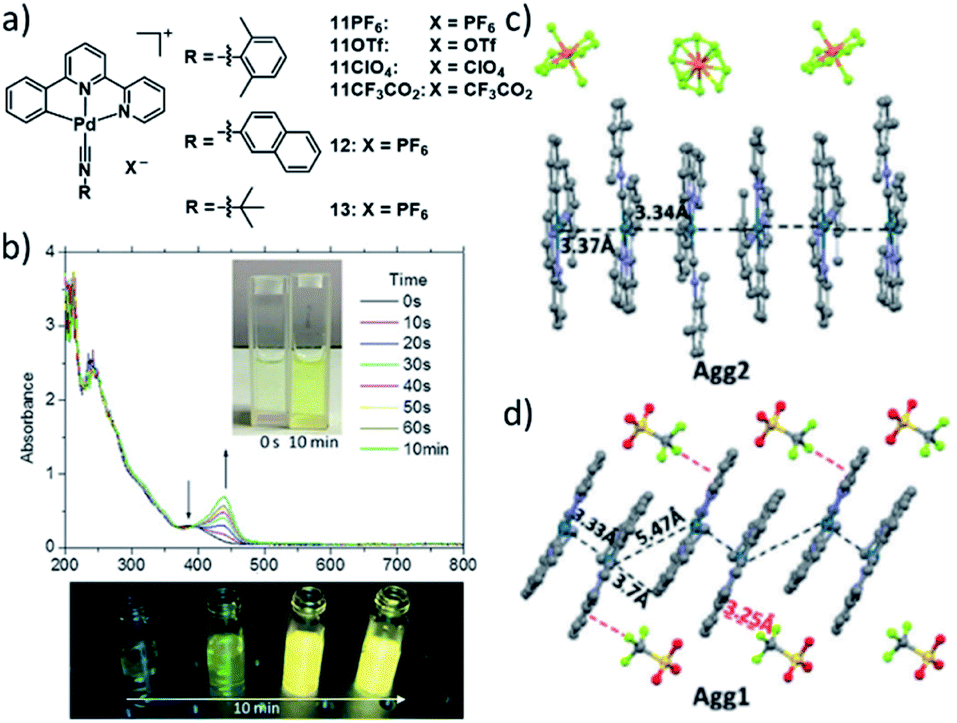 | ||
| Fig. 6 (a) Chemical structures of pincer-isocyanide PdII complexes 11–13. (b) Top: time-dependent absorption spectra of 11PF6 and photographs of the solution. Bottom: photographs showing the corresponding changes of the emission of 11PF6 over time. (c and d) Crystal structures of 11PF6 and 11OTF6 illustrating the two possible aggregation modes with (11PF6) and without (11OTF6) metal contacts. Panel (b)–(d) adapted with permission from ref. 62. Copyright 2018 John Wiley and Sons. | ||
Interestingly, the control over emissive properties is not strictly limited to different aggregated states, but can also be controlled in a more complex self-assembly energy landscape encompassing more than one configuration of the monomer species. This was recently demonstrated in an example by Yam and coworkers, who designed a series of PtII-based molecular hinges in which two cyclometalated PtII complexes (the hinges' wings/flaps) are connected by a rigid aromatic alkynyl group (axis).77 Molecular hinge 14 can exist in an open and a closed conformation with distinct emissive properties (Fig. 7). The more stable, red-phosphorescent, closed form is present in good solvents such as DCM. A molecular rotation to the less stable open form of the hinge causes a distinct, green phosphorescence. Computational optimization (DFT) of ground and excited state geometries/energies was used to corroborate the assigned triplet emissions. The thermodynamically less stable open form can be favoured by decreasing the solvent polarity: to compensate the reduced solvation, aggregation of the open form into 1D fibres occurs via intermolecular π–π interactions, which is not possible in the closed conformation. Additionally, variation of the temperature was exploited to reversibly switch between the open and closed form in apolar media: at high temperatures, the SPs were disrupted causing a transformation to the closed species and vice versa.
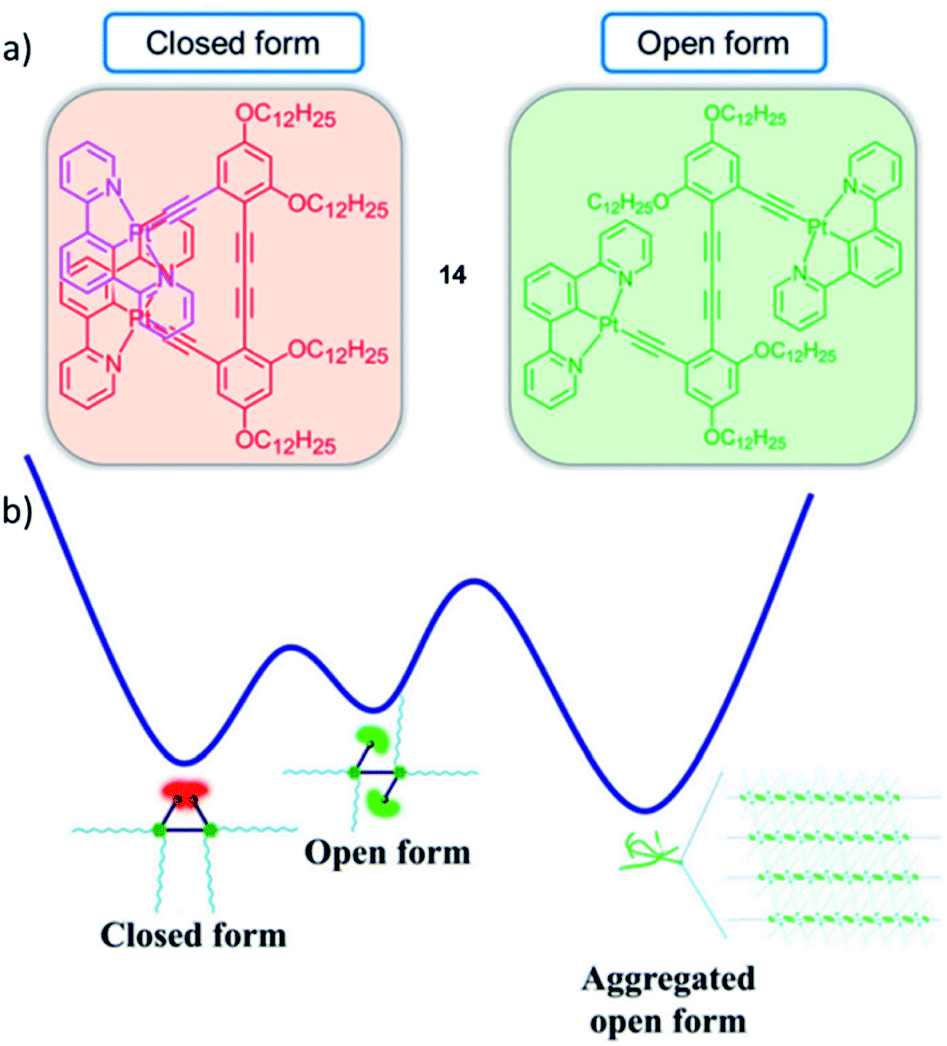 | ||
| Fig. 7 (a) Closed and open forms of PtII molecular hinge 14 investigated by the group of Yam. (b) Qualitative energy landscape illustrating the conversion processes between the different species. Adapted with permission from ref. 77. Copyright 2019 National Academy of Sciences. | ||
The selected examples highlighted in this section bring to light the versatile photophysical properties of self-assembled metal complexes, which can be tuned both by molecular design and by careful control of sample preparation techniques. Additionally, the examples demonstrate the feasibility of computational chemistry methods not only to elucidate energy landscapes and packing modes, but also in correlating them with the observed photophysical phenomena.
4 Tuning morphologies and size distribution
Controlling the nanoscale morphology of self-assembled structures is a key requirement for the development of functional materials, which has attracted significant research interest in the past decades. In accordance with the large variety of self-assembling molecular building blocks, the morphological diversity of the corresponding self-assembled structures is equally immense, covering different size regimes and dimensionalities. In the following section, we wish to demonstrate some elegant examples where precise morphology control was achieved by tuning both primary (i.e. interactions driving the formation of a molecular stack) as well as secondary interactions (i.e. interactions directing the spatial arrangement of multiple polymer stacks into higher order structures) in the molecular design. We will also emphasize how the previously described control over energy landscapes and pathway selection can be employed to achieve desired morphologies.The high potential of controlled secondary interactions has been recently highlighted by the groups of Yagai as well as the groups of Takeuchi and Sugiyasu. Yagai and co-workers demonstrated the formation of concentric toroids as the starting point for secondary nucleation events.78–80 Based on the specific size of these toroids, the nucleation process led to the formation of polycatenanes, unique hierarchical structures that had remained previously elusive at the supramolecular level.
Another highly relevant recent example in this regard is a family of ZnII porphyrin based complexes reported by the groups of Takeuchi and Sugiyasu (Fig. 8).81–84 Derivatives with short alkyl substituents in the R′ position initially form J-type nanoparticles (NPs), which rearrange over time to H-type 1D fibres (Fig. 8b).82 More strikingly, the stepwise increase of the length of the alkyl side chain from methyl to heptyl resulted in an entirely new self-assembled morphology. Compounds with intermediate alkyl chain lengths (hexyl 15HH and heptyl) could form a slipped packing which resulted in a lamellar arrangement of multiple 1D stacks, forming 2D sheet structures (Fig. 8). This drastic change in the energy landscape and supramolecular morphology could be rationalized by additional lateral van der Waals (vdW) interactions. Nevertheless, the formation of the previously observed H-type 1D fibres was also achieved by application of mechanical agitation. Thus, the authors could selectively direct the self-assembly in this “extended” pathway complexity system along one of the two pathways of the energy landscape, in both cases starting from the kinetically formed J-type nanoparticles. The authors envisaged that a constant increase in vdW interactions could be used to further modulate the morphology towards desired aspect ratios based on the balance between 1D slipped stacking and lateral alkyl chain interactions. However, above a critical chain length, the self-assembly behaviour reverted to the simpler two-species system featuring only J-type NPs and H-type nanofibres.84 This initially unexpected structure/property relationship could be correlated to unfavourable enthalpic contributions based on the pronounced conformational freedom of alkyl chains longer than heptyl.
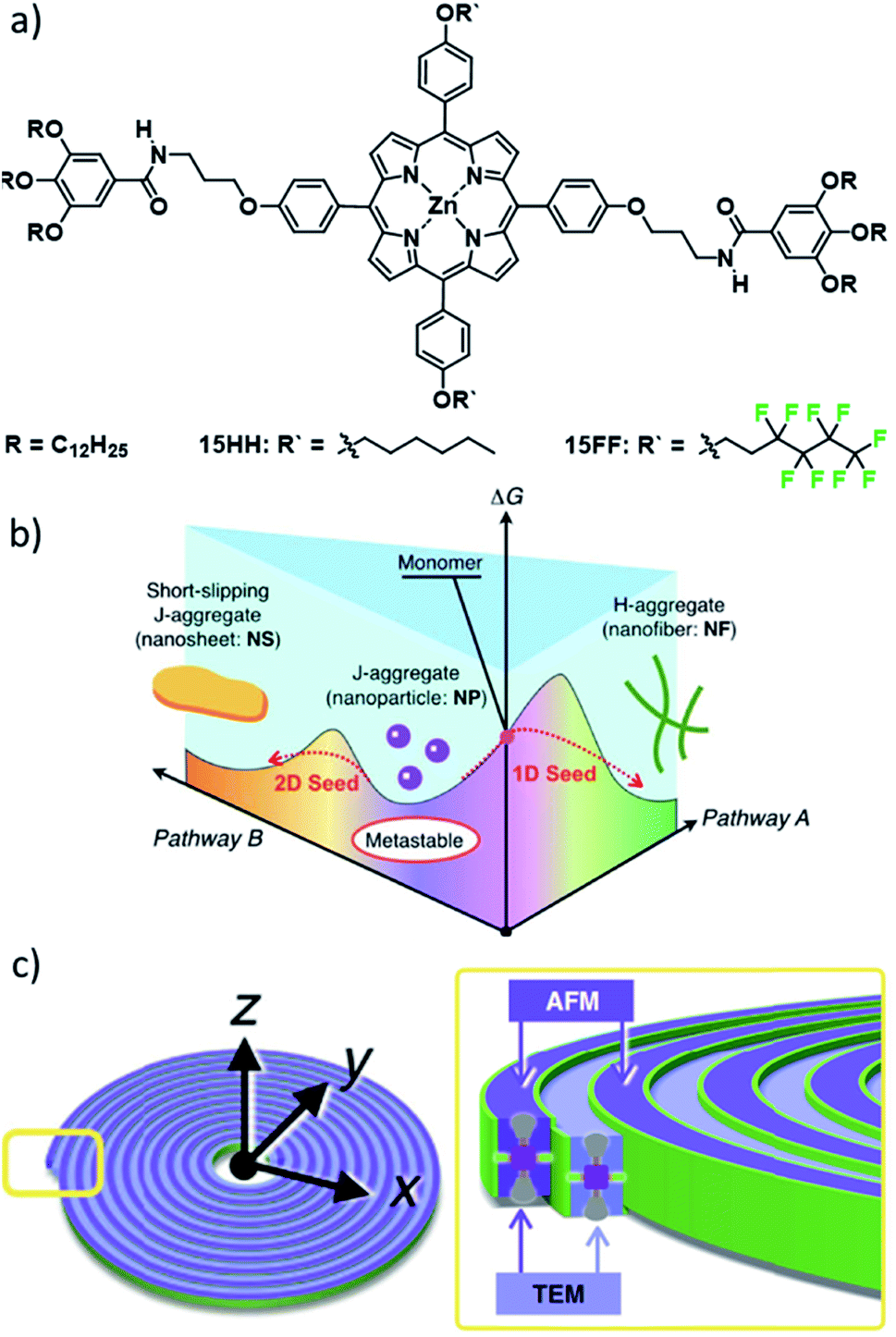 | ||
| Fig. 8 (a) Molecular structures of ZnII porphyrins 15 investigated by the groups of Sugiyasu and Takeuchi. (b) Complex energy landscape of the self-assembly into different morphologies. (c) Schematic representation of the self-assembled Archimedean spirals formed by 15FF. Panels (b) and (c) adapted with permission from ref. 82 and 81, respectively. Copyright 2020 John Wiley and Sons and Copyright 2020 Springer Nature. | ||
An even more striking change in morphology was achieved by replacing the hexyl side chains of 15HH with fluorinated side chains to give 15FF (Fig. 8).81 The spectroscopic observations for the aggregated state (hypsochromic shift) indicated the formation of the known H-type 1D fibres. Surprisingly, a combined morphological elucidation by AFM and TEM revealed the presence of unusual double-stranded Archimedean spirals. The authors attributed this morphology difference to a further stabilization of the 1D fibres by a fluorophilic effect, producing the spirals. Additionally, thorough thermodynamic analysis unveiled a different self-assembled structure at low concentrations. This notion could by confirmed by AFM analysis showing the formation of layered concentric toroids. This slightly altered morphology could be correlated to the initial formation of a concentric toroid with subsequent growth in the second dimension based on secondary nucleation events followed by elongation around the existing toroid in a process reminiscent of templated self-assembly. This intriguing behaviour highlights that control over self-assembled morphologies based on molecular design remains a major challenge, as secondary nucleation events or newly emerging hierarchical aggregation pathways can lead to unexpected outcomes. Additionally, these studies have shown that one molecular packing can coincide with multiple morphologies, as complementary secondary interactions can lead to distinct topologies, which are difficult to analyse based on spectroscopy alone.
This series of complexes highlights how balancing secondary interactions with primary intermolecular binding can lead to diverse supramolecular morphologies based on subtle changes in molecular design. Particularly, the flexibility within a one-dimensional assembly can dictate the superstructure morphology by engaging in efficient secondary bonding. A particularly elegant example of this fine balance has been recently reported by the group of Yam, who investigated a series of PtII based double complex salts 16–18 (DCS, Fig. 9).85 The double salts consist of a positively charged complex with a neutral 2,6-bis(benzimidazol-2′-yl)pyridine (bzimpy) ligand appended with TEG chains and a negatively charged PtII complex with a bzimpy ligand appended with anionic sulfonate groups. Both species of the double salt feature an additional anionic ligand (either Cl− or phenyl alkynyl). In the comparative study, the steric demand of the smaller anionic ligand was systematically altered by a stepwise replacement of the smaller Cl− with the sterically more demanding phenyl alkynyl. The self-assembly of DCS 16 exhibiting the lowest steric bulk yields isolated 1D architectures with minor flexibility. In contrast, DCS 17 self-assembles into highly flexible 1D fibres, which can further bundle leading to interwoven architectures. While in their report this behaviour was not desirable for the investigated application (semiconductivity), the results emphasize that flexibility can be introduced by a fine modulation of repulsive interactions. However, the investigations of DCS 18 reflect that considerable care should be taken when enforcing steric bulk, as the replacement of both Cl− ligands leads to another change in morphology from elongated fibres to short bundled nanorods. While inter-strand secondary interactions between the 1D polymers become more prevalent, an overall decrease in the elongation of the individual 1D fibres as a result of accumulated steric bulk, is responsible for this behaviour.
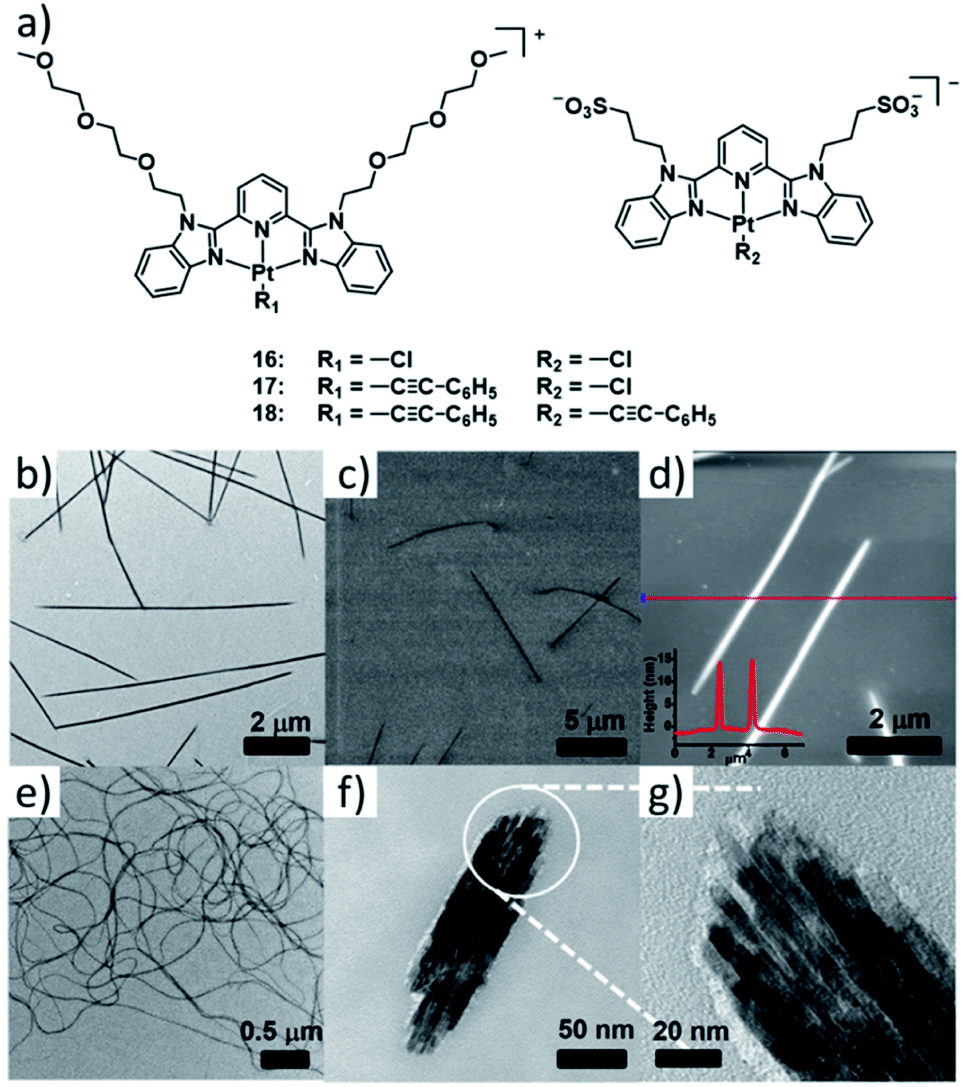 | ||
| Fig. 9 (a) Structure of complex double salts 16–18. (b) TEM, (c) SEM and (d) AFM images of the aggregate of 16 formed in water. (d–f) TEM images of the aggregates formed by 17 (e) and 18 (f and g). Panels (b)–(g) adapted with permission from ref. 85. Copyright 2018 American Chemical Society. | ||
These case studies have demonstrated the importance of controlling intermolecular interactions by targeted molecular design in the context of morphology control. However, sample preparation protocols are at least equally influential to regulate the outcome of supramolecular polymerization.24 In this regard, time-dependent experiments can be recognized as the simplest method to achieve diverse polymer architectures.86,87 Additionally, various other sample preparation methods to switch between supramolecular packings and self-assembled morphologies include the fine-tuning of solvent polarity,88 modulation of self-assembly dynamics by good/bad solvent ratios,86,89,90 as well as differing cooling rates, mechanical agitation11,82 or quenching methods. However, while all these examples offer a certain level of morphology control, they are typically unable to fully control its dimensions, such as overall size, aspect ratio, etc. Crystallization-driven self-assembly (CDSA) or living supramolecular polymerization (LSP) approaches are arguably the most commonly used sample preparation techniques to obtain precise control over multiple facets of the morphology, such as length, aspect ratio or polydispersity. Inspired by classical, covalent polymerization, the group of Manners has successfully established living crystallization driven self-assembly (LCDSA, Fig. 10).91 This approach is based on a stepwise growth of self-assembled structures through addition of monomers to aggregate seed-crystals and has produced promising systems for a number of applications.92 Using LCDSA, targeted block sequences,93 as well as diverse heteromorphologies94 or controlled branching95 can be achieved. LSP is based on the same principles as LCDSA, namely the successive addition of a kinetically trapped species (monomers or even aggregates) to a solution of seeds of a thermodynamically more stable species, causing a stepwise growth at the termini of the seeds.83,96,97 Usually, the species used for seed formation is the thermodynamically most stable one. While maintaining a narrow polydispersity, the increase in size is dictated by the ratio between the number of seeds initially used (typically obtained by sonication) and the amount of monomers that can be released from the aliquots of trapped species added. From this simplified description of supramolecular polymer growth in a living manner, some important requirements/limitations can be derived:
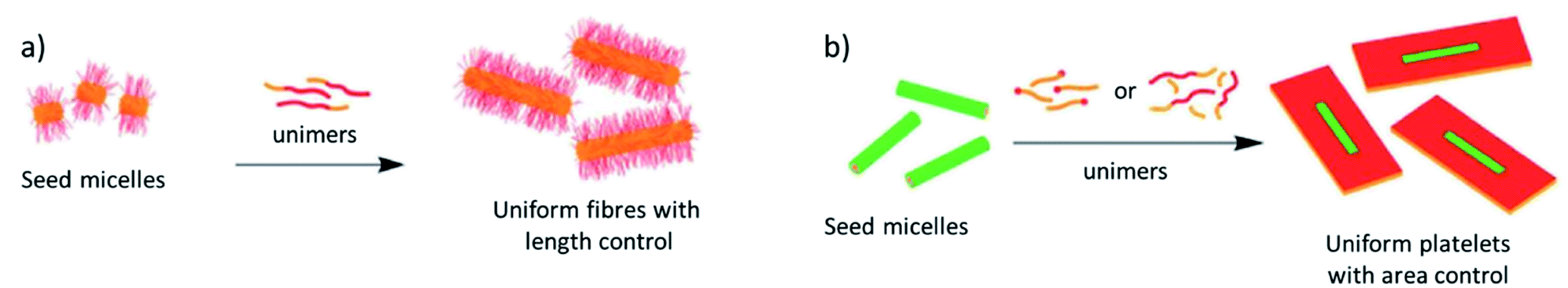 | ||
| Fig. 10 Representative examples of living crystallization driven self-assembly (LCDSA) in 1 (a) and 2 dimensions (b). Adapted with permission from ref. 92. Copyright 2021 Royal Society of Chemistry. | ||
(1) The approach is limited to systems where the monomer can be trapped either in a “dormant” monomer or a kinetically controlled aggregate, from which spontaneous elongation to the thermodynamically stable state is retarded.
(2) The final thermodynamic product needs to exhibit a certain stability to allow seed formation through mechanical agitation.
(3) Apart from some exceptions,11,82 only the most stable supramolecular structure is accessible. Thus, control over size is possible, but typically not over shape.
(4) Even if the prerequisite of a kinetically trapped state is fulfilled, a variety of literature examples show that the energy difference between the desired structure and the trapped one also plays an important role. Species that are close in energy (i.e. polymorphs)58,66 may not be susceptible to LSP. Furthermore, additional kinetic effects (such as topological ones) may also prevent efficient elongation upon seeding.98
Accordingly, while living approaches allow supramolecular chemists to effectively control size distributions, particularly at the mesoscale, precise control at the nanoscale remains challenging. In particular, the seed formation is a rather poorly controlled process and the factors governing this important step, apart from a rough correlation between the sonication time and the size of the seeds, remain to be understood. We hypothesize that size restricted self-assembly can be an ideal addition to LSP to control the nanoscale size regime. In this regard, distinct topologies, templated self-assembly99 as well as the use of end-capping compounds56,100,101 represent approaches that can enforce a definitive size limit. This potential has further been realized in detailed studies of particularly arranged and elongated oligomeric dye assemblies.102,103
Additionally, we argue that anticooperative self-assembly, which remains underrepresented in the literature beyond its mechanistic foundation, represents an ideal tool to achieve aggregates of defined size at the nanoscale.104–106 Build-up of steric demand, or increased electrostatic repulsion are some strategies towards the design of anticooperative SPs. The main restriction in these approaches has been the limited understanding of structure–property relationships to hamper extended growth. In this regard, MSPs represent particularly relevant model systems, as the diverse coordination geometries, potential ionic charges and increased sterics endowed by the metal centre and its various possible ligands, can be exploited to drive self-assembly towards an anticooperative process.
We hypothesize that this approach can serve as an ideal complement to LSP, which is particularly powerful at the mesoscale, while anticooperative self-assembly typically leads to only a small number of monomers in an oligomer stack, as well as slightly more extended short polymers.58 Ultimately, the recent surge in the understanding of secondary nucleation events and aggregate stabilization by secondary interactions serves as an ideal starting point to control the macroscale morphology of self-assembled structures with defined nanoscale size (either by size-limited growth or LSP).
Furthermore, we expect theoretical approaches to become a driving force for progress towards controlling or limiting supramolecular growth in the following years. Automatization in combination with large data analysis and machine learning, which have advanced considerably in the last few years, are a necessity to overcome the hurdles imposed by limited design options.107 In other fields, such as crystal structure prediction or porous materials design, this prospect has already become reality and we expect a similar development for supramolecular polymerization.108–111 Particularly, aspects of molecular design strategies that can be difficult to estimate beforehand, such as the formation of macrodipoles, the accumulation or delocalization of charge and synergistic effects upon self-assembly, will become more accessible with the combination of synthetic work and simulations. This dual approach requires a more intimate collaboration between research groups working on molecular self-assembly in the lab and in silico. While these collaborative projects have become more regular in the recent past, complementary experimental and theoretical work should be encouraged whenever possible.
5 Co-assembly/multicomponent metallosupramolecular self-assembly
Co-assembly represents a promising approach to combine the advantageous properties of different monomer units within one polymer structure or to even achieve emerging properties, not present in the individual components. Whereas in classical covalent polymerization, methods to control the type of copolymerization (i.e. statistical, alternating, gradient, block, graft copolymers etc.) are well established, control over different co-polymerization mechanisms in their supramolecular counterparts remains limited due to the inherent dynamic nature of reversible self-association. Apart from statistical supramolecular copolymerization, which requires a lesser degree of control, increasing attention has been recently devoted to block supramolecular copolymers (BSPs) and, to a lesser extent, to graft copolymers. In particular, LSP approaches have been vastly exploited to create BSPs. For more detailed information on different theoretical and practical aspects of multicomponent SPs, the reader is referred to excellent reviews recently published by the groups of Meijer, Palmans and Vantomme.112,113 In this section, we will present selected examples to highlight the potential of multicomponent self-assembly to achieve desired supramolecular and photophysical properties focused on metal-based monomers.Apart from the co-assembly of different molecular building blocks with complementary binding/interaction sites (e.g. H-bonding arrays,16 coulombic interactions,85,114,115 donor–acceptor units,116,117 host–guest moieties118etc.), where self-assembly is preferred as a copolymer, predicting the extent of copolymerization of different constituents in solution remains a challenging task. Despite the seemingly straightforward preparation of statistical/random copolymers by simple mixing of different building blocks in a monomeric state and subsequently inducing self-assembly, the segregated SP of the individual building blocks needs to be circumvented. A general approach to tackle this obstacle is the design of structurally similar building blocks,119 in which the intermolecular interactions anticipated for the components involved are the same or at least well compatible (e.g. matching/complementary H-bonding moieties or π-surfaces of similar size).
In 2019, our group investigated mechanistic aspects of the statistical co-assembly of metal complexes combining PdII (19) and PtII complexes (20) based on the same molecular design (Fig. 11).120 In isolation, the complexes self-assemble in n-decane into a similar pseudo-parallel arrangement, however resulting in different morphologies (fibres for PdIIvs. disks for PtII) due to a different rotational offset in the packing. Co-assembly of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of both complexes results in a two-step process: after a statistical co-nucleation event, social-self-sorting into Pd-enriched fibres takes place, followed by a self-organization of the residual Pt-monomers after full consumption of the Pd-monomers. Variation of the ratios of the two components on the one hand resulted in a more efficient copolymerization (single-step polymerization curves), whereas the elongation temperature and morphology of the resulting SPs was dictated by the component in excess. This illustrates how the degree of co-assembly and, thus, the material properties can depend on the molar ratio of two structurally similar complexes, although one might intuitively expect a more efficient statistical co-assembly if both species are present in equal amounts.
:1 mixture of both complexes results in a two-step process: after a statistical co-nucleation event, social-self-sorting into Pd-enriched fibres takes place, followed by a self-organization of the residual Pt-monomers after full consumption of the Pd-monomers. Variation of the ratios of the two components on the one hand resulted in a more efficient copolymerization (single-step polymerization curves), whereas the elongation temperature and morphology of the resulting SPs was dictated by the component in excess. This illustrates how the degree of co-assembly and, thus, the material properties can depend on the molar ratio of two structurally similar complexes, although one might intuitively expect a more efficient statistical co-assembly if both species are present in equal amounts.
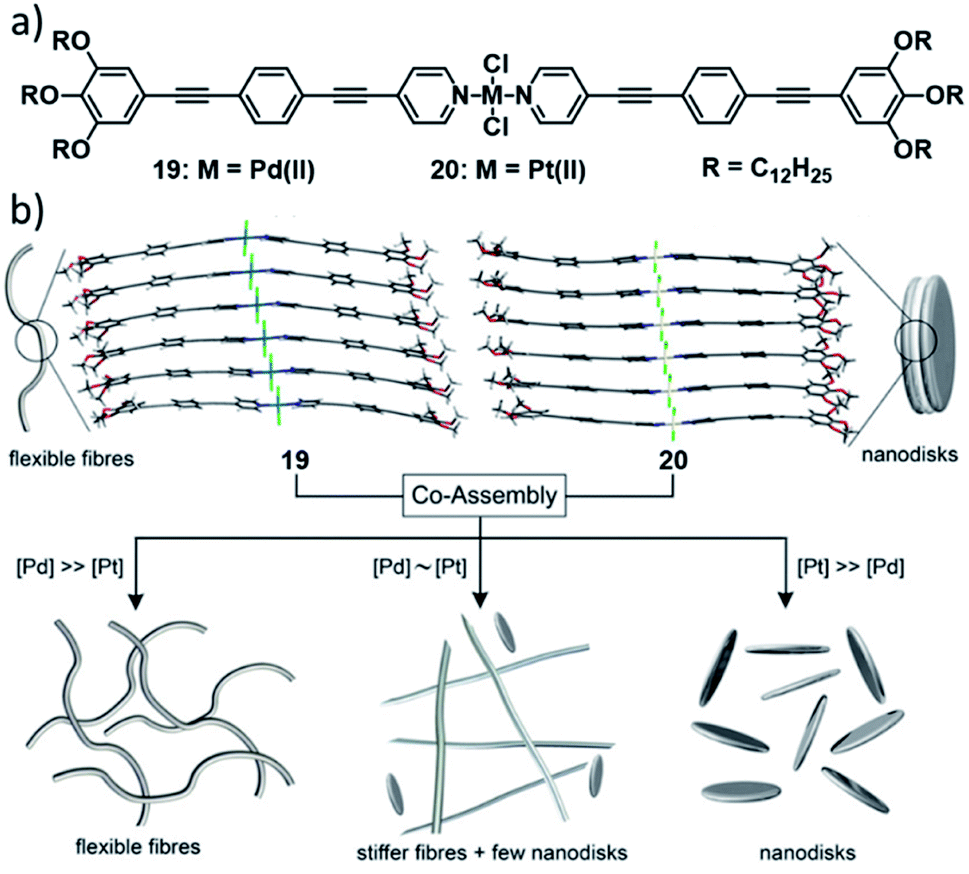 | ||
| Fig. 11 (a) Structure of complexes 19 and 20 which statistically co-assemble into nanofibres or nanodiscs when one component (which dictates the morphology) is in large excess, or into a mixture of Pd-enriched, co-assembled fibres and Pt-only nanodiscs at intermediate ratios (b). Panel (b) adapted with permission from ref. 120. Copyright 2019 John Wiley and Sons. | ||
Whereas a number of examples of statistical and block copolymers have been reported, examples on supramolecular graft polymers remain scarce. Only recently, the group of Das outlined the important role of the orthogonality of secondary interactions to achieve graftable supramolecular (co)polymers. The authors first polymerized a benzene-1,3,5-tricarboxamide (BTA, 21) motif into 1D SPs in aqueous media driven by threefold H-bonding and π-stacking (Fig. 12).121 Subsequently, a hydrophilic ethylene glycol shell was grafted to the molecule's periphery via orthogonal halogen bonding interactions to pendant pyridyl groups. The authors demonstrated that only the orthogonality of the H-bonding motif driving 1D self-assembly and the X-bonding interactions opened the door to post-assembly functionalization by graft copolymerization.
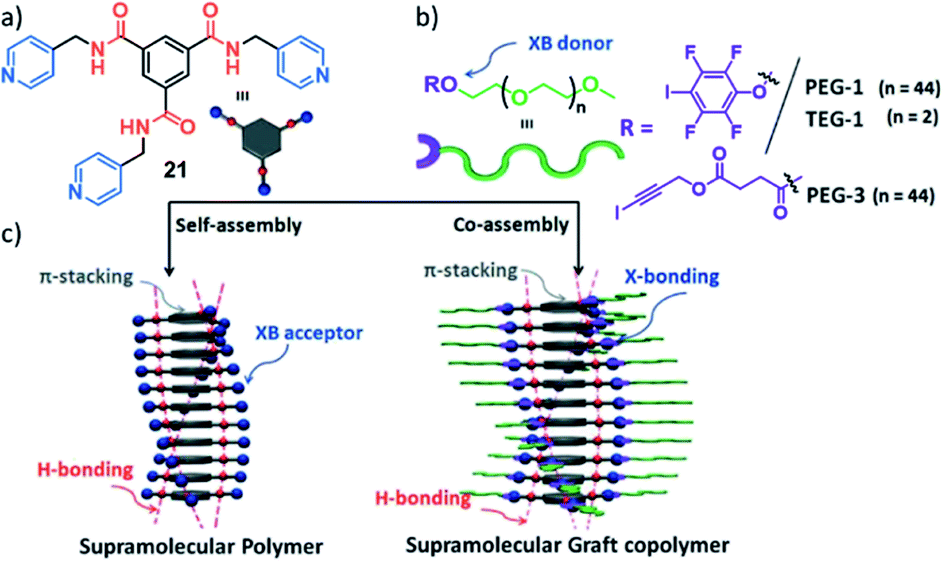 | ||
| Fig. 12 (a and b) Molecular structure of BTA 21 (a) and the halogen bonding donors (b) used for orthogonal secondary binding for graftable supramolecular (co)polymers. (c) Schematic representation of the self-assembly of 21 in the absence (left) and presence (right) of orthogonal halogen bonding donors. Adapted with permission from ref. 121. Copyright 2020 Royal Society of Chemistry. | ||
Especially in aqueous media, strong hydrophobic interactions and competing H-bonding with solvent molecules often times impose significant challenges on the controlled formation of supramolecular species with particular properties. This post-assembly supramolecular grafting-approach could be beneficial for the successful design of future applications in aqueous media.
Transferring the idea of graftable SPs to supramolecular polymers of metal complexes could represent a versatile approach to achieve tailored solubility and processability, which are key requirements and challenges in the fabrication of functional nanomaterials. For example, MSPs with extended M⋯M contacts in the core of the structure could be combined with orthogonal binding sites at the periphery suitable for grafting through various non-covalent interactions.
Emergent properties from heterometallic co-assembly
The previously mentioned prerequisite of geometrical complementarity and compatibility of non-covalent interactions in co-assembly can be easily met by employing the same ligand for different types of metal ions. The combination of multiple metal ions in SPs can not only bestow the resulting assemblies with a superposition of properties of the individual building blocks, but also amplify existing behaviours or give rise to emergent properties unobtainable by homometallic assemblies. In this section, we would like to highlight examples of co-assemblies of different metal ions and emphasize the potential of employing a heterometallic approach.In this regard, the group of Che published a remarkable example showcasing the potency of co-assembly with respect to controlling various aspects of self-assembly (Fig. 13). They took advantage of the LSP behaviour of the metal complexes previously described in Chapter 3 to construct supramolecular block copolymers.122 The group employed seeds of thermodynamic aggregates (Agg2) of a PtII or PdII complex and subsequently added a suitable kinetically trapped species (Agg1) of the other metal, respectively. In this way, they constructed block copolymers with up to seven segments and different dimensionalities/shapes (Fig. 13b and c). The distinct emission properties of complexes with PdII (green) or PtII (red) metal centres enabled a visualization of the block copolymers by confocal fluorescence microscopy. Apart from the ability to attach segments with different luminescence color, the authors were also able to fine tune the absorption and emission wavelengths through modulation of the ratio between the two complexes in the initial non-segmented co-nucleation/copolymerization. Application of mixed metal Agg2 seeds in subsequent LSP experiments adding segments of pristine PdII or PtII complexes yielded block copolymers with three different colours (Fig. 13b). Moreover, doping minor amounts (<5 mol%) of PtII complexes into PdII assemblies during supramolecular polymerization could amplify the phosphorescence efficiency of PdII assemblies: emission intensity and quantum yield (<4% to >75%), as well as lifetime (0.12 μs to 1.3 μs) were dramatically increased (Fig. 13d). The observed photophysical phenomena could be explained with the help of TD-DFT calculations in terms of a mixing of PtII 5dz2 and PdII 4dz2 orbitals upon formation of extended metal–metal contacts in the copolymers (→ 3MMLCT excited state), causing an external heavy-atom spin–orbit coupling.
 | ||
| Fig. 13 (a) Molecular structures of complex series 22 & 23. (b and c) Confocal fluorescence microscopy images of: (b) 1D 5-block yellow-green-red ([Pd:Pt 2:1]–Pd–Pt segments), (c) flower-shaped triblock green-red-green (Pd–Pt–Pd segments) copolymers. (d) Photograph comparing the emission of Agg2 of pure Pt-complex (left), Pt-doped Pd-copolymer (middle) and pure Pd complex (right). Scale bars: 5 μm. Panel (b)–(d) adapted with permission from ref. 122. Copyright 2020 Elsevier. | ||
Apart from manipulation and control of photophysical properties, supramolecular copolymerization can also have implications for the chemical stability/inertness of SPs. The groups of Pavan, Takeuchi and Sugiyasu reported on the stabilization of SPs of ZnII porphyrin 24-Zn against depolymerization by Lewis bases (4-dimethylaminopyridine, DMAP), through formation of supramolecular block copolymers with their copper homolog 24-Cu (Fig. 14).119 The SPs of pristine CuII porphyrin, are by nature inert to depolymerization by DMAP due to the low affinity of CuII porphyrins for DMAP coordination. Coarse grained molecular modelling/dynamics (CG-MD) simulations brought to light that the depolymerization through DMAP coordination in the ZnII porphyrin assemblies primarily happens at the termini of the SPs, in competition with monomer exchange. Thus, the end-capping of those active (de-)polymerization sites through attachment of CuII porphyrin blocks effectively hampers disassembly. Apparently, this “compartmentalization” into block segments is an important factor in the protection from Lewis acid-induced perturbation of the self-assembly, as Meijer and co-workers were able to selectively remove chiral Zn-porphyrin sergeants in a random copolymer with achiral Cu-porphyrin soldiers, after their coaggregation.123,124 Hence, the kinetic compartmentalization in the BSP is reminiscent of biomolecular compartmentalization, and could thus be a key strategy to obtain functional multicomponent synthetic supramolecular systems.
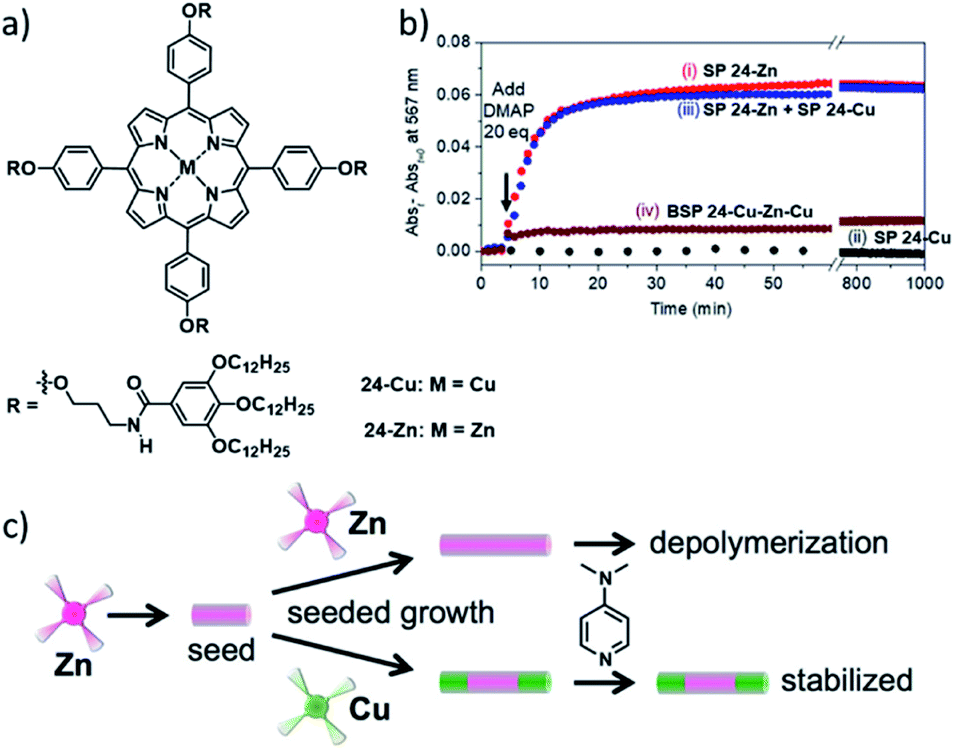 | ||
| Fig. 14 (a) Molecular structures of the investigated metal-porphyrins 24-Cu and 24-Zn. (b) Kinetic profiles after the addition of DMAP to SPs of pure 24-Zn (i), SPs of pure 24-Cu (ii), a mixture of SPs of pure 24-Zn and 24-Cu (iii), the 24-Cu–Zn–Cu block-copolymer (iv). (c) Schematic illustration of the depolymerization of 1D aggregates of ZnII porphyrin 24-Zn through DMAP coordination and its suppression by “end-capping” the 24-Zn SPs with blocks of SPs of 24-Cu. Panel (b) and (c) adapted with permission from ref. 119. Copyright 2018 American Chemical Society. | ||
Emergent properties in homometallic co-assemblies based on different ligand design
Although many co-assembly strategies targeting a modulation of the SP properties rely on the co-assembly of complexes with different metals, a significant alteration of the self-assembly characteristics can also be achieved through co-assembly of complexes bearing the same metal, but distinct ligands.Wang and coworkers demonstrated the feasibility of this approach for various material properties: PtII complex 25 by itself non-covalently polymerizes into 1D fibres in a cooperative manner in MCH/DCE 95/5.125 Within the supramolecular stacks, the complexes organize in an alternating manner, dominantly driven by π–π stacking (Fig. 15). The UV/Vis and fluorescence spectra in the monomer and aggregated state show the typical IL and MLCT absorption and emission bands known for this class of compounds. Complex 26, on the other hand, was insoluble in the same solvent mixture due to the lack of alkoxy side chains. However, when the two molecules were combined in equal amounts, a clear solution with a dark colour emerged. Spectroscopy revealed a new, low-energy absorption band (560–800 nm) and a near-infrared (NIR) emission, which were not observed for either 25 or 26 in isolation. The authors could attribute these characteristics to the surge of MMLCT in the copolymer due to intermolecular Pt⋯Pt interactions together with donor–acceptor charge transfer effects. Although the co-assembly proceeds via an isodesmic mechanism, the negative Gibbs free energy associated with the process is higher than that of pure 25, resulting in a preferred co-assembly rather than a narcissistic self-sorting.
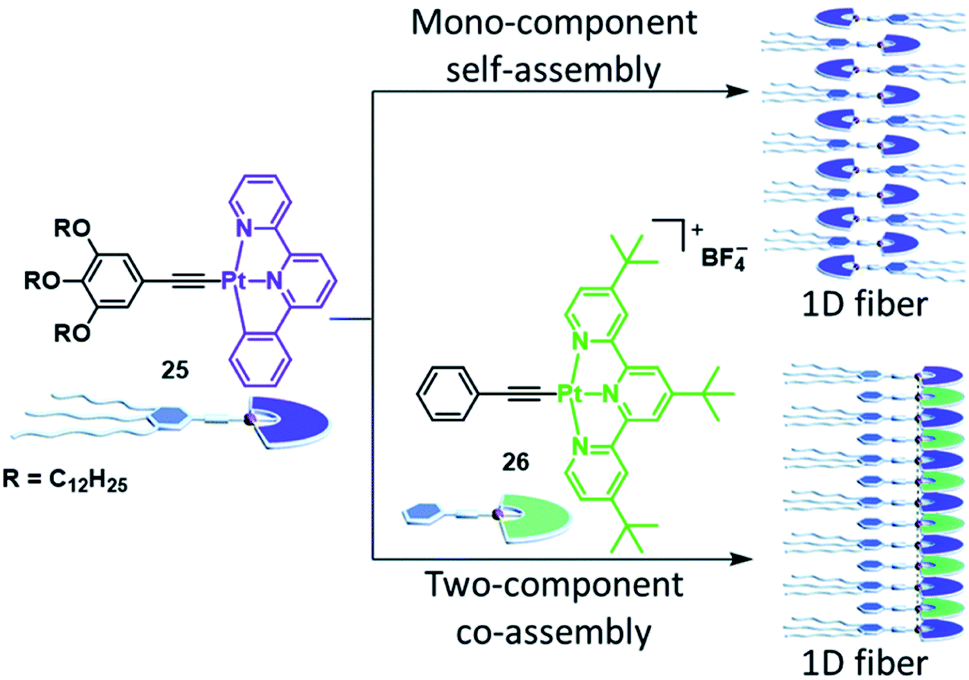 | ||
| Fig. 15 Comparison of the mono-component self-assembly of complex 25 and its co-assembly with complex 26. Whereas the SP stacks of isolated 25 lack metal–metal interactions, the co-assembly induces heteromeric Pt⋯Pt interactions, giving rise to an MMLCT transition. Adapted with permission from ref. 125. Copyright 2018 Royal Society of Chemistry. | ||
Another remarkable example of co-assembly of two metal complexes with modified ancillary ligands was very recently published by the groups of Pavan and de Cola.126 The authors took advantage of the different luminescence properties of aggregates of PtII complexes 27 and 28 to monitor the templated co-assembly of 28 around fibres of 27 (Fig. 16), reminiscent of graft copolymerization. Despite the intricate supramolecular polymerization pathways of the two species (27 forms three aggregated species, 28 forms two aggregates), careful selection of the good/bad solvent ratio allowed the selective formation of the blue-emitting species of 27 (27C), which upon addition of metastable aggregates of 28 (28A) were gradually coated with a layer of 28 in its yellow-emitting form 28B. The higher critical good solvent volume fraction for the disassembly of 27C (i.e. its higher stability) was then exploited to selectively remove the coated layer of 28B, recovering the initial fibres of 27C with unaltered properties. Remarkably, the wrapping of 27C with 28B could even be achieved at higher 1,4-dioxane percentages, where self-assembly of pristine 28B was not possible. On the contrary, at higher water fractions, where 28B is sufficiently stabilized, homo-assemblies of 28B emerged in competition with the wrapping of 27C. Thus, the authors hypothesized that packing defects at the surface of 27C, facilitated by an increase in the good solvent fraction, are the origin of the wrapping phenomenon, generating vacant superficial π-stacking sites. These can be occupied by monomers of 28, allowing for a co-nucleation, inducing a seed-like propagation of the co-assembly. Sophisticated CG-MD simulations were developed to compare the strength of the incorporation of each individual monomer in a stack of 27C with the solvent accessible surface area (SASA) of each monomer, i.e. the degree of exposition to the solvent. The calculations revealed the defect sites in the stack, which indeed were the positions at which the wrapping phenomenon originated in subsequent simulations of the co-assembly process.
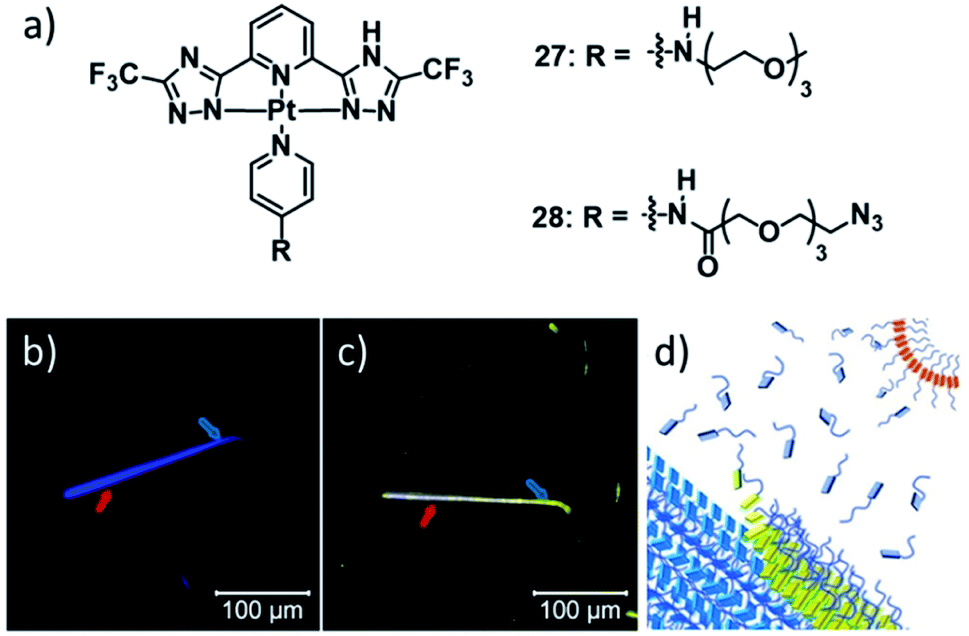 | ||
| Fig. 16 (a) Molecular structure of PtII complexes 27 & 28 whose co-assembly renders a reversible supramolecular wrapping of fibres formed by 27 through aggregates of 28. The lower panel (b and c) shows the confocal microscopy images visualizing the time-progression of the co-assembly and a schematic illustration of the molecular processes (d). Adapted with permission from ref. 126. Copyright 2021 John Wiley and Sons. | ||
These examples demonstrate that precise control over (homometallic) co-assembly still remains a challenge, as not only aspects of molecular design but also solution conditions as well as sample preparation have to be finely tuned to achieve the desired co-assembly. However, as a side effect of the new possibilities opened up by metallosupramolecular co-assembly, the larger number of molecular building blocks automatically increases the complexity of the supramolecular system, exceeding that of a simple superposition of the individual components. On the other hand, the examples by the groups of Che, Pavan and de Cola demonstrate that the increased complexity of multicomponent metallosupramolecular systems can be met by the development of increasingly more sophisticated computational models.
6 (Future) applications
Self-assembled functional materials derived from metal complexes have attracted considerable interest for multiple applications.127,128 In particular, light emitting supramolecular assemblies have been investigated and thoroughly reviewed.21,129 Additionally, the potential of semiconductive SPs has been recognized.130 Accordingly, we will not devote this section to these fields but instead showcase some examples that have recently demonstrated other potential applications for MSPs. Additionally, we aim at discussing potentially interesting areas, such as e.g. catalysis, which to date have received little attention in the context of MSP research.Biological imaging and therapy
Multiple self-assembled structures with various intriguing properties have found applications in biomedicine, such as in the field of drug delivery.30,131,132 For instance, the enhanced permeability and retention (EPR) effect has been exploited for precise drug delivery using self-assembled structures with precisely controlled morphology and size for selective cancer treatment.133,134 Additionally, emissive assemblies have been used for bioimaging applications.135–137 In contrast, metallosupramolecular polymerization directed by biologically relevant structures has remained relatively underdeveloped.138 A recent notable progress has been made by the group of Yam, who successfully designed a bis-(benzimidazole)pyridine derived anionic PtII complex for the sensing of insulin amyloid (Fig. 17).139 As the formation of amyloids is linked to a number of neurodegenerative diseases and many common stains have drawbacks to their applicability, the need for new sensors is apparent. Upon addition of insulin amyloid, a MSP of 29 is formed, driven by the accumulation of molecules on the amyloid surface. This co-assembly exhibiting desirable strong red emission could be exploited for amyloid fibrillation detection as well as inhibitor screening.137,140,141 Later, based on a slight modification of their molecular design (changing the overall charge from minus one to plus two), the group successfully developed a luminescent self-assembled structure that can efficiently detect RNA.142 The MSP could be successfully employed for nucleus imaging as well as RNA synthesis inhibitor screening.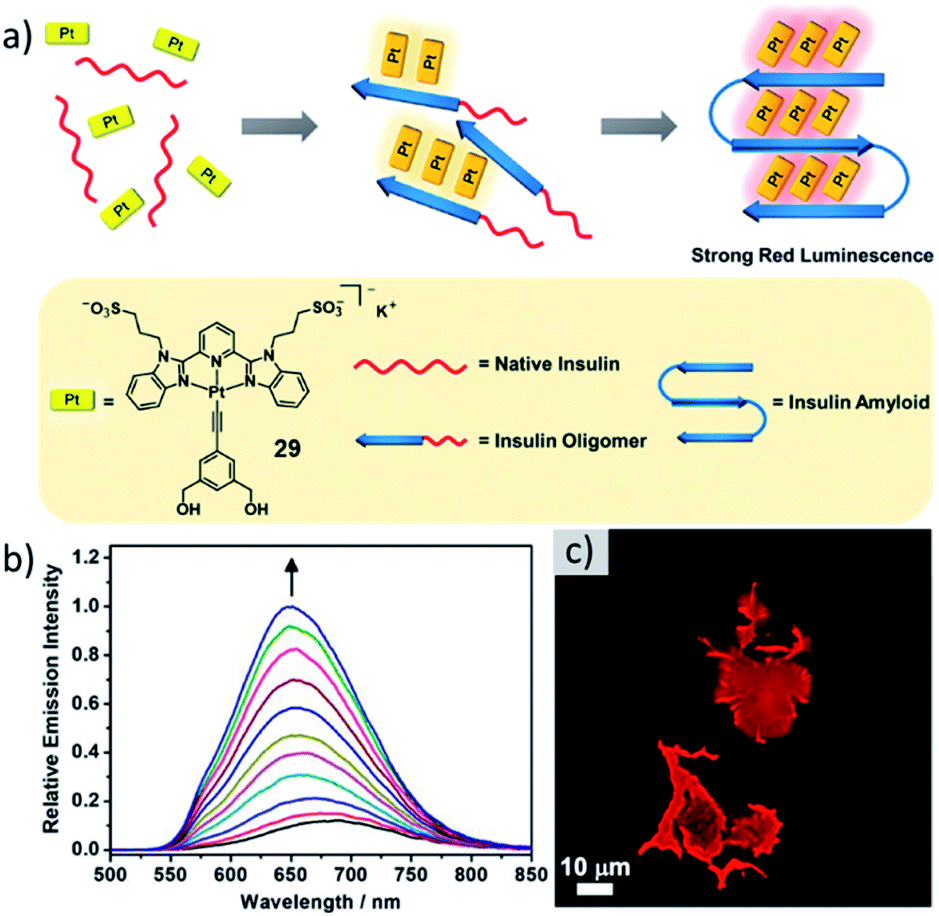 | ||
| Fig. 17 (a) Schematic representation of the insulin amyloid directed self-assembly of 29 with its molecular structure depicted below. (b) Corrected emission spectra of 29 in PBS buffer upon addition of different amounts of insulin amyloid. (c) Fluorescence microscopy image of aggregates of 29 formed on amyloid insulin fibrils. Adapted with permission from ref. 139. Copyright 2019 American Chemical Society. | ||
Likewise, Strassert, Viappiani and co-workers have exploited self-assembled PtII complexes as labels for other biomolecules, such as bovine serum albumin (BSA, Fig. 18).143 In aqueous media (e.g. PBS buffer) PtII complex 30 forms aggregates, which exhibit an increased triplet emission intensity and lifetime compared to non-emissive monomers. In the presence of BSA, self-assembly is controlled by hydrophobic interactions of the complex with the protein, i.e. templated self-assembly at BSA occurs. Furthermore, the aggregate's photophysical properties are insensitive to oxygen, which indicates an enhanced biocompatibility, as other triplet emitters commonly form exciplexes with O2, generating cytotoxic reactive oxygen species. The long-lived triplet emission of the aggregates of 30 on the protein can be selectively detected in more complex biological matrices, where autofluorescence in the same wavelength region occurs, by means of time-gated detection. Moreover, the aggregates are capable to enhance the emission lifetime of other fluorophores through energy transfer (ET) by up to 1000 times, which could be useful in imaging of living systems. Thus, the templated-aggregation of 30 can be exploited to label and selectively image biomolecules. These recent examples highlight the strong potential of MSPs, particularly based on d8 metal complexes in highly selective biological imaging.
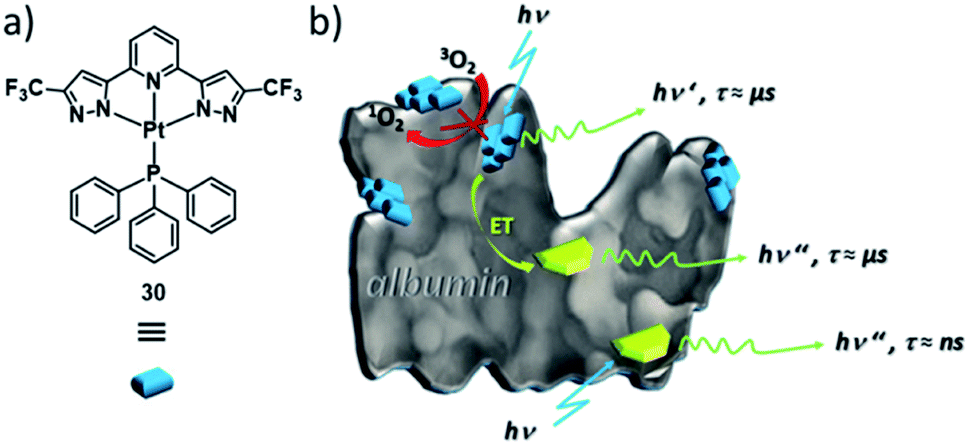 | ||
| Fig. 18 (a) Molecular structure of triplet emitter PtII complex 30. (b) Schematic illustration of the self-assembly of 30 (blue labels) at fluorescein-labelled (green labels) BSA. Panel (b) adapted with permission from ref. 143. Copyright 2018 American Chemical Society. | ||
Catalysis
In striking contrast to fields such as optoelectronics and biomedicine, SPs have received little attention in the field of catalysis, although the potential of precisely controlling a confined microenvironment is profitable for catalysis. For discrete supramolecular structures (such as macrocycles or metallacages)41,144,145 or self-assembled structures with long range order, for instance metal–organic or covalent organic frameworks,146,147 this beneficial property has been acknowledged.148,149 The potentially available reactions to be catalysed by incorporating metal centres into confined microenvironments are highly diverse and can likewise be expanded to MSPs.In this regard, Wang and co-workers designed a discrete metallosupramolecular tetranuclear complex-pair capable of efficient intercalation driven by metallophilic interactions.150 Based on strong Pt–Pt interactions, the photoinduced formation of singlet oxygen was used for the oxidation of amines. In a later report, the same group advanced this approach from a discrete supramolecular entity to a metallosupramolecular copolymer (Fig. 19). The alternate arrangement of a sterically demanding PtII acceptor 31 and a sterically unhindered PtII donor molecule 32 leads to the emergence of 3MMLCT transitions, which are expressed as a low energy emission in the visible/NIR region. The dynamic nature of the co-assembly was utilized to selectively trigger or suppress the photocatalytic activity of the polymer.151
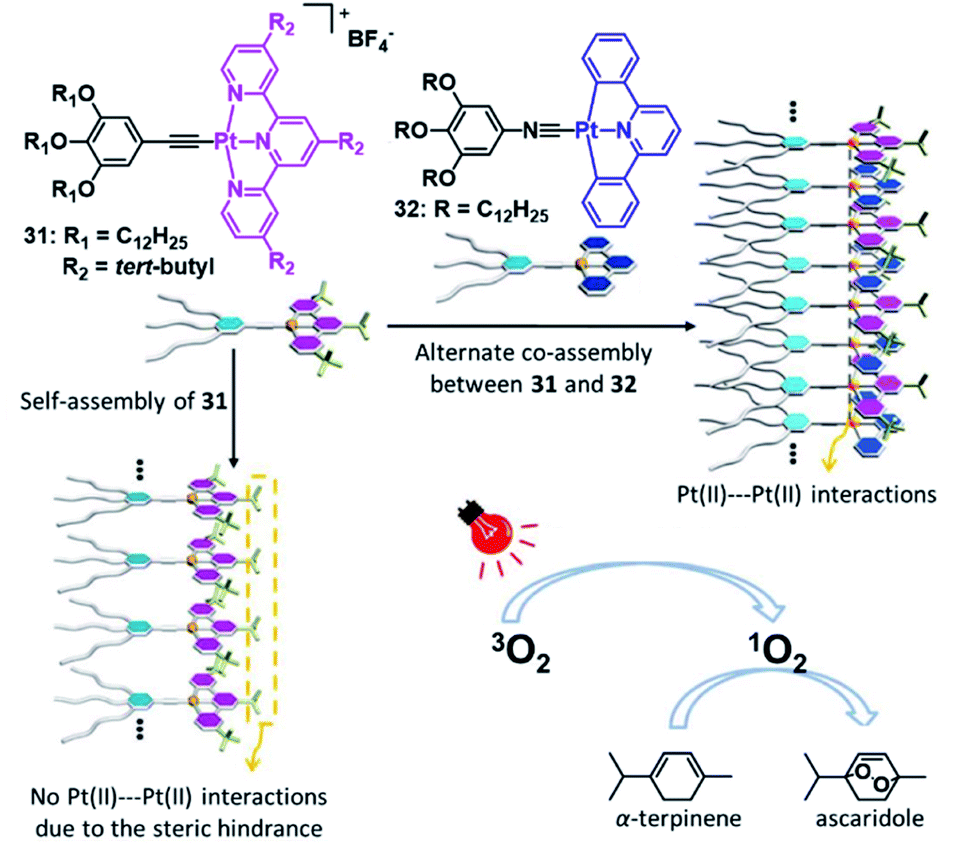 | ||
| Fig. 19 Schematic representation of a photocatalytically active co-assembly based on donor–acceptor interactions, exhibiting strong Pt–Pt interactions. Adapted with permission from ref. 150. Copyright 2018 Royal Society of Chemistry. | ||
Despite these important advances, the full catalytic potential of MSPs remains to be explored. This lack of examples may come as a surprise considering some key properties of metallosupramolecular assemblies to be used in catalysis (Fig. 20):
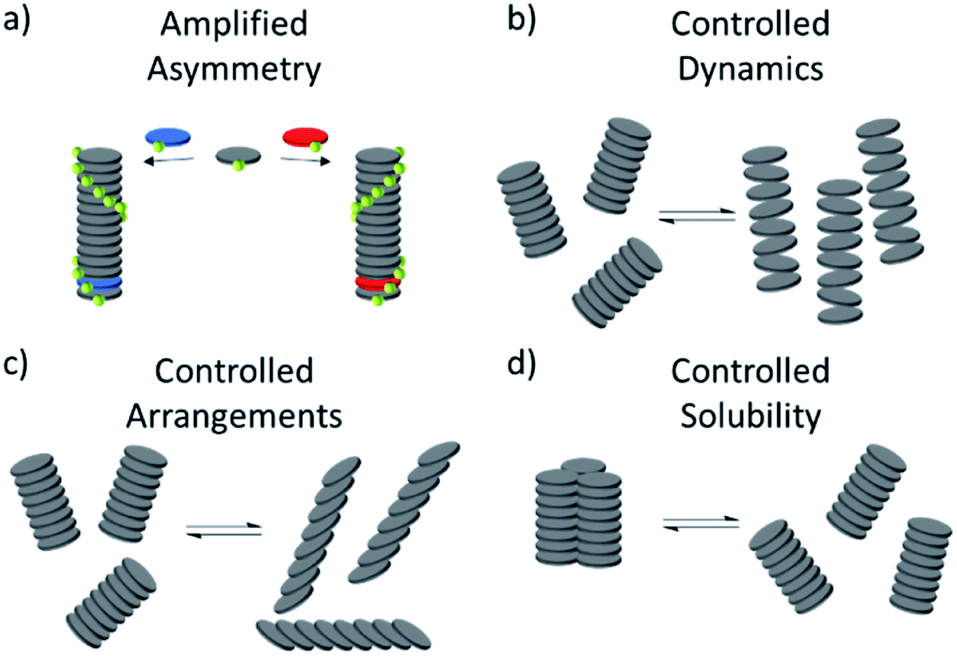 | ||
| Fig. 20 (a–d) Schematic representations of the potentially viable properties of supramolecular assemblies for applications in catalysis. | ||
(1) The chirality of a SP can be controlled and at times switched based on sample preparation protocols or amplification of asymmetry.
(2) Aggregate dynamics and molecular arrangements and in turn catalytic activity can be precisely regulated by external conditions such as temperature.
(3) The solubility of aggregates can be controlled through secondary interactions, allowing for an effective removal of the aggregates from the reaction solution, which could be exploited for efficient and convenient recyclability.
We envisage that a supramolecular approach where the SP structure or its molecular constituents act as active catalysts could offer great versatility and allow to switch between different chiral products based on MSP-catalyst preparation.152 To this end, a balance between controlled molecular dynamics to achieve accessibility of the active site within a SP while preserving a chiral microenvironment of the active site needs to be maintained. Ultimately, we envisage that the inherent dynamics and adaptivity of SPs in combination with the catalytic activities of metal complexes could be exploited to tailor potent chiral metallosupramolecular catalyst systems.
Nevertheless, we acknowledge that a system fulfilling all or even only a few of the above conditions represents a formidable challenge for any researcher. Therefore, we believe that combined theoretical and experimental efforts may lead to feasible applications in the field in a not too distant future.153–155
7 Conclusions
The increasing interest in kinetic aspects of complex self-assembly phenomena has inspired researchers to expand this knowledge to MSPs. Finely controlling the balance between repulsive and attractive non-covalent interactions associated with the incorporation of a metal-containing moiety has produced fascinating ways to tune supramolecular energy landscapes, packing motifs and photophysical properties. Control over aggregate morphologies has concomitantly matured into an exciting field of research. The surge of molecular design strategies as well as sample preparation protocols to control primary as well as secondary interactions has considerably broadened the toolbox of supramolecular chemists. However, even the most elaborate recent examples show that great research effort remains to be made to fully understand and predict supramolecular morphologies: in many cases, predicting complex energy landscapes of SPs by molecular design remains a hard task.The recent progress in multicomponent self-assembly has shed light on its high potential in achieving unprecedented supramolecular properties and amplifying existing properties not present in the individual components. However, this research area is still in its infancy and the full potential remains to be explored. While the potential of MSPs in various applications cannot be denied, actual or industrial applications remain scarce. In particular, we argue that the field of catalysis represents a great frontier for researchers working on metallosupramolecular chemistry. However, to overcome the challenges imposed by the complexity of reversible self-assembly, we believe that a more intimate collaboration between experimental and theoretical scientists could render outstanding innovations in the future of MSP chemistry. Given the substantial development of the field in the past decade, we anticipate a bright future for metal-based supramolecular systems in terms of unprecedented properties and new applications.
Author contributions
All authors contributed to the preparation of the perspective article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC-StG-2016 SUPRACOP-715923) and the Verband der chemischen Industrie for funding.Notes and references
- P. R. Ashton, C. L. Brown, E. J. T. Chrystal, T. T. Goodnow, A. E. Kaifer, K. P. Parry, D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 634 RSC.
- J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422 RSC.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS PubMed.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312 CrossRef CAS PubMed.
- L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed.
- T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171 CrossRef CAS PubMed.
- N. Grabicki, O. Dumele, H. Sai, N. E. Powers-Riggs, B. T. Phelan, M. H. Sangji, C. T. Chapman, J. V. Passarelli, A. J. Dannenhoffer, M. R. Wasielewski and S. I. Stupp, Chem. Mater., 2021, 33, 706 CrossRef CAS.
- R. V. Kazantsev, A. Dannenhoffer, T. Aytun, B. Harutyunyan, D. J. Fairfield, M. J. Bedzyk and S. I. Stupp, Chem, 2018, 4, 1596 CAS.
- N. J. Hestand, R. V. Kazantsev, A. S. Weingarten, L. C. Palmer, S. I. Stupp and F. C. Spano, J. Am. Chem. Soc., 2016, 138, 11762 CrossRef CAS PubMed.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- T. D. Clemons and S. I. Stupp, Prog. Polym. Sci., 2020, 111, 101310 CrossRef CAS PubMed.
- C. Shahar, Y. Tidhar, Y. Jung, H. Weissman, S. R. Cohen, R. Bitton, I. Pinkas, G. Haran and B. Rybtchinski, Beilstein J. Org. Chem., 2021, 17, 42 CrossRef CAS PubMed.
- E. Cohen, Y. Soffer, H. Weissman, T. Bendikov, Y. Schilt, U. Raviv and B. Rybtchinski, Angew. Chem., Int. Ed., 2018, 57, 8871 CrossRef CAS PubMed.
- C. Fouquey, J.-M. Lehn and A.-M. Levelut, Adv. Mater., 1990, 2, 254 CrossRef CAS.
- J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1994, 116, 2655 CrossRef CAS.
- J. S. Moore, Acc. Chem. Res., 1997, 30, 402 CrossRef CAS.
- V. Wing-Wah Yam and K. Kam-Wing Lo, Chem. Soc. Rev., 1999, 28, 323 RSC.
- M. W. Cooke, D. Chartrand and G. S. Hanan, Coord. Chem. Rev., 2008, 252, 903 CrossRef CAS.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042 RSC.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. de Greef and E. W. Meijer, Nature, 2012, 481, 492 CrossRef CAS PubMed.
- M. F. J. Mabesoone, A. J. Markvoort, M. Banno, T. Yamaguchi, F. Helmich, Y. Naito, E. Yashima, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7810 CrossRef CAS PubMed.
- K. Zhang, M. C.-L. Yeung, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 9594 CrossRef CAS PubMed.
- S. Otto, Acc. Chem. Res., 2012, 45, 2200 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825 CrossRef CAS PubMed.
- B. Rieß, R. K. Grötsch and J. Boekhoven, Chem, 2020, 6, 552 Search PubMed.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600 RSC.
- S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244 CrossRef CAS PubMed.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769 CrossRef CAS PubMed.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1154 CrossRef.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Paik Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715 CAS.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005 CrossRef CAS PubMed.
- S. Samantray, S. Krishnaswamy and D. K. Chand, Nat. Commun., 2020, 11, 880 CrossRef CAS PubMed.
- N. Ahmad, A. H. Chughtai, H. A. Younus and F. Verpoort, Coord. Chem. Rev., 2014, 280, 1 CrossRef CAS.
- A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19 CrossRef CAS.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed.
- Y. Sun, F. Zhang, S. Jiang, Z. Wang, R. Ni, H. Wang, W. Zhou, X. Li and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 17297 CrossRef CAS PubMed.
- Y. Sun, S. Li, Z. Zhou, M. L. Saha, S. Datta, M. Zhang, X. Yan, D. Tian, H. Wang, L. Wang, X. Li, M. Liu, H. Li and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 3257 CrossRef CAS PubMed.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889 RSC.
- J. Zhao, Z. Zhou, G. Li, P. J. Stang and X. Yan, Natl. Sci. Rev., 2021, 8 DOI:10.1093/nsr/nwab045.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342 CrossRef CAS PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952 CrossRef CAS PubMed.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397 RSC.
- J.-M. Lehn, Science, 2002, 295, 2400 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- J.-P. Sauvage, J.-P. Collin, S. Durot, J. Frey, V. Heitz, A. Sour and C. Tock, C. R. Chim., 2010, 13, 315 CrossRef CAS.
- Y. Tidhar, H. Weissman, S. G. Wolf, A. Gulino and B. Rybtchinski, Chem.–Eur. J., 2011, 17, 6068 CrossRef CAS PubMed.
- M. J. Mayoral, C. Rest, V. Stepanenko, J. Schellheimer, R. Q. Albuquerque and G. Fernández, J. Am. Chem. Soc., 2013, 135, 2148 CrossRef CAS PubMed.
- N. A. M. S. Caturello, Z. Csók, G. Fernández and R. Q. Albuquerque, Chem.–Eur. J., 2016, 22, 17681 CrossRef CAS.
- M. Chen, C. Wei, J. Tao, X. Wu, N. Huang, G. Zhang and L. Li, Chem.–Eur. J., 2014, 20, 2812 CrossRef CAS PubMed.
- N. Bäumer, K. K. Kartha, N. K. Allampally, S. Yagai, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 15626 CrossRef PubMed.
- N. Bäumer, K. K. Kartha, S. Buss, I. Maisuls, J. P. Palakkal, C. A. Strassert and G. Fernández, Chem. Sci., 2021, 12, 5236 RSC.
- J. Matern, N. Bäumer and G. Fernández, J. Am. Chem. Soc., 2021, 143, 7164 CrossRef CAS.
- L. Li, N. Zhou, H. Kong and X. He, Polym. Chem., 2019, 10, 5465 RSC.
- M. Chen, C. Wei, X. Wu, M. Khan, N. Huang, G. Zhang and L. Li, Chem.–Eur. J., 2015, 21, 4213 CrossRef CAS PubMed.
- Y. Atoini, E. A. Prasetyanto, P. Chen, S. Silvestrini, J. Harrowfield and L. de Cola, Chem.–Eur. J., 2018, 24, 12054 CrossRef CAS PubMed.
- Q. Wan, W.-P. To, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 3089 CrossRef CAS PubMed.
- A. Langenstroer, Y. Dorca, K. K. Kartha, M. J. Mayoral, V. Stepanenko, G. Fernández and L. Sánchez, Macromol. Rapid Commun., 2018, 39, 1800191 CrossRef PubMed.
- K. K. Kartha, N. K. Allampally, A. T. Politi, D. D. Prabhu, H. Ouchi, R. Q. Albuquerque, S. Yagai and G. Fernández, Chem. Sci., 2019, 10, 752 RSC.
- A. Langenstroer, K. K. Kartha, Y. Dorca, J. Droste, V. Stepanenko, R. Q. Albuquerque, M. R. Hansen, L. Sánchez and G. Fernández, J. Am. Chem. Soc., 2019, 141, 5192 CrossRef CAS PubMed.
- J. Matern, K. K. Kartha, L. Sánchez and G. Fernández, Chem. Sci., 2020, 11, 6780 RSC.
- M. J. Mayoral, C. Rest, V. Stepanenko, J. Schellheimer, R. Q. Albuquerque and G. Fernández, J. Am. Chem. Soc., 2013, 135, 2148 CrossRef CAS PubMed.
- N. K. Allampally, M. J. Mayoral, S. Chansai, M. C. Lagunas, C. Hardacre, V. Stepanenko, R. Q. Albuquerque and G. Fernández, Chem.–Eur. J., 2016, 22, 7810 CrossRef CAS PubMed.
- S. Sengupta and F. Würthner, Acc. Chem. Res., 2013, 46, 2498 CrossRef CAS PubMed.
- S. Sengupta, D. Ebeling, S. Patwardhan, X. Zhang, H. von Berlepsch, C. Böttcher, V. Stepanenko, S. Uemura, C. Hentschel, H. Fuchs, F. C. Grozema, L. D. A. Siebbeles, A. R. Holzwarth, L. Chi and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6378 CrossRef CAS PubMed.
- V. Huber, M. Katterle, M. Lysetska and F. Würthner, Angew. Chem., Int. Ed., 2005, 44, 3147 CrossRef CAS PubMed.
- S. Uemura, S. Sengupta and F. Würthner, Angew. Chem., Int. Ed., 2009, 48, 7825 CrossRef CAS PubMed.
- S. Ogi, C. Grzeszkiewicz and F. Würthner, Chem. Sci., 2018, 9, 2768 RSC.
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579 RSC.
- V. W.-W. Yam and A. S.-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS.
- V. W.-W. Yam, A. K.-W. Chan and E. Y.-H. Hong, Nat. Rev. Chem., 2020, 4, 528 CrossRef CAS.
- Y. Ai, M. H.-Y. Chan, A. K.-W. Chan, M. Ng, Y. Li and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13856 CrossRef CAS PubMed.
- Y. Kitamoto, Z. Pan, D. D. Prabhu, A. Isobe, T. Ohba, N. Shimizu, H. Takagi, R. Haruki, S. Adachi and S. Yagai, Nat. Commun., 2019, 10, 4578 CrossRef PubMed.
- S. Yagai, Y. Kitamoto, S. Datta and B. Adhikari, Acc. Chem. Res., 2019, 52, 1325 CrossRef CAS PubMed.
- S. Datta, Y. Kato, S. Higashiharaguchi, K. Aratsu, A. Isobe, T. Saito, D. D. Prabhu, Y. Kitamoto, M. J. Hollamby, A. J. Smith, R. Dalgliesh, N. Mahmoudi, L. Pesce, C. Perego, G. M. Pavan and S. Yagai, Nature, 2020, 583, 400 CrossRef CAS PubMed.
- N. Sasaki, M. F. J. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki, N. Shimizu, S. Adachi, E. W. Meijer, M. Takeuchi and K. Sugiyasu, Nat. Commun., 2020, 11, 3578 CrossRef PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188 CrossRef CAS PubMed.
- N. Sasaki, J. Yuan, T. Fukui, M. Takeuchi and K. Sugiyasu, Chem.–Eur. J., 2020, 26, 7840 CrossRef CAS PubMed.
- V. C.-H. Wong, C. Po, S. Y.-L. Leung, A. K.-W. Chan, S. Yang, B. Zhu, X. Cui and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 657 CrossRef CAS PubMed.
- A. Aliprandi, M. Mauro and L. de Cola, Nat. Chem., 2016, 8, 10 CrossRef CAS PubMed.
- Y. Tsarfati, S. Rosenne, H. Weissman, L. J. W. Shimon, D. Gur, B. A. Palmer and B. Rybtchinski, ACS Cent. Sci., 2018, 4, 1031 CrossRef CAS PubMed.
- A. Isobe, D. D. Prabhu, S. Datta, T. Aizawa and S. Yagai, Chem.–Eur. J., 2020, 26, 8997 CrossRef CAS PubMed.
- I. Helmers, N. Bäumer and G. Fernández, Chem. Commun., 2020, 56, 13808 RSC.
- N. Bäumer, K. K. Kartha, J. P. Palakkal and G. Fernández, Soft Matter, 2020, 16, 6834 RSC.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644 CrossRef CAS PubMed.
- L. MacFarlane, C. Zhao, J. Cai, H. Qiu and I. Manners, Chem. Sci., 2021, 12, 4661 RSC.
- Z. M. Hudson, D. J. Lunn, M. A. Winnik and I. Manners, Nat. Commun., 2014, 5, 3372 CrossRef PubMed.
- Y. Liu, Y. Gong, Y. Guo, W. Xiong, Y. Zhang, J. Zhao, Y. Che and I. Manners, Chem.–Eur. J., 2019, 25, 13484 CrossRef CAS PubMed.
- Y. Zhang, S. Pearce, J.-C. Eloi, R. L. Harniman, J. Tian, C. Cordoba, Y. Kang, T. Fukui, H. Qiu, A. Blackburn, R. M. Richardson and I. Manners, J. Am. Chem. Soc., 2021, 143, 5805 CrossRef CAS PubMed.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646 CrossRef CAS PubMed.
- A. Suzuki, K. Aratsu, S. Datta, N. Shimizu, H. Takagi, R. Haruki, S. Adachi, M. Hollamby, F. Silly and S. Yagai, J. Am. Chem. Soc., 2019, 141, 13196 CrossRef CAS PubMed.
- P. Picchetti, G. Moreno-Alcántar, L. Talamini, A. Mourgout, A. Aliprandi and L. de Cola, J. Am. Chem. Soc., 2021, 143, 7681 CrossRef CAS PubMed.
- G. M. ter Huurne, P. Chidchob, A. Long, A. Martinez, A. R. A. Palmans and G. Vantomme, Chem.–Eur. J., 2020, 26, 9964 CrossRef CAS PubMed.
- C. Naranjo, Y. Dorca, G. Ghosh, R. Gómez, G. Fernández and L. Sánchez, Chem. Commun., 2021, 57, 4500 RSC.
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, J. Am. Chem. Soc., 2021, 143, 4500 CrossRef CAS PubMed.
- E. Kirchner, D. Bialas, F. Fennel, M. Grüne and F. Würthner, J. Am. Chem. Soc., 2019, 141, 7428 CrossRef CAS PubMed.
- L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 11344 CrossRef CAS PubMed.
- K. Cai, J. Xie, D. Zhang, W. Shi, Q. Yan and D. Zhao, J. Am. Chem. Soc., 2018, 140, 5764 CrossRef CAS PubMed.
- R. Appel, J. Fuchs, S. M. Tyrrell, P. A. Korevaar, M. C. A. Stuart, I. K. Voets, M. Schönhoff and P. Besenius, Chem.–Eur. J., 2015, 21, 19257 CrossRef CAS PubMed.
- F. Noé, A. Tkatchenko, K.-R. Müller and C. Clementi, Annu. Rev. Phys. Chem., 2020, 71, 361 CrossRef PubMed.
- C. M. Aitchison, C. M. Kane, D. P. McMahon, P. R. Spackman, A. Pulido, X. Wang, L. Wilbraham, L. Chen, R. Clowes, M. A. Zwijnenburg, R. S. Sprick, M. A. Little, G. M. Day and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7158 RSC.
- E. Berardo, R. L. Greenaway, M. Miklitz, A. I. Cooper and K. E. Jelfs, Mol. Syst. Des. Eng., 2020, 5, 186 RSC.
- P. Cui, D. P. McMahon, P. R. Spackman, B. M. Alston, M. A. Little, G. M. Day and A. I. Cooper, Chem. Sci., 2019, 10, 9988 RSC.
- R. J. Kearsey, B. M. Alston, M. E. Briggs, R. L. Greenaway and A. I. Cooper, Chem. Sci., 2019, 10, 9454 RSC.
- E. Weyandt, M. F. J. Mabesoone, L. N. J. de Windt, E. W. Meijer, A. R. A. Palmans and G. Vantomme, Org. Mater., 2020, 02, 129 CrossRef CAS.
- B. Adelizzi, N. J. van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110 CrossRef CAS PubMed.
- H. Frisch, E.-C. Fritz, F. Stricker, L. Schmüser, D. Spitzer, T. Weidner, B. J. Ravoo and P. Besenius, Angew. Chem., Int. Ed., 2016, 55, 7242 CrossRef CAS PubMed.
- S. Engel, D. Spitzer, L. L. Rodrigues, E.-C. Fritz, D. Straßburger, M. Schönhoff, B. J. Ravoo and P. Besenius, Faraday Discuss., 2017, 204, 53 RSC.
- K. Jalani, M. Kumar and S. J. George, Chem. Commun., 2013, 49, 5174 RSC.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038 CrossRef CAS PubMed.
- Z. Gao, Y. Han, Z. Gao and F. Wang, Acc. Chem. Res., 2018, 51, 2719 CrossRef CAS PubMed.
- S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570 CrossRef CAS PubMed.
- J. P. Coelho, J. Matern, R. Q. Albuquerque and G. Fernández, Chem.–Eur. J., 2019, 25, 8960 CrossRef CAS PubMed.
- A. Jamadar and A. Das, Polym. Chem., 2020, 11, 385 RSC.
- Q. Wan, W.-P. To, X. Chang and C.-M. Che, Chem, 2020, 6, 945 CAS.
- F. Helmich, C. C. Lee, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 16753 CrossRef CAS PubMed.
- F. Helmich, M. M. J. Smulders, C. C. Lee, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 12238 CrossRef CAS PubMed.
- Z. Gao, P. A. Korevaar, R. Zhong, Z. Wu and F. Wang, Chem. Commun., 2018, 54, 9857 RSC.
- G. Moreno-Alcántar, A. Aliprandi, R. Rouquette, L. Pesce, K. Wurst, C. Perego, P. Brüggeller, G. M. Pavan and L. de Cola, Angew. Chem., Int. Ed., 2021, 60, 5407 CrossRef PubMed.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176 CrossRef CAS PubMed.
- Y.-S. Wong, M. Ng, M. C.-L. Yeung and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 973 CrossRef CAS PubMed.
- K. Li, G. S. Ming Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653 RSC.
- Y. Han, Z. Gao, C. Wang, R. Zhong and F. Wang, Coord. Chem. Rev., 2020, 414, 213300 CrossRef CAS.
- A. Casini, B. Woods and M. Wenzel, Inorg. Chem., 2017, 56, 14715 CrossRef CAS PubMed.
- C.-N. Lok, T. Zou, J.-J. Zhang, I. W.-S. Lin and C.-M. Che, Adv. Mater., 2014, 26, 5550 CrossRef CAS PubMed.
- A. Sampedro, Á. Ramos-Torres, C. Schwöppe, C. Mück-Lichtenfeld, I. Helmers, A. Bort, I. Díaz-Laviada and G. Fernández, Angew. Chem., Int. Ed., 2018, 57, 17235 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271 CrossRef CAS PubMed.
- S. S. Liow, H. Zhou, S. Sugiarto, S. Guo, M. L. S. Chalasani, N. K. Verma, J. Xu and X. J. Loh, Biomacromolecules, 2017, 18, 886 CrossRef CAS PubMed.
- P. Wang, C. Zhang, H.-W. Liu, M. Xiong, S.-Y. Yin, Y. Yang, X.-X. Hu, X. Yin, X.-B. Zhang and W. Tan, Chem. Sci., 2017, 8, 8214 RSC.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. de Cola, Chem. Soc. Rev., 2014, 43, 4144 RSC.
- Y. Tian, X. Yan, M. L. Saha, Z. Niu and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 12033 CrossRef CAS PubMed.
- A. S.-Y. Law, L. C.-C. Lee, M. C.-L. Yeung, K. K.-W. Lo and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 18570 CrossRef CAS PubMed.
- K. Mitra, C. E. Lyons and M. C. T. Hartman, Angew. Chem., Int. Ed., 2018, 57, 10263 CrossRef CAS PubMed.
- A. Colombo, F. Fiorini, D. Septiadi, C. Dragonetti, F. Nisic, A. Valore, D. Roberto, M. Mauro and L. de Cola, Dalton Trans., 2015, 44, 8478 RSC.
- A. S.-Y. Law, L. C.-C. Lee, K. K.-W. Lo and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 5396 CrossRef CAS PubMed.
- P. Delcanale, A. Galstyan, C. G. Daniliuc, H. E. Grecco, S. Abbruzzetti, A. Faust, C. Viappiani and C. A. Strassert, ACS Appl. Mater. Interfaces, 2018, 10, 24361 CrossRef CAS PubMed.
- M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 30, 1909842 CrossRef CAS.
- B. Sun, B. Shen, A. Urushima, X. Liu, X. Feng, E. Yashima and M. Lee, Angew. Chem., Int. Ed., 2020, 59, 22690 CrossRef CAS PubMed.
- J. Jiang, Y. Zhao and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 3255 CrossRef CAS PubMed.
- H. V. Babu, M. G. M. Bai and M. Rajeswara Rao, ACS Appl. Mater. Interfaces, 2019, 11, 11029 CrossRef CAS PubMed.
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256 CrossRef CAS PubMed.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433 RSC.
- Z. Li, Y. Han, Z. Gao and F. Wang, ACS Catal., 2017, 7, 4676 CrossRef CAS.
- Z. Gao, Z. Li, Z. Gao and F. Wang, Nanoscale, 2018, 10, 14005 RSC.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948 CrossRef CAS PubMed.
- J. Gebhardt, M. Kiesel, S. Riniker and N. Hansen, J. Chem. Inf. Model., 2020, 60, 5319 CrossRef CAS PubMed.
- Y. Wang, J. M. Lamim Ribeiro and P. Tiwary, Curr. Opin. Struct. Biol., 2020, 61, 139 CrossRef CAS PubMed.
- V. Botu and R. Ramprasad, Int. J. Quantum Chem., 2015, 115, 1074 CrossRef CAS.
Footnote |
| † Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |