
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAllosteric inhibition of SARS-CoV-2 3CL protease by colloidal bismuth subcitrate†
Xuan
Tao
a,
Lu
Zhang
b,
Liubing
Du
c,
Ruyan
Liao
b,
Huiling
Cai
b,
Kai
Lu
a,
Zhennan
Zhao
a,
Yanxuan
Xie
a,
Pei-Hui
Wang
d,
Ji-An
Pan
c,
Yuebin
Zhang
*e,
Guohui
Li
*e,
Jun
Dai
*b,
Zong-Wan
Mao
 *a and
Wei
Xia
*a
*a and
Wei
Xia
*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry School of Chemistry, Sun Yat-Sen University, Guangzhou, 510275, China
bGuangzhou Customs District Technology Center, No. 66 Huacheng Avenue, Zhujiang New Town, Tianhe District, Guangzhou, 510700, China
cThe Center for Infection and Immunity Study, School of Medicine, Sun Yat-sen University, Guangming Science City, Shenzhen, 518107, China
dKey Laboratory for Experimental Teratology of Ministry of Education, Advanced Medical Research Institute, Cheeloo College of Medicine, Shandong University, Jinan, 250012, China
eState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China
First published on 24th September 2021
Abstract
The SARS-CoV-2 3-chymotrypsin-like protease (3CLpro or Mpro) is a key cysteine protease for viral replication and transcription, making it an attractive target for antiviral therapies to combat the COVID-19 disease. Here, we demonstrate that bismuth drug colloidal bismuth subcitrate (CBS) is a potent inhibitor for 3CLpro in vitro and in cellulo. Rather than targeting the cysteine residue at the catalytic site, CBS binds to an allosteric site and results in dissociation of the 3CLpro dimer and proteolytic dysfunction. Our work provides direct evidence that CBS is an allosteric inhibitor of SARS-CoV-2 3CLpro.
Introduction
COVID-19 is a highly infectious viral disease caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2).1 Since last December, this respiratory disease has spread rapidly to more than 200 countries and territories. To date, more than 180 million people around the world were infected and global deaths have exceeded 3.9 million.2 Although US FDA has issued emergency use authorization for three COVID-19 vaccines recently, no specific antiviral drugs are currently available for the treatment of viral infections.Similar to other coronaviruses, SARS-CoV-2 encodes a cysteine protease, the 3-chymotrypsin-like protease (3CLpro or Mpro, encoded by nsp5) for proteolytic cleavage of non-structural proteins essential for viral replication and thus enables viral spread. 3CLpro functions as a homo-dimer and cleaves two large overlapping viral polyproteins pp1a and pp1ab at at least 11 conserved sites, including its own N-terminal and C-terminal auto-processing sites.3,4 The absolute requirement of proper function of 3CLpro for SARS-CoV-2 viral replication, together with the absence of a homologous human protease, makes 3CLpro an attractive target for designing specific anti-viral drugs. Therefore, development of inhibitors against 3CLpro is a current focus of global academic and pharma efforts for the treatment of COVID-19 disease.5–8 A series of small molecules or metal complexes have been reported to inhibit 3CLpro activity, including boceprevir, calpain inhibitor XII, GRL-0496, GC376 and Re(I) tricarbonyl complexes.9–11 Typically, a metallodrug used to treat duodenal ulcer, ranitidine bismuth citrate has recently been identified to suppress SARS-CoV-2 replication, implying the potential to repurpose the old metallodrugs as therapies for treatment of COVID-19.12 However, the detailed molecular mechanism has not been fully elucidated. Herein, we identified that the essential SARS-CoV-2 protease 3CLpro is one of the targets for bismuth drug, colloidal bismuth subcitrate (CBS). CBS allosterically inhibited the proteolytic activity of 3CLpro and suppressed SARS-CoV-2 viral replication in mammalian cell.
Results
Since 3CLpro contains a Cys residue at the active site, we envisioned that thiophilic metal ions could coordinate to the sulfur atom and abrogate its protease function. Therefore, we performed a primary evaluation of the enzyme inhibition properties of a series of metallodrugs and their active derivatives, including two Bi(III)-based drugs, colloidal bismuth subcitrate (CBS) and bismuth gluconate, Sb(V)-based drug (sodium stibogluconate, SSG), Pt(II)-based drug (cis-platinum), Au(I)-based drug (Auranofin and its metabolite AuP(CH2CH3)3Cl) and four reported Au(I) active complexes (Fig. S1†).13,14In brief, 0.5 μM purified 3CLpro was incubated with 50 μM metal complexes for 20 min. The protease activity of 3CLpro was then measured using the quenched fluorescence resonance energy transfer (FRET) assays as previously described.15,16 Among all the examined metal complexes, the two Bi(III)-based drugs, CBS and bismuth gluconate were the most potent inhibitors. After incubation with 50 μM CBS or bismuth gluconate, the protease activity of 3CLpro decreased more than 90% (Fig. 1A). The inhibition properties of Bi(III)-based drugs are attributed to the binding of Bi(III) to the proteases, since the complex ligands, citrate and gluconate displayed no inhibitory effects (Fig. S2†). The half-maximum inhibitory concentration (IC50) value of CBS against 3CLpro protease activity was subsequently measured. A feline coronavirus drug GC376 was recently reported as a highly potent inhibitor of 3CLpro from both SARS-CoV and SARS-CoV-2 with IC50 values in the nanomolar range.17–19 Therefore, we used GC376 as a positive control. As shown in Fig. 1B, the IC50 value of CBS against 3CLpro was calculated to be 0.93 ± 0.04 μM. Similar IC50 value was also obtained for bismuth gluconate (Fig. S3†). The calculated sub-micromolar IC50 values are comparable to the potent small-molecule inhibitor GC376,17 which has a calculated IC50 value of 0.47 ± 0.03 μM in our proteolytic activity assay (Fig. S4†). Furthermore, the Michaelis–Menten kinetics of 3CLpro was also examined before and after Bi(III)-binding (Fig. S5†). Unexpectedly, although both Km and Vmax of 3CLpro decreased, the kcat/Km value kept unchanged after Bi(III)-binding, implying that Bi(III) is probably an uncompetitive inhibitor for 3CLpro. The results indicated that Bi(III)-based drugs were highly potent protease inhibitors for SARS-CoV-2 3CLpro in vitro.
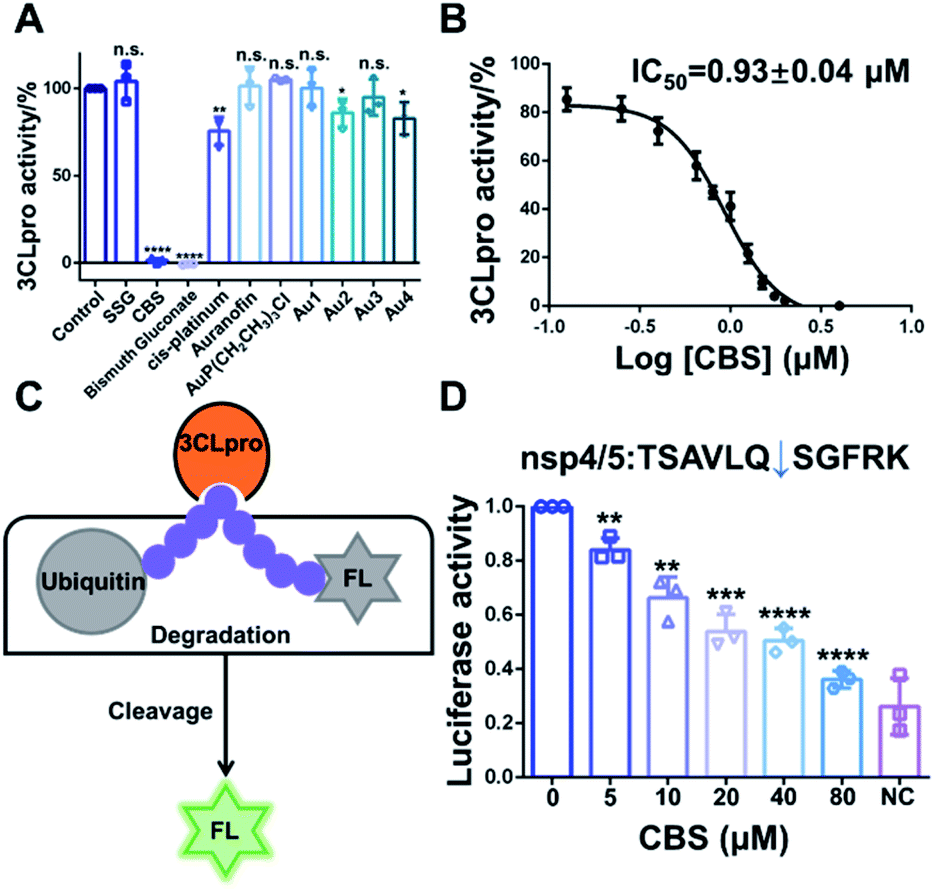 | ||
| Fig. 1 (A) Identification of inhibitory metallodrugs for SARS-CoV-2 3CLpro. Bar charts show the relative protease activities of 3CLpro after incubation with 50 μM sodium stibogluconate (SSG), colloidal bismuth subcitrate (CBS), bismuth gluconate, cis-platinum, Auranofin, AuP(CH2CH3)3Cl and four Au(I)-based complexes. (B) Inhibition of the protease activities of 3CLpro by CBS at varying concentrations in vitro. Dose–response curves for half-maximum inhibitory concentration (IC50) values were determined by nonlinear regression. (C) Inhibition of SARS-CoV-2 proteases by CBS in cellulo. Schematic illustration of the cell-based luciferase reporter assay. (D) The cells that expressed 3CLpro are treated with varying concentrations of CBS. NC represents negative control. Bar charts show the relative luciferase activity of cell lysate. Each experiment were performed in triplicates. The data are shown in mean ± sd. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
To further examine the inhibitory activity of Bi(III)-based drugs against 3CLpro activity in cellulo, a luciferase reporter activity assay was developed. In brief, firefly luciferase (FL) reporter gene is fused with polyubiquitin (UB), which leads to the quick degradation of luciferase in proteasome. A 3CLpro cleavage site is inserted between FL and UB. In the presence of 3CLpro, proteolytic cleavage on this site will set free the luciferase and the luciferase activity can be detected (Fig. 1C). Therefore, the relative luciferase activity was in direct proportion to the activity of 3CLpro in cellulo. The small-molecule inhibitor GC376 was first used to validate the in cellulo luciferase assay. Three different 3CLpro cleavage sites with the lowest protein sequence similarity (nsp4/5, nsp5/6 and nsp8/9) were examined in the experiment. Indeed, GC376 could potently inhibit 3CLpro proteolytic activity for all the three cleavage sites with concentrations of 5–20 μM, indicating that the luciferase assay could be used to examine the 3CLpro activity in cellulo (Fig. S6†). We subsequently employed the assay to examine the inhibitory effect of CBS. The inhibitory effect of CBS was more potent than GC376. CBS remarkably retarded the proteolytic cleavage for all the three sites as low as 5 μM (Fig. 1D and S7†). Whereas 10–20 μM GC376 are required to inhibit 3CLpro proteolytic activity for nsp5/6 and nsp8/9 site, respectively. Collectively, the results clearly demonstrated that CBS can abrogate 3CLpro activity both in vitro and in mammalian cells.
Subsequently, we would like to elucidate the molecular mechanism of 3CLpro activity inhibition by Bi(III)-based drugs. First, we employed inductively coupled plasma mass spectrometry (ICP-MS) coupled with protein BCA assay to investigate the Bi(III) binding capability of 3CLpro. The experiment results indicated that 3CLpro was capable to bind 2.13 ± 0.11 molar equivalents (eq.) of Bi(III) per monomer (Fig. S8†). Given the highly thiophilic property of Bi(III), we speculated that Bi(III) could bind to Cys residues of 3CLpro to interfere with the protease activity. Similar to the reported homologue of SARS-CoV, the SARS-CoV-2 3CLpro structure is a homodimer with multiple Cys residues, consisting of a N-terminal finger (residues 1–8), a catalytic domain (residues 8–184) and a C-terminal dimerization domain (residues 201–306).5,7
In order to identify the potential Bi(III)-binding sites, the solvent accessible Cys residues in 3CLpro structure (PDBID: 6M2Q) were identified using the AREAIMOL software in CCP4 package.20 The top three ranking Cys residues accessible to solvent are Cys300, Cys156 and Cys145 (Table S2†). Cys300 is close to the C-terminus and located at the dimerization domain of 3CLpro, while Cys156 is a surface exposed residue at the catalytic domain. Cys145 is one of the conserved residues in the 3CLpro catalytic dyad (His41 and Cys145) (Fig. 2A). The three Cys residues were subsequently mutated to serine individually. In vitro proteolytic assay results demonstrated that the activities of 3CLproD155A/C156S and 3CLproC300S mutants were similar to wild-type (WT) 3CLpro, suggesting that the overall conformation of 3CLpro was not significantly altered by mutations (Fig. S9†). Subsequently, the Bi(III)-binding capabilities of the two mutants were examined. Unexpected, the binding stoichiometries of 3CLproC145S and 3CLproD155A/C156S mutants were almost the same as wild-type (WT) 3CLpro, with 2.04 ± 0.07 and 2.29 ± 0.01 eq. of Bi(III) per monomer respectively, indicating that both Cys145 and Cys156 are not involved in Bi(III) binding. In contrast, the 3CLproC300S mutant had significantly reduced Bi(III) binding capability with 1.28 ± 0.01 eq. of Bi(III) per monomer, suggesting that the Cys300 is essential at least for one Bi(III) binding site. Protein electrostatic potential map of 3CLpro revealed that the molecular surfaces around Cys145 and Cys156 residues are positively charged. In contrast, the surrounding amino acids of Cys300 residue were mostly negatively charged (Fig. S10†). The different surface electrostatic potential could at least partially explain why Bi(III) preferred Cys300 rather than two other exposed Cys residues.
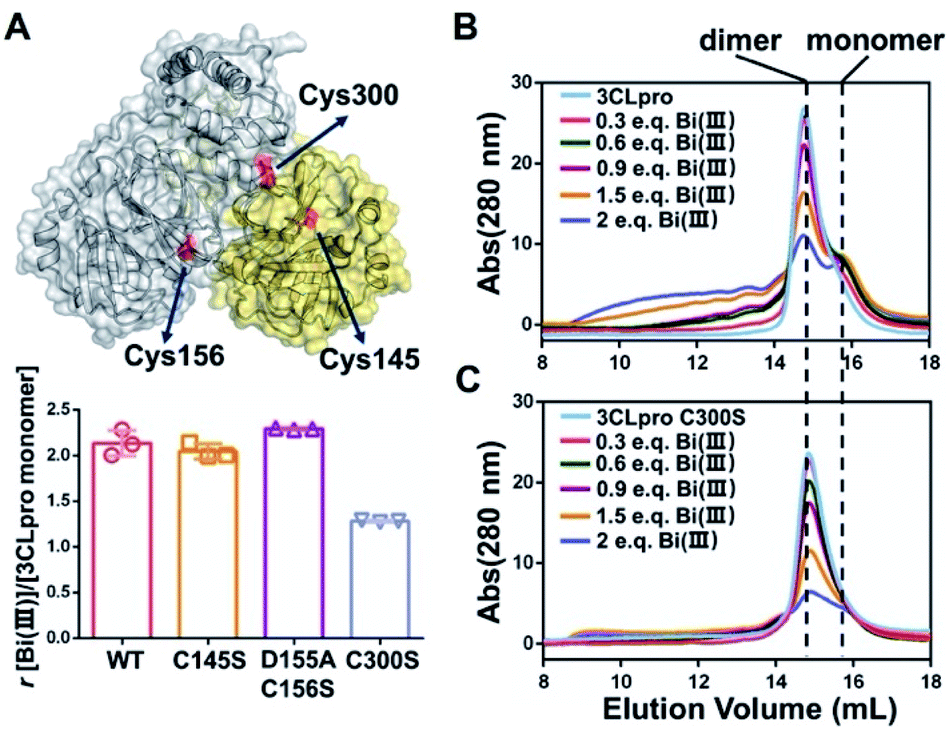 | ||
| Fig. 2 SARS-CoV-2 3CLpro dimer dissociation induced by colloidal bismuth subcitrate (CBS). (A) Structure of SARS-CoV-2 3CLpro dimer is shown in cartoon and surface model (PDBID: 6M2Q). The cysteine residues with the highest solvent accessibility are highlighted in red. Bar chart shows the Bi(III)-binding capability of wild-type 3CLpro and mutants. Each experiment were performed in triplicates. The data are shown in mean ± sd. Size exclusion chromatography analysis of (B) 3CLpro and (C) 3CLproC300S mutant after incubation with different molar ratios of Bi(III). | ||
It is worth noting that the Cys300 is located at the C-terminal domain of 3CLpro, which is essential to stabilize the dimeric state of 3CLpro. A recent study demonstrated that small molecule binding at the C-terminal domain would lead to 3CLpro dimer dissociation.21 Therefore, we are prompted to investigate whether Bi(III)-binding could abrogate 3CLpro dimerization. In brief, approximately 10 μM 3CLpro was incubated with increasing concentrations of CBS and the oligomerization states were analyzed by size-exclusion chromatography (SEC). As shown in Fig. 2B, a shoulder peak appeared in SEC of 3CLpro after incubation with low dosage of CBS, indicative of monomeric 3CLpro formation. In contrast, no monomeric shoulder peak was observed for 3CLproC300S mutant (Fig. 2C), suggesting that Bi(III) coordination to Cys300 led to 3CLpro dimer dissociation. Typically, higher dosage of Bi(III) incubation caused larger molecular weight aggregation of 3CLpro, implying that Bi(III)-binding to the second metal site could further induce oligomerization of 3CLpro.
Previous studies demonstrated that the N-terminal finger and C-terminal dimerization domain are essential to stabilize 3CLpro dimer. To further elucidate the underlying molecular mechanisms of Bi(III)-induced 3CLpro dimer dissociation, all-atom molecular dynamics (MD) simulations were performed to compare the structures and flexibility of 3CLpro in the presence of Na(I) and Bi(III) ions, respectively. The details about the MD simulations are described in the ESI.† The MM/GBSA free energy analysis indicated that the protomer–protomer interactions of 3CLpro dimer in the presence of Na(I) are quite stable, with a binding free energy of −75.36 kcal mol−1 between the two protomers. In contrast, Bi(III) binding to the Cys300 residue significantly destabilized the dimeric 3CLpro structure with a calculated free energy of −52.80 kcal mol−1 (Fig. S11†). Detailed MD trajectories analysis revealed that stable binding of Bi(III) ion with Cys300 would significantly alter the conformational state of the C-terminal helical segment of 3CLpro, which exhibited a more solvent-exposed conformation. Particularly, the solvent accessible surface area (SASA) of the C-terminal helical segment (residue 292 to 303) increased by about 100 Å2 compared to that in the presence of Na(I) (Fig. S12†).
The induced structurally changes of the C-terminal helical segment upon Bi(III) binding, hence, influenced the conformational state of Arg298 and disrupted the hydrogen bond between the guanidino group of Arg298 and the backbone carbonyl oxygen atom of Met6 (Fig. 3A and S13†). The breakage of Arg298-Met6 hydrogen bond increased the flexibility of the N-finger, which destabilized the salt bridge between Arg4 and Glu290* on the other protomer (Fig. 3). In all our five 1000 ns MD trajectories, the Arg4–Glu290* salt bridge was disrupted in the presence of Bi(III) ions. In contrast, this salt bridge was well maintained in all the MD trajectories with Na(I) (Fig. 3B and S14†), implying the essential role of these residues to maintain 3CLpro dimeric state. Therefore, the disruption of SARS-CoV-2 3CLpro dimer is probably due to the conformational distortion of these key residues upon Bi(III) binding. Previous studies showed that R4A, E290A or R298A mutations of SARS-CoV 3CLpro lead to the dissociation of dimeric structure.22,23 Since the highly structural similarity between SARS-CoV-2 and SARS-CoV 3CLpro, it is reasonable to assume that the dissociation of SARS-CoV-2 3CLpro dimer would also cause the collapse of substrate binding pocket, resulting in loss of protease activity as observed for SARS-CoV 3CLpro.24
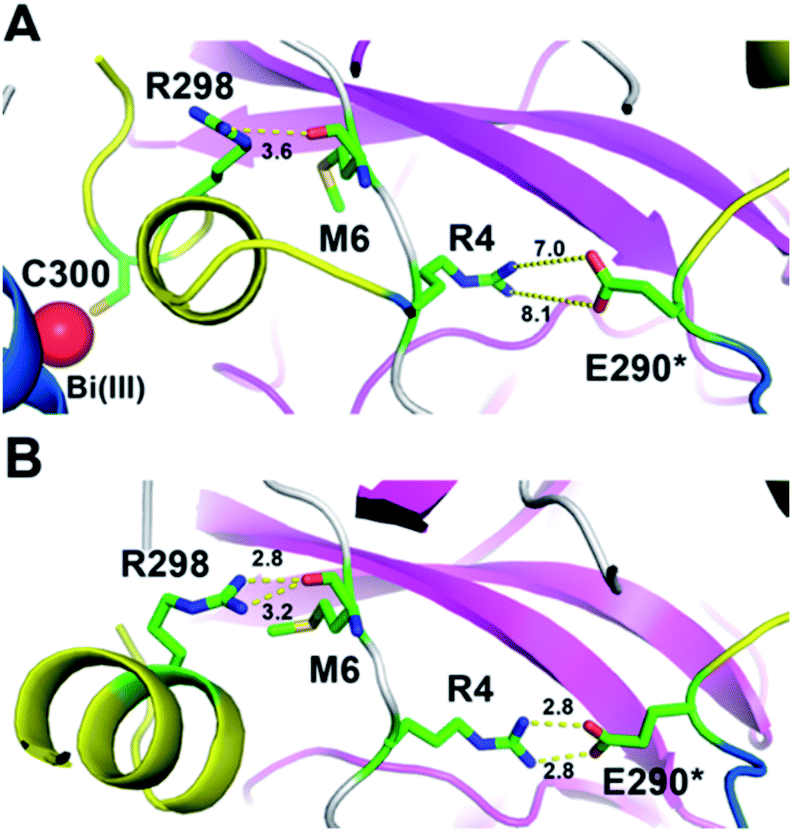 | ||
| Fig. 3 A snapshot of 3CLpro dimer in MD simulations in the presence of Bi(III) and Na(I). The C-terminal helical segment was colored in yellow cartoon and Bi(III) ion was shown in red sphere. (A) Both the hydrogen bonds between R298-M6 and R4-E290* are disrupted in the presence of Bi(III). (B) The hydrogen bonds between R298-M6 and R4-E290* are well maintained in the presence of Na(I). The distances between two residues are labeled in Å. | ||
To further confirm the results, we constructed two 3CLpro mutants, 3CLproE290A and 3CLproR298A to break the essential hydrogen bonds between two protomers of 3CLpro. Indeed, SEC results showed that the elution volumes of the two mutants shifted backwards approximately 1 mL, corresponding to the molecular weight of a monomeric 3CLpro (Fig. S15†). Moreover, in vitro proteolytic assays indicated that the protease activity of the two mutants were completely abolished compared to dimeric 3CLpro (Fig. S16†).
Finally, virus infection experiments were performed to evaluate the suppression effect of Bi(III)-based drug on SARS-CoV-2 replication as described previously.25 In brief, Vero E6 cells were infected with SARS-CoV-2 at an MOI of 0.05 with a treatment of different concentrations of CBS for 24 h. Virus nucleocapsid (N protein) in the infected Vero E6 cells were then blotted and quantified by immunofluorescence as described in the ESI.† In line with previous studies, bismuth drug CBS efficiently suppressed the SARS-CoV-2 replication as evidenced by the significantly decrease of the expression of viral nucleocapside protein in CBS treated groups compared to the control group (Fig. 4A). The half maximum effective doses (EC50) of CBS against SARS-CoV-2 replication was calculated to be 177.3 ± 32.0 μM and no significant cytotoxicity was observed for the applied CBS (Fig. 4B).
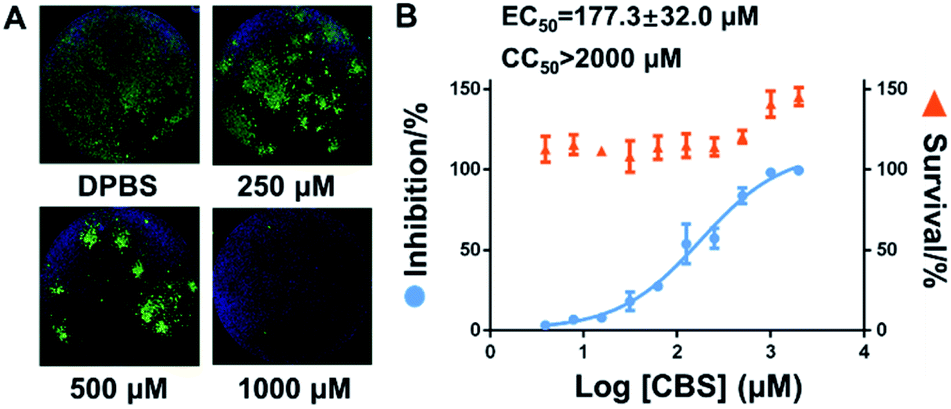 | ||
| Fig. 4 Inhibition of SARS-CoV-2 replication in Vero E6 cell by colloidal bismuth subcitrate (CBS). (A) Representative immunofluorescence staining images of Vero E6 cells infected with SARS-CoV-2. The nucleocapsid protein of SARS-CoV-2 (N protein) antigens and cell nuclei (DAPI) were stained in green and blue, respectively. (B) Inhibition of SARS-CoV-2 replication and cytotoxicity of increasing concentrations of colloidal bismuth subcitrate (CBS). Each experiment was performed in triplicates. The data are shown in mean ± sd. | ||
Conclusions
In conclusion, we demonstrated that colloidal bismuth subcitrate (CBS) remarkably inhibited the SARS-CoV-2 3CLpro activity in vitro and in cellulo, indicating that 3CLpro was a biological target of CBS to suppress viral replication. Although the catalytic residue Cys145 of 3CLpro is solvent accessible, Bi(III) did not bind to catalytic site. Instead, biochemical analysis together with MS simulations revealed that Bi(III) first bound to the metal site at the C-terminal domain of 3CLpro and caused dimeric 3CLpro to dissociate into monomers, thereby resulting in protease dysfunction. Additional binding of Bi(III) to the second metal site of 3CLpro could further induce the aggregation of the protease (Fig. 5). Our work not only revealed that CBS functions as a unique allosteric inhibitor of 3CLpro but also provided valuable insight into the anti-SARS-CoV-2 mechanisms of bismuth drugs.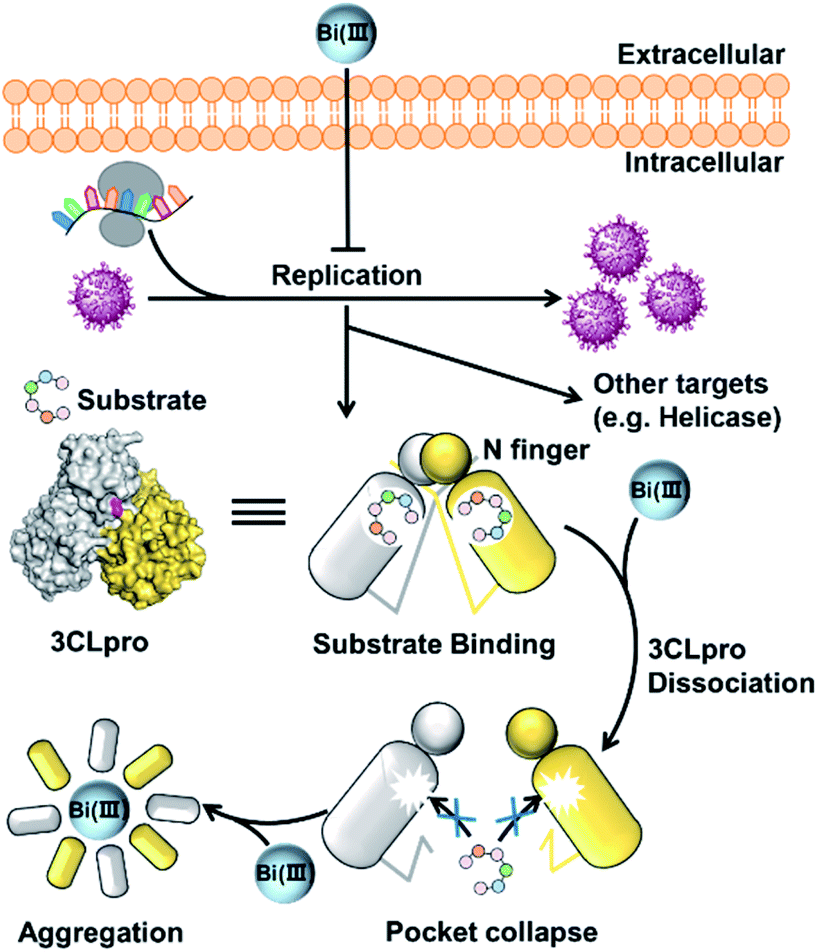 | ||
| Fig. 5 Schematic illustration of the inhibition of SARS-CoV-2 replication in cell. Bismuth-binding induced dissociation of 3CLpro dimer leads to the collapse of substrate-binding site and proteolytic dysfunction. | ||
It is worth noting that the required concentrations of CBS for inhibition in cellular-based experiments were much higher than that in the proteolytic assay. This discrepancy is probably due to the multiple-targeting property of bismuth drugs. Indeed, a recent study demonstrated that ranitidine bismuth citrate and its related compounds exhibited inhibition towards the activity of the SARS-CoV-2 helicase.12 And our preliminary data also showed that the activity of SARS-CoV-2 papain-like protease (PLpro) was abrogated in the presence of CBS. Therefore, a systematic identification of potential targets in SARS-CoV-2 is warranted in future, which could provide further insight into the anti-viral mechanism of bismuth drugs.
Author contributions
X. T., W. X. and Z.-W. M conceived the project and designed the experiments. X. T., K. L., Z. Z., Y. X. and P.-H. W. carried out the protein purification and in vitro proteolytic assays. L. D. carried out in cellulo inhibition experiment. L. Z., R. L. and H. C. carried out virus inhibition experiments and statistical analyses. Y. Z. and G. L. carried out molecular dynamics studies. X. T., L. Z., L. D., J.-A. P., W. X., Y. Z., and Z.-W. M. wrote and revised the paper. J.-A. P., W. X., G. L, J. D. and Z.-W. M. supervised the study. All authors proofread, commented on, and approved the final submitted version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22077142, 22022706, 21837006, 91953117 and 21877131), Natural Science Foundation of Guangdong Province, China (2019A1515011156), the Ministry of Education of China (IRT-17R111) and the Fundamental Research Funds for the Central Universities. The Au(I) compounds are generous gifts from Professor Taotao Zou (School of Pharmaceutical Sciences, Sun Yat-Sen University).Notes and references
- P. Zhou, X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li, C.-L. Huang, H.-D. Chen, J. Chen, Y. Luo, H. Guo, R.-D. Jiang, M.-Q. Liu, Y. Chen, X.-R. Shen, X. Wang, X.-S. Zheng, K. Zhao, Q.-J. Chen, F. Deng, L.-L. Liu, B. Yan, F.-X. Zhan, Y.-Y. Wang, G.-F. Xiao and Z.-L. Shi, Nature, 2020, 588(7836), E6 CrossRef CAS PubMed.
- F. U. Hartl and M. Hayer-Hartl, Nat. Struct. Mol. Biol., 2009, 16(6), 574–581 CrossRef CAS PubMed.
- Y. M. Baez-Santos, S. E. St John and A. D. Mesecar, Antiviral Res., 2015, 115, 21–38 CrossRef CAS PubMed.
- K. Anand, G. J. Palm, J. R. Mesters, S. G. Siddell, J. Ziebuhr and R. Hilgenfeld, EMBO J., 2002, 21(13), 3213–3224 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582(7811), 289–293 CrossRef CAS PubMed.
- Z. M. Jin, Y. Zhao, Y. Sun, B. Zhang, H. F. Wang, Y. Wu, Y. Zhu, C. Zhu, T. Y. Hu, X. Y. Du, Y. K. Duan, J. Yu, X. B. Yang, X. N. Yang, K. L. Yang, X. Liu, L. W. Guddat, G. F. Xiao, L. K. Zhang, H. T. Yang and Z. H. Rao, Nat. Struct. Mol. Biol., 2020, 27(6), 529–532 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368(6489), 409–412 CrossRef CAS PubMed.
- W. H. Dai, B. Zhang, X. M. Jiang, H. X. Su, J. A. Li, Y. Zhao, X. Xie, Z. M. Jin, J. J. Peng, F. J. Liu, C. P. Li, Y. Li, F. Bai, H. F. Wang, X. Cheng, X. B. Cen, S. L. Hu, X. N. Yang, J. Wang, X. Liu, G. F. Xiao, H. L. Jiang, Z. H. Rao, L. K. Zhang, Y. C. Xu, H. T. Yang and H. Liu, Science, 2020, 368(6497), 1331–1335 CrossRef CAS PubMed.
- J. Karges, M. Kalaj, M. Gembicky and S. M. Cohen, Angew. Chem., Int. Ed., 2021, 60(19), 10716–10723 CrossRef CAS PubMed.
- Y.-W. Zhou, Y. Xie, L.-S. Tang, D. Pu, Y.-J. Zhu, J.-Y. Liu and X.-L. Ma, Signal Transduction Targeted Ther., 2021, 6(1), 317 CrossRef CAS PubMed.
- J. M. O. Rawson, A. Duchon, O. A. Nikolaitchik, V. K. Pathak and W.-S. Hu, Viruses, 2021, 13(2), 173 CrossRef CAS PubMed.
- S. Yuan, R. Wang, J. F.-W. Chan, A. J. Zhang, T. Cheng, K. K.-H. Chik, Z.-W. Ye, S. Wang, A. C.-Y. Lee, L. Jin, H. Li, D.-Y. Jin, K.-Y. Yuen and H. Sun, Nat. Microbiol., 2020, 5(11), 1439–1448 CrossRef CAS PubMed.
- T. T. Zou, C. T. Lum, C. N. Lok, W. P. To, K. H. Low and C. M. Che, Angew. Chem., Int. Ed., 2014, 53(23), 5810–5814 CrossRef CAS PubMed.
- N. V. Tzouras, M. Saab, W. Janssens, T. Cauwenbergh, K. Van Hecke, F. Nahra and S. P. Nolan, Chem.–Eur. J., 2020, 26(24), 5541–5551 CrossRef CAS PubMed.
- Y.-S. Han, G.-G. Chang, C.-G. Juo, H.-J. Lee, S.-H. Yeh, J. T.-A. Hsu and X. Chen, Biochemistry, 2005, 44(30), 10349–10359 CrossRef CAS PubMed.
- H. Yang, W. Xie, X. Xue, K. Yang, J. Ma, W. Liang, Q. Zhao, Z. Zhou, D. Pei, J. Ziebuhr, R. Hilgenfeld, K. Y. Yuen, L. Wong, G. Gao, S. Chen, Z. Chen, D. Ma, M. Bartlam and Z. Rao, PLoS Biol., 2005, 3(10), e324 CrossRef PubMed.
- C. L. Ma, M. D. Sacco, B. Hurst, J. A. Townsend, Y. M. Hu, T. Szeto, X. J. Zhang, B. Tarbet, M. T. Marty, Y. Chen and J. Wang, Cell Res., 2020, 30(8), 678–692 CrossRef CAS PubMed.
- S. Iketani, F. Forouhar, H. Liu, S. J. Hong, F.-Y. Lin, M. S. Nair, A. Zask, Y. Huang, L. Xing, B. R. Stockwell, A. Chavez and D. D. Ho, Nat. Commun., 2021, 12(1), 2016 CrossRef CAS PubMed.
- Y. Kim, H. Liu, A. C. Galasiti Kankanamalage, S. Weerasekara, D. H. Hua, W. C. Groutas, K.-O. Chang and N. C. Pedersen, PLoS Pathog., 2016, 12(3), e1005531 CrossRef PubMed.
- B. Lee and F. M. Richards, J. Mol. Biol., 1971, 55(3), 379–400 CrossRef CAS PubMed.
- T. J. El-Baba, C. A. Lutomski, A. L. Kantsadi, T. R. Malla, T. John, V. Mikhailov, J. R. Bolla, C. J. Schofield, N. Zitzmann, I. Vakonakis and C. V. Robinson, Angew. Chem., Int. Ed., 2020, 59(52), 23544–23548 CrossRef CAS PubMed.
- C.-Y. Chou, H.-C. Chang, W.-C. Hsu, T.-Z. Lin, C.-H. Lin and G.-G. Chang, Biochemistry, 2004, 43(47), 14958–14970 CrossRef CAS PubMed.
- J. Shi, J. Sivaraman and J. Song, J. Virol., 2008, 82(9), 4620 CrossRef CAS PubMed.
- S. Chen, T. Hu, J. Zhang, J. Chen, K. Chen, J. Ding, H. Jiang and X. Shen, J. Biol. Chem., 2008, 283(1), 554–564 CrossRef CAS PubMed.
- M. L. Wang, R. Y. Cao, L. K. Zhang, X. L. Yang, J. Liu, M. Y. Xu, Z. L. Shi, Z. H. Hu, W. Zhong and G. F. Xiao, Cell Res., 2020, 30(3), 269–271 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03526f |
| This journal is © The Royal Society of Chemistry 2021 |