
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Genomics-driven discovery of chiral triscatechol siderophores with enantiomeric Fe(III) coordination†
Parker R.
Stow
 a,
Zachary L.
Reitz
a,
Timothy C.
Johnstone
b and
Alison
Butler
*a
a,
Zachary L.
Reitz
a,
Timothy C.
Johnstone
b and
Alison
Butler
*a
aDepartment of Chemistry and Biochemistry, University of California Santa Barbara, CA 93106-9510, USA. E-mail: butler@chem.ucsb.edu
bDepartment of Chemistry and Biochemistry, University of California Santa Cruz, CA 95064, USA
First published on 25th August 2021
Abstract
Ferric complexes of triscatechol siderophores may assume one of two enantiomeric configurations at the iron site. Chirality is known to be important in the iron uptake process, however an understanding of the molecular features directing stereospecific coordination remains ambiguous. Synthesis of the full suite of (DHBL/DLysL/DSer)3 macrolactone diastereomers, which includes the siderophore cyclic trichrysobactin (CTC), enables the effects that the chirality of Lys and Ser residues exert on the configuration of the Fe(III) complex to be defined. Computationally optimized geometries indicate that the Λ/Δ configurational preferences are set by steric interactions between the Lys sidechains and the peptide backbone. The ability of each (DHBL/DLysL/DSer)3 diastereomer to form a stable Fe(III) complex prompted a genomic search for biosynthetic gene clusters (BGCs) encoding the synthesis of these diastereomers in microbes. The genome of the plant pathogen Dickeya chrysanthemi EC16 was sequenced and the genes responsible for the biosynthesis of CTC were identified. A related but distinct BGC was identified in the genome of the opportunistic pathogen Yersinia frederiksenii ATCC 33641; isolation of the siderophore from Y. frederiksenii ATCC 33641, named frederiksenibactin (FSB), revealed the triscatechol oligoester, linear-(DHBLLysLSer)3. Circular dichroism (CD) spectroscopy establishes that Fe(III)–CTC and Fe(III)–FSB are formed in opposite enantiomeric configuration, consistent with the results of the ferric complexes of the cyclic (DHBL/DLysL/DSer)3 diastereomers.
Introduction
Chirality is universally significant in biological reactions, including the essential microbial process of iron acquisition mediated by small-molecule chelators known as siderophores. The specific three-dimensional structure of an Fe(III)–siderophore complex plays a role in the ability of a bacterium to recognize, acquire, and extract iron from it.1,2 The triscatechol siderophores enterobactin (Ent) and bacillibactin (BB) each coordinate Fe(III) with three 2,3-dihydroxybenzoyl (DHB) ligands framed on a macrolactone derived from three LSer or LThr residues, respectively (ESI Fig. S1†). Unlike Ent, BB also contains a glycine residue inserted between the macrolactone core and DHB. In stark contrast to Fe(III)–Ent3−, which forms exclusively in the Δ configuration,3,4 Fe(III)–BB3− adopts the opposing Λ configuration.3 Several related triscatechol siderophores are further distinguished from Ent and BB by the presence of a chiral amino acid inserted between DHB and the oligoester backbone, including cyclic trichrysobactin [CTC; Dickeya chrysanthemi EC16] with DLys,5 as well as the linear tris-LSer scaffolds of trivanchrobactin with DArg,6 and turnerbactin with LOrn.7 Structurally, the influence amino acids exert on the configuration at the Fe(III) site is incompletely understood; evolutionarily, these structural differences hint that chirality confers a competitive advantage in microbial iron uptake.To understand the factors controlling stereospecific Fe(III) coordination of the expanded triscatechol–triserine siderophore CTC, (DHBDLysLSer)3, we synthesized the full suite of cyclic (DHBL/DLysL/DSer)3 diastereomers of CTC (Fig. 1). We report that circular dichroism (CD) spectroscopic measurements of the Fe(III) complexes of these ligands allow the relationship between siderophore chirality and the configuration at the Fe(III) site to be defined. Computational modeling of the ferric complexes reveals steric interactions between the Lys sidechains and the peptide backbone dictate the configurational preference. Fe(III) complexation by each (DHBL/DLysL/DSer)3 diastereomer prompted microbial genome mining that led to the discovery of a new triscatechol siderophore, frederiksenibactin (FSB). FSB features LLys residues inserted between a linear triserine backbone and chelating DHB units. The genomics data, in combination with our spectroscopic investigation of isolated FSB, establish that it is a natural diastereomer of linear trichrysobactin. Circular dichroism (CD) spectra reveal that FSB and CTC coordinate Fe(III) in opposing enantiomeric configuration, consistent with the setting of the handedness of Fe(III) coordination by the stereochemistry at Lys.
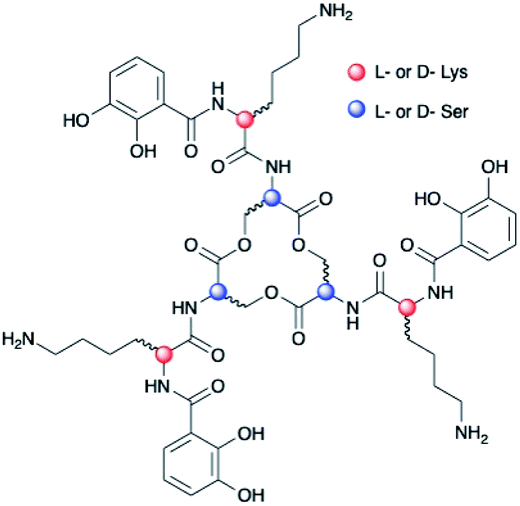 | ||
| Fig. 1 Diastereomers of (DHBL/DLysL/DSer)3. | ||
Results and discussion
Chirality of Fe(III)–[(DHBL/DLysL/DSer)3] diastereomers
Chiral triscatechol siderophores and synthetic analogs are capable of coordinating labile metal ions with a thermodynamic preference for a specific stereochemistry at the metal center.8 The presence of chirality at both the metal center and in the ligand renders the Δ and Λ stereoisomers diastereomeric and energetically inequivalent. To establish the relationship between the chirality at the amino acid adjacent to the catecholamide, the stereochemistry of the triserine macrolactone, and the stereochemistry at Fe(III), we synthesized the four C3-symmetric cyclic diastereomers (DHBLLysLSer)3, (DHBDLysLSer)3, (DHBLLysDSer)3, and (DHBDLysDSer)3 (Scheme S1†) of which (DHBDLysLSer)3 is structurally identical to CTC. Well-established methodology to construct the cyclic triserine macrolactone (1 in Scheme S1†),9 provided a convenient synthetic platform to access CTC and related diastereomers (Fig. S2–S6, Tables S1 and S2†). In the absence of crystallographic information, circular dichroism (CD) spectroscopy can provide information on the stereochemical configuration of optically-active metal complexes.10As expected, enantiomeric pairs of ligands, such as (DHBLLysLSer)3 and (DHBDLysDSer)3, coordinate Fe(III) with opposite handedness, as indicated by the CD spectra (Fig. 2). Two prominent CD bands at 435 nm and 545 nm arise from characteristic LMCT transitions and are therefore sensitive to the chirality at the iron center. When comparing diastereomeric ligands, we observed that the CD spectra of Fe(III)–[(DHBLLysLSer)3] and Fe(III)–[(DHBLLysDSer)3] are similar in both the sign and the intensity. The analogous correspondence was observed for the CD bands of Fe(III)–[(DHBDLysLSer)3] (i.e., CTC) and Fe(III)–[(DHBDLysDSer)3] (Fig. 2, Table 1).‡
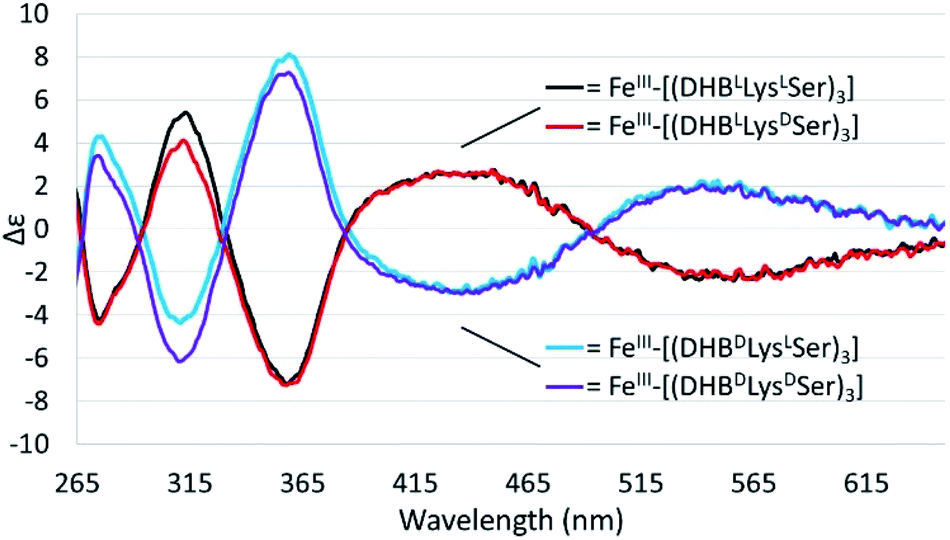 | ||
| Fig. 2 CD spectra of 40 mM solutions of the Fe(III)–(DHBL/DLysL/DSer)3] complexes in citrate–phosphate buffer, pH 7.40. | ||
| Ligand | Cyclic (C) or linear (L) | Configuration | λ max (nm) | Δε (M−1 cm−1) |
|---|---|---|---|---|
| a 40 μM Fe(III) complexes in citrate–phosphate buffer, pH 7.40. b (DHBLLysLSer)3 is the cyclic analog of FSB. c Synthetic (DHBDLysLSer)3 shown here is indistinguishable from CTC isolated from D. chrysanthemi EC16. | ||||
| Ent = (DHBLSer)3 (ref. 3) | C | Δ | 553 | −2.2 |
| BB = (DHB-GlyLThr)3 (ref. 3) | C | Λ | 545 | +1.7 |
| (DHBLLysLSer)3b | C | Δ | 563 | −2.3 |
| (DHBDLysLSer)3c (CTC) | C | Λ | 559 | +2.2 |
| (DHBLLysDSer)3 | C | Δ | 561 | −2.3 |
| (DHBDLysDSer)3 | C | Λ | 558 | +2.1 |
| Frederiksenibactin (FSB) = linear-(DHBLLysLSer)3 | L | Δ | 556 | −2.2 |
Comparison of the signs of the Cotton effects for the Fe(III)–[(DHBL/DLysL/DSer)3] complexes with those of Fe(III)–Ent3− and Fe(III)–BB3−, for which the chirality at the metal center is known, allows the configuration of the new complexes to be determined. (DHBLLysLSer)3 and (DHBLLysDSer)3 both form Δ complexes, whereas (DHBDLysDSer)3 and (DHBDLysLSer)3 both form Λ complexes. These results suggest that the handedness of metal-ion chelation is set by the chirality of the Lys unit, and not the chirality of the triserine macrolactone. In comparison, the Δ configuration of Fe(III)–Ent3− and Λ configuration of Fe(III)–enantioEnt3− has been attributed to nonbonding interactions within the chiral triserine macrolactone.11–13
Computational modeling
To better understand the mechanism by which amino acid chirality dictates the configurational preferences of the Fe(III)–[(DHBL/DLysL/DSer)3] complexes, the structures and energies of the four enantiomeric pairs of diastereomers were optimized computationally (PBE0/6-311++G(d,p)) (Fig. 3). | ||
| Fig. 3 Energies for the optimized structures of the eight Fe(III)–[(DHBL/DLysL/DSer)3] diastereomers. | ||
Comparing the energies of the Fe(III) complexes of a given ligand with different handedness, we observe complete agreement with the CD spectroscopic results. For example, the energy of Λ-Fe(III)–[(DHBDLysLSer)3] is lower than that of Δ-Fe(III)–[(DHBDLysLSer)3], consistent with the formation of the Λ complex in aqueous solution (Fig. 2).
Insight into the origin of the differential stabilities of the Fe(III)–[(DHBL/DLysL/DSer)3] complexes comes directly from the optimized geometries (Fig. 4). In this molecular framework, the Lys sidechains are able to wrap around the complex so as to allow each terminal ammonium group to hydrogen-bond with the carbonyl of the DHB unit of an adjacent arm. We observe, however, that this interaction is present in all of the optimized geometries, preferred and non-preferred.
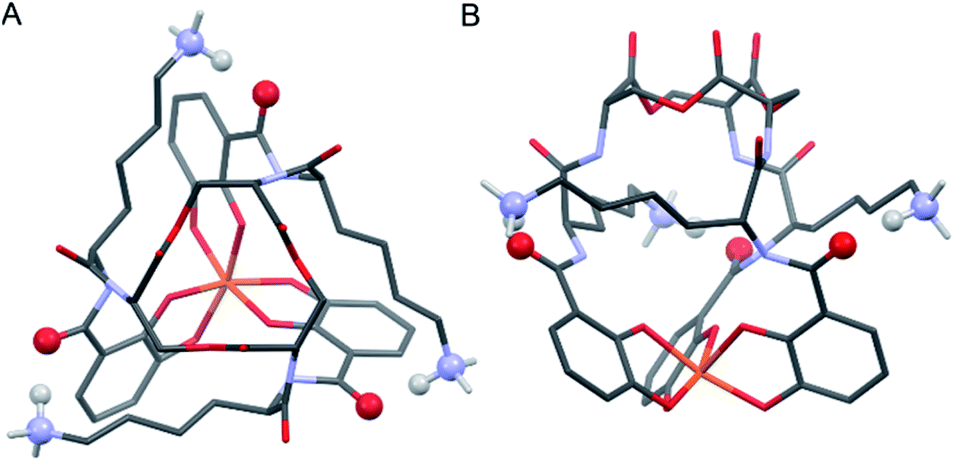 | ||
| Fig. 4 Stick representations of the computationally (PBE0/6-311++G(d,p)) optimized structure of Λ-Fe(III)–[(DHBDLysLSer)3] from the top (left) and side (right). H atoms removed for clarity except on Lys NH3 units. Color code: Fe orange, O red, N blue, C grey, H white. The atoms engaging in Lys-DHB intramolecular hydrogen bonding are shown as balls. | ||
Closer analysis revealed that the prime influence of the Lys residue chirality is the impact that it has on ψ (N–Ccarbonyl–Cα–N torsion angle). It is well established that certain values of ψ are unfavorable for polypeptides, contributing, for example, to the characteristic distribution of protein dihedral angles in Ramachandran plots. Specifically, favorable ψ values are those that prevent the amino acid side chain from eclipsing the adjacent carbonyl.14 In the Fe(III)–[(DHBL/DLysL/DSer)3] complexes, combination of either Δ configuration at Fe(III) and DLys, or Λ and LLys, produce ψ angles near ±60°, which introduces a steric clash between the carbonyl O atom and the Lys side chain (Fig. 5). In contrast, the combination of Λ and DLys, as occurs in CTC, produces no such clash.
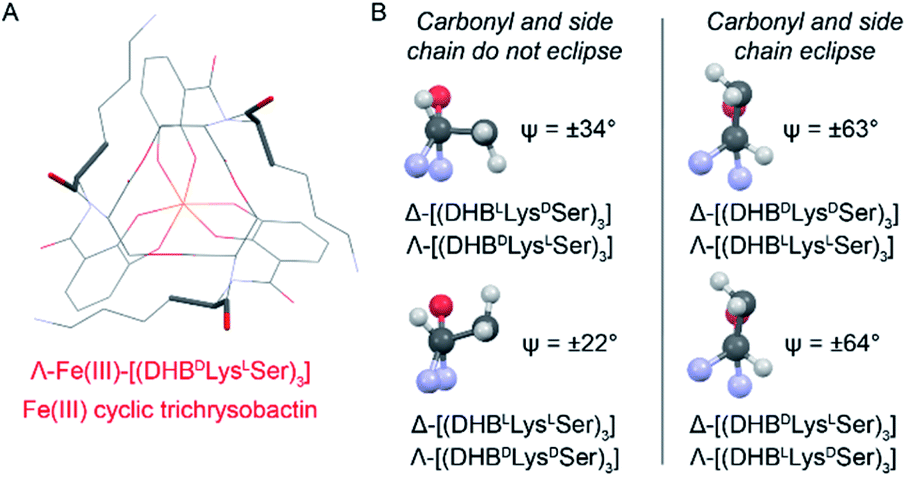 | ||
| Fig. 5 Left: Line diagram of Λ-Fe(III)–[(DHBDLysLSer)3] from the top with the carbonyl and first Lys side chain length shown as sticks. Right: Views along the Cα–Ccarbonyl bonds of Lys residues from the indicated Fe(III) complexes. Ψ values are averaged across the three present in each structure. Positive and negative Ψ values correspond to the compounds with DLys and LLys, respectively. | ||
It is noteworthy that, of all the diastereomeric combinations of metal chelation handedness and amino acid chirality, our calculations predict that the most stable structures are those assumed by Δ-Fe(III)–[(DHBLLysDSer)3] and its enantiomer Λ-Fe(III)–[(DHBDLysLSer)3]. Organisms have adopted this stability by using (DHBDLysLSer)3, which is the siderophore CTC, for iron acquisition. The fact that the other diastereomers that we investigated also form Fe(III) complexes gives rise to the question of whether they too might be used biologically.
Genomic screen for catechol-based siderophores
Inspired by the discovery of other naturally occurring siderophores with D- and L-amino acids – that is, trivanchrobactin (DArg), and turnerbactin (LOrn) – we initiated a search for biosynthetic gene clusters (BGCs) encoding diastereomers of CTC. The biosynthesis of chrysobactin (i.e., DHBDLysLSer) in D. dadantii 3937 requires genes encoding 2,3-DHB synthesis, as well as the non-ribosomal peptide synthetase (NRPS) CbsF with an epimerization, E, domain to convert LLys to DLys.15,16 In contrast to D. dadantii 3937, the plant pathogen D. chrysanthemi EC16 produces not only the monocatechol chrysobactin, but also the triscatechol macrolactone CTC. We found that the genome of the D. chrysanthemi EC16 contains a BGC homologous to the cbs locus of D. dadantii 3937 (genome sequence reported herein; Tables S3 and S4†). Genome mining revealed similar but distinct BGCs in several Yersinia genomes, including the BGC freABCEF of opportunistic pathogen Yersinia frederiksenii ATCC 33641 (Tables S5 and S6†). The fre locus contains genes encoding 2,3-DHB synthesis, as well as the NRPS FreF with adenylation domains selecting for LLys and LSer. However, FreF lacks an E domain, implicating biosynthesis of a siderophore comprised of DHBLLysLSer units (Fig. 6).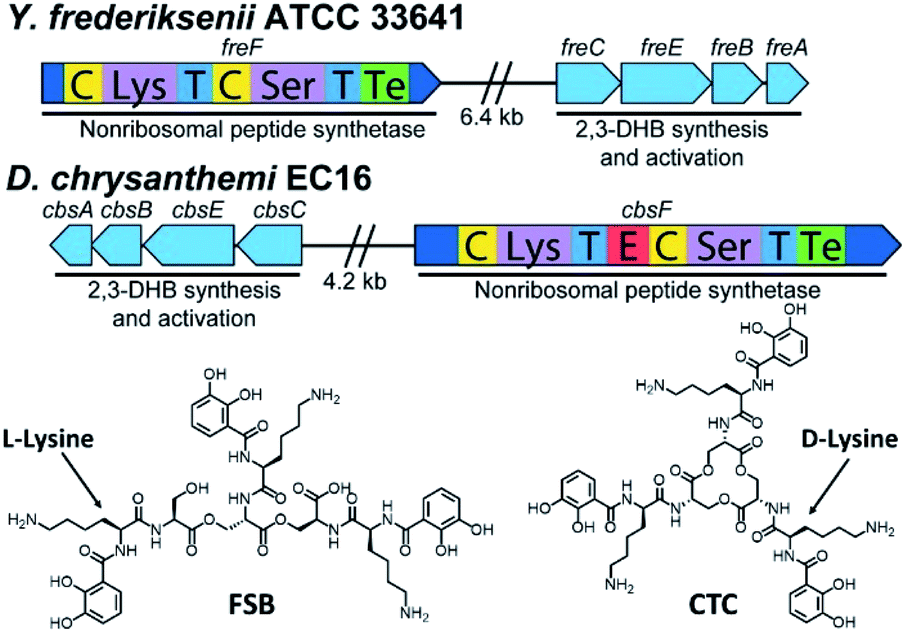 | ||
| Fig. 6 Biosynthetic gene clusters and structures of frederiksenibactin, FSB (Y. frederiksenii ATCC 33641) and cyclic trichrysobactin, CTC (D. chrysanthemi EC16). | ||
Frederiksenibactin and cyclic trichrysobactin siderophores
Siderophores from Y. frederiksenii ATCC 33641 were extracted and purified from a low-iron culture (Fig. S7†). Three compounds with m/z of 370.17, 721.31 and 1072.44 were identified by UPLC-ESIMS. These signals are equivalent to the [M + H]+ signals for chrysobactin, linear dichrysobactin, and linear trichrysobactin, which are DHB-DLys-LSer, linear-(DHB-DLys-LSer)2, and linear-(DHB-DLys-LSer)3, respectively.5 In contrast to D. chrysanthemi EC16, which produces trichrysobactin in both cyclic and linear forms,5 we have only been able to detect a linear triscatechol siderophore (m/z 1072.44; Fig. S8†) in the culture supernatant of Y. frederiksenii ATCC 33641. We have named this new siderophore frederiksenibactin (FSB). We note that the related triscatechol siderophores trivanchrobactin and turnerbactin are also linear and that their cyclic forms have not been detected in biological systems.6,7Marfey's analysis17 establishes the presence of LLys and LSer in FSB, consistent with the genomic prediction (Fig. S9†). The proposed structure of FSB was confirmed by 1H and 13C NMR spectroscopic data, which were assigned through 1H–1H COSY, 1H–13C HSQC, and 1H–13C HMBC NMR data (Fig. S10–S14†). While the NMR spectral data of FSB (Table S7†) are similar to those of CTC, several features confirm the mass spectrometric results indicating that FSB is a linear compound. Specifically, the three Ser residues are inequivalent (Fig. S11†). The Ser methylene protons involved in the backbone ester linkages, C16/C16′, at 4.25–4.46 ppm are shifted significantly downfield relative to the corresponding protons on C16′′ at 3.67 ppm and 3.78 ppm, which are adjacent to the unmodified Ser hydroxyl group. Additionally, the protons on the three methine carbons (C15, 4.59 ppm; C15′, 4.69 ppm; C15′′, 4.41 ppm) are inequivalent, as are the protons on the chiral methine carbons derived from Lys (C9, C9′ and C9′′, 4.50–4.65 ppm). The 1H NMR spectrum of FSB is consistent with related asymmetric linear triscatechol siderophores trivanchrobactin6 and turnerbactin.7 Thus, FSB is a novel siderophore and a natural diastereomer of linear trichrysobactin.
Chirality of Fe(III)–FSB and Fe(III)–CTC
The CD spectra of Fe(III)–CTC and Fe(III)–FSB (Fig. 7) appear as near mirror-images of each other, indicating an opposite configurational preference around iron. Through comparison to the CD spectra of Fe(III)–Ent3−, Fe(III)–BB, and the Fe(III)–[(DHBL/DLysL/DSer)3] complexes (Table 1)3,4 Fe(III)–FSB is assigned a Δ configuration. The comparison of Fe(III)–FSB to Fe(III)–[(DHBLLysLSer)3] (Fig. 2, Table 1) also establishes that linearization of the trilactone does not significantly affect the configuration of the ferric complex. Earlier work revealed that linearization of Ent also does not invert its overall configurational preference, however, a small fraction of the Λ enantiomer is formed.11,13 Our earlier work with the cyclic Fe(III)–[(DHBL/DLysL/DSer)3] complexes suggests that the opposing chirality observed for ferric complexes of FSB and CTC is likely due to the stereochemistry of the Lys residue adjacent to the catecholamide and not due to the linear or cyclic nature of the triserine backbone.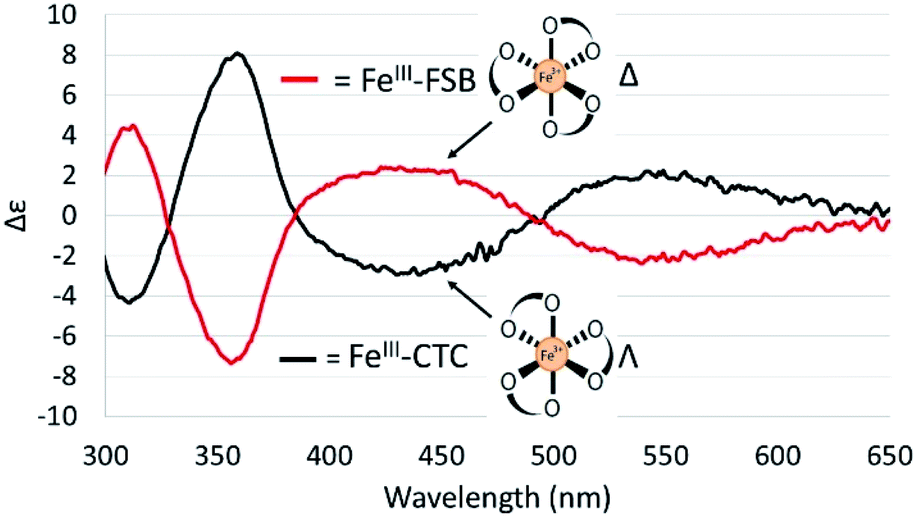 | ||
| Fig. 7 CD spectra of 40 μM Fe(III)–FSB and 40 μM Fe(III)–CTC in citrate–phosphate buffer (pH 7.40). | ||
Fe(III) exchange between FSB and CTC
Surprisingly little is known about the exchange of Fe(III) among triscatechol siderophores. CD spectroscopy is uniquely poised to monitor Fe(III) exchange between optically-active siderophores. The intensity of the Λ-Fe(III)–CTC CD bands decrease upon addition of equimolar FSB as a result of formation of nearly equimolar Λ-Fe(III)–CTC and Δ-Fe(III)–FSB (Fig. 8A). Moreover the equivalent equilibration approached from reaction of Δ-Fe(III)–FSB with CTC is also observed (Fig. 8B). Interestingly, a weak negative band at 435 nm and a weak positive band at 550 nm are formed after four hours of equilibration (Fig. 8B), suggestive of a slight preference favoring formation of Fe(III)–CTC over Fe(III)–FSB, consistent with the increased stability constant of macrocyclic ligands.13 The intensity of this band diminishes upon further equilibration, potentially due to hydrolysis of the labile macrolactone. Under neutral pH conditions with 100 μM Fe(III)–CTC and 100 μM FSB, the magnitude of the CD signal decreases within hours of mixing, indicating that exchange occurs on a relatively short, biologically relevant time scale.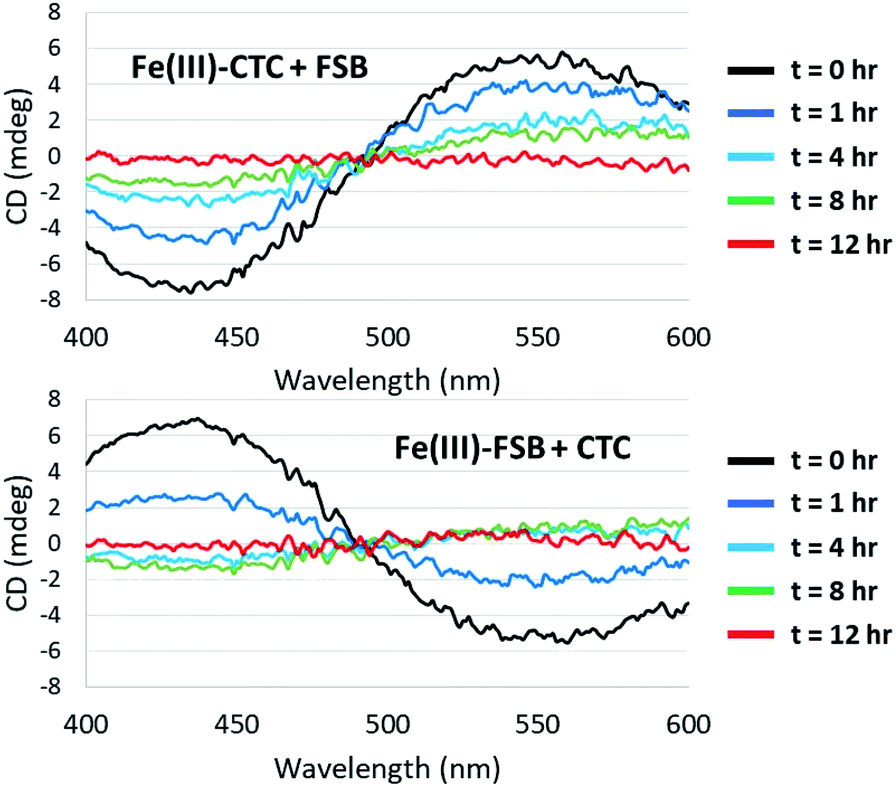 | ||
| Fig. 8 Equilibration of Fe(III)–CTC and Fe(III)–FSB with FSB and CTC, respectively. (Top) Reaction of 100 μM Fe(III)–CTC with 100 μM FSB in 50 mM citrate–phosphate, pH 7.40. (Bottom) Reaction of 100 μM Fe(III)–FSB with 100 μM CTC in 50 mM citrate–phosphate, pH 7.40. | ||
Conclusions
In sum, BGCs encoding synthesis of the triscatechol siderophores CTC and FSB were identified and the structure of FSB was elucidated. The opposing configurations of Δ-Fe(III)–FSB and Λ-Fe(III)–CTC are established by the stereochemistry at Lys. The most stable configuration by computational modeling is Λ-Fe(III)–[(DHBDLysLSer)3], which, strikingly, has been adopted for microbial iron acquisition as Fe(III)–CTC. The next most stable conformation corresponds to the cyclic form of FSB, Δ-Fe(III)–[(DHBLLysLSer)3], raising questions about the predicted low energy conformations of the corresponding linear triscatechol siderophores.The suite of cyclic and linear (DHBL/DLysL/DSer)3 siderophores and analogs raises further significant questions regarding the effect of a mismatched Δ- and Λ-Fe(III) configuration on microbial iron uptake and growth. For example, discrimination at the outer membrane receptor protein could prevent uptake of the wrong Fe(III)–enantiomer as has been observed with Fe(III) complexes of pyochelin and enantiopyochelin.18–20 If iron uptake is insensitive to the Fe(III)–enantiomer chirality, discrimination could still occur at other points including the iron-release process, as is observed in Bacillus subtilis in the Fes-catalyzed hydrolysis of the macrolactone of Fe(enantioEnt)3− which is required for release of iron.1 Additionally, it may be possible for the relevant siderophore-interacting proteins to invert the configuration of a mismatched Fe(III)–siderophore complex upon binding, as has been observed for the periplasmic binding protein CeuE of Campylobacter jejuni.21–23
Siderophores are primarily extracellular metabolites and facile Fe(III) exchange observed between triscatechol siderophores is likely of biological consequence within complex microbial communities. Certainly, the rate of release of the newly synthesized apo siderophores during growth of Y. frederiksenii ATCC 33641 and D. chrysanthemi EC16, which is occurring over the time scale of hours to days, could be exchanging Fe(III) within hours with other triscatechol siderophores, as evinced by the CD results (Fig. 8). In fact Fe(III) exchange between the triscatecholate siderophores is orders of magnitude faster than Fe(III) exchange between hydroxamate siderophores or between hydroxamate and catecholate siderophores.24
Experiments addressing the questions raised above are in progress, as well as the question of whether BGCs encoding the synthesis of the diastereomers of trivanchrobactin, (DHBD/LArgL/DSer)3, and turnerbactin, (DHBL/DOrnL/DSer)3, are present in microbial genomes. The discovery of frederiksenibactin and its relationship to CTC exemplifies the structural variability of microbial siderophores and provides a natural system to determine the significance of chirality within siderophore-mediated microbial iron-uptake pathways.
Experimental
General experimental procedures
UV-visible absorbance and circular dichroism spectroscopy were measured on an Agilent Cary 300 UV Vis spectrophotometer and a Jasco J-1500 CD spectrophotometer, respectively. 13C NMR spectroscopy was performed on a Bruker Advanced Neo 500 MHz spectrometer equipped with a prodigy cryoprobe at RT. All 1H, COSY, HMBC, HSQC NMR spectroscopy was performed on a Varian Unity 600 MHz spectrometer at RT. Chemical shifts were referenced through residual solvent peaks [1H (DMSO-d6) 2.50 ppm, 13C (DMSO-d6) 39.51 ppm]. Mass spectrometry analysis of Y. frederiksenii ATCC 33641 supernatant extracts and purified FSB was carried out on a Waters Xevo G2-XS QToF with positive mode electrospray ionization coupled to an ACQUITY UPLC H-Class system with a waters BEH C18 column. Y. frederiksenii ATCC 33641 culture extracts were analyzed using a linear gradient of 0–30% CH3CN (+0.1% formic acid) in ddH2O (+0.1% formic acid) over 10 min. For MS/MS analysis, a collision energy of 15 eV was employed. HR-ESIMS analysis of synthetic compounds was carried out on a Waters LCT Premier ESI TOF introduced into the ESI by direct infusion via a syringe pump.General synthetic procedures
All reactions performed under an argon atmosphere were carried out using a high-vacuum line, standard Schlenk techniques, and dry solvents. DMF, DCM, and DMSO-d6 were stored over 3 Å molecular sieves for at least 72 h prior to use. N,N′-Diisopropylethylamine (DIPEA) was purified by distillation over ninhydrin (×3) and was subsequently stored over 3 Å molecular sieves. Nα-Boc-Nε-Cbz-L-lysine and Nα-Boc-Nε-Cbz-D-lysine were acquired from Bachem. All other reagents (including those used for Marfey's analysis) were purchased from Sigma-Aldrich.Synthesis of the cyclic (DHBL/DLysL/DSer)3 diastereomers
Established peptide coupling methodology25 was employed to construct the two key amide bonds in 4 (Scheme S1†). Reaction of chiral triamine 1 with HATU (3 eq.), Boc–Lys(Z)–OH (3 eq.), and DIPEA (9 eq.) cleanly affords intermediate 2 (Step a, Scheme S1†). Removal of the Nα-Boc protecting groups (step b, Scheme S1†) and subsequent coupling to benzyl-protected 2,3-dihydroxybenzoic acid (step c, Scheme S1†) yields 3 in an 81% yield over two steps. Global deprotection by hydrogenolysis over 10% Pd/C (step d, Scheme S1†) yields (DHBL/DLysL/DSer)3, (4) as an enantiopure product. Initial synthetic efforts in which the direction of peptide coupling was reversed were highly susceptible to epimerization at the Lys stereocenter, consistent with the observed chiral instability of Nα-acylated amino acids upon activation as a HOBT or HOAT ester.26Nα-Boc–Nε-Cbz-L-lysine was substituted for Nα-Boc-Nε-Cbz-D-lysine in the synthesis of (DHBDLysLSer)3 and (DHBDLysDSer)3. N-Trityl-L-serine was substituted for N-trityl-D-serine in the synthesis of 1 to yield (DHBLLysDSer)3 and (DHBDLysDSer)3.Amino acid analysis of frederiksenibactin and synthetic cyclic (DHBL/DLysL/DSer)3 analogs by Marfey's method
(DHBL/DLysL/DSer)3 or FSB (2 mg) was dissolved in 2 M HCl and heated at 110 °C in a sealed glass ampule under argon for 24 h. The hydrolysis mixture was evaporated to dryness under a stream of air and redissolved in 100 μL ddH2O. 1-Fluoro-2,4-dinitrophenyl-L-alanine amide (FDAA, 1 M in acetone, 150 μL) and NaHCO3 (1 M, 20 μL) were added and the solution was briefly vortexed and placed on a heating block (40 °C) for 1 h. 10 μL of 2 M HCl was then added to quench the reaction and solutions were stored at −20 °C in the dark prior to analysis. Amino acid standards were derivatized according to the same procedure. Derivatized hydrolysis products of FSB were separated by HPLC on a YMC 4.6 × 250 mm C18-AQ column using a gradient from 10% CH3CN in ddH2O (0.05% trifluoroacetic acid) to 40% CH3CN in ddH2O (0.05% trifluoroacetic acid) over 60 min. Derivatized hydrolysis products of (DHBL/DLysL/DSer)3 were separated by HPLC on a YMC 4.6 × 250 mm C18-A column using a gradient from 10% CH3CN in TEAP buffer (50 mM, pH 3.00) to 40% CH3CN in TEAP buffer over 60 min. Derivatized hydrolysis products were co-injected with derivatized amino acid standards to determine the constituent amino acids of FSB and to determine the extent of epimerization during synthesis of synthetic (DHBL/DLysL/DSer)3. Three peaks corresponding to FDAA-derivatized lysine were observed, corresponding to products derivatized at either the α-amine, ε-amine, or both amines. FDAA-derivatized DSer co-eluted with LLys and DLys derivatized at the ε-amine under the conditions used for Marfey's analysis of FSB (YMC C18-AQ column).Preparation of Fe(III)-complexes and circular dichroism spectroscopy
Fe(III)-complexes of the (DHBL/DLysL/DSer)3 diastereomers and FSB for CD spectroscopy were prepared in citrate–phosphate buffer (50 mM, pH 7.40) by mixing a solution of FeCl3 [2.45 mM, 0.1 M HCl (aq)] with 1.0 equivalent of the desired apo-ligand. Formation of the Fe(III)-complex was tracked by UV-visible spectroscopy by observing the absorbance at 498 nm. The resulting solution was equilibrated for 30 min in the dark prior to analysis by CD spectroscopy.Full CD spectra were acquired using the following parameters: 4 s D.I.T., 1 nm bandwidth, 50 nm s−1 scanning speed, with 3 accumulations. Fe(III) exchange assays were performed by preparing pre-equilibrated Fe(III)-complexes of either FSB or CTC as described above. At time t = 0, an equimolar amount of the opposing apo-ligand was added to the Fe(III)-complex and the resulting solution was gently vortexed. CD spectra were acquired as a single accumulation at 20 min intervals using the following parameters: 400–600 nm; 2 s D.I.T., 1 nm bandwidth, and 100 nm s−1 scan speed.
Computational modeling
Electronic structure calculations were performed using Gaussian 16.28 The structures of the following four complexes were optimized: Δ-Fe(III)–[(DHBLLysLSer)3], Δ-Fe(III)–[(DHBLLysDSer)3], Δ-Fe(III)–[(DHBDLysLSer)3], and Δ-Fe(III)–[(DHBDLysDSer)3]. Note that the structures of the corresponding Λ isomers were not optimized, because each is an enantiomer of one of the four Δ complexes listed above, and therefore energetically equivalent. The input geometries were generated manually. The Fe(III) centers were treated as high-spin (S = 5/2) and the Lys residues were protonated to afford neutral complexes (z = 0). Optimizations were performed at the PBE0/6-311++G(d,p) level of theory with Grimme's D3 empirical dispersion correction and tight convergence criteria.29–32Implicit aqueous solvation was included using a conductor-like polarizable continuum model (CPCM). The energy values presented in Fig. 3 are electronic energies that have not been zero-point corrected. Optimized coordinates are collected in Tables S8–S11.† Representations of Λ-Fe(III)–[(DHBDLysLSer)3] (i.e., Fe(III)–CTC) were generated by inverting the optimized coordinates of Δ-Fe(III)–[(DHBLLysDSer)3]. Geometric analyses were performed using Mercury.33
Genome sequence of Dickeya chrysanthemi EC16 (ATCC 11662)
Dickeya chrysanthemi EC16 (ATCC 11662) was obtained from the ATCC and maintained on Difco Luria–Bertani (LB) agar plates at 30 °C. A liquid LB culture was inoculated from a single colony and incubated for 18 h at 30 °C and 180 rpm. Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer's instructions for Gram-negative bacteria. Extracted DNA was quantified by a Qubit 2.0 fluorometer (Invitrogen). Library preparation and sequencing were performed by the Microbial Genome Sequencing Center (Pittsburgh, PA): paired-end libraries were prepared according to Baym et al.34 and sequenced on the NextSeq 550 platform (Illumina), generating 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 232664 pairs of 2 × 150 bp reads.
232664 pairs of 2 × 150 bp reads.
Read quality was assessed with FastQC v0.11.9 (https://github.com/s-andrews/FastQC). Low-quality reads were trimmed with trimmomatic using the settings “LEADING:10 TRAILING:10 SLIDINGWINDOW:4:20 MINLEN:80”. Trimmed reads were assembled using SPAdes v3.14.1 with the flags “--isolate -k 21,33,55,77” recommended for bacterial isolates.35 Scaffold quality was assessed by QUAST v5.1.0rc1.36 Scaffolds over 500 bp in length were retained and quality was assessed by QUAST. The final assemblies were annotated using the NCBI Prokaryotic Genome Annotation Pipeline v5.0.37 Default parameters were used, except where otherwise noted. Assembly statistics are given in Table S3.† Taxonomic classification was determined by comparing average-nucleotide identity (ANI) against type strain Genbank sequences using OrthoANIu.38 By this metric, the previous assignment of strain EC16 as D. chrysanthemi is strongly supported, with a 99.97% ANI with D. chrysanthemi NCPPB 402T.
Bacterial growth and siderophore isolation
Yersinia frederiksenii ATCC 33641, obtained from the American Type Culture Collection (ATCC), was cultured on Difco Luria Bertani (LB) Miller (BD biosciences) medium plates. A single colony of Y. frederiksenii ATCC 33641 was inoculated into 50 mL of Difco LB Miller (BD biosciences) media and grown overnight at 30 °C, shaking at 180 rpm. A portion of the overnight culture (5 mL) was then inoculated into low-iron minimal media (2 L, pH 7.0) containing sodium succinate (4 g L−1), K2HPO4 (6 g L−1), KH2PO4 (3 g L−1), NH4Cl (1 g L−1), CaCl2·2H2O (20 mg L−1), and MgSO4·7H2O (200 mg L−1) in an acid-washed 4 L Erlenmeyer flask. The culture was shaken at RT, 180 rpm for 72 h. Cultures were harvested in the late log phase of growth by centrifugation (SLA-3000 rotor, ThermoScientific) at 6000 rpm for 30 min at 4 °C. Culture supernatants were decanted into a clean, acid-washed Erlenmeyer flask containing 100 g of Amberlite XAD-4 polystyrene resin, which was shaken at 120 rpm for 4 h at 4 °C. The resin was filtered from the supernatant, rinsed with 100 mL of 90/10% ddH2O/MeOH, and then eluted with 250 mL of 95:5% MeOH/ddH2O. The eluent was concentrated under reduced pressure to a volume of 30 mL and stored at 4 °C prior to analysis. Frederiksenibactin and the related monocatechol and dicatechol compounds were purified by semi-preparative RP-HPLC on a YMC-Actus 20 × 250 mm C18 ODS-AQ column using a linear gradient of 15% MeOH in ddH2O (+0.1% trifluoroacetic acid) to 40% MeOH in ddH2O (+0.1% trifluoroacetic acid) over 25 min.
Data availability
The draft genome sequence of Dickeya chrysanthemi EC16 was deposited at NCBI under the BioProject ID PRJNA690813.Author contributions
P. R. S. synthesized the compounds, isolated the FSB siderophore and carried out its structural and CD characterization; Z. L. R. carried out the bioinformatics analysis and biosynthetic gene cluster prediction; T. C. J. carried out the computational investigations. A. B. directed the project. All authors were involved in writing the manuscript and all authors provided feedback on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for support from NSF CHE-1710761 (A. B.) and a fellowship from the Hellman Foundation (T. C. J.). The research reported here made use of the shared facilities of the UCSB MRSEC (NSF DMR 172056), a member of the Materials Research Facilities Network (http://www.mrfn.org). The authors acknowledge the use of the Biological Nanostructures Laboratory within the California NanoSystems Institute, supported by UCSB and the University of California, Office of the President.Notes and references
- R. J. Abergel, A. M. Zawadzka, T. M. Hoette and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 12682–12692 CrossRef CAS PubMed.
- J. B. Neilands, T. J. Erickson and W. H. Rastetter, J. Biol. Chem., 1981, 256, 3831–3832 CrossRef CAS.
- M. E. Bluhm, S. S. Kim, E. A. Dertz and K. N. Raymond, J. Am. Chem. Soc., 2002, 124, 2436–2437 CrossRef CAS PubMed.
- T. C. Johnstone and E. M. Nolan, J. Am. Chem. Soc., 2017, 139, 15245–15250 CrossRef CAS.
- M. Sandy and A. Butler, J. Nat. Prod., 2011, 74, 1207–1212 CrossRef CAS PubMed.
- M. Sandy, A. Han, J. Blunt, M. Munro, M. Haygood and A. Butler, J. Nat. Prod., 2010, 73, 1038–1043 CrossRef CAS PubMed.
- A. W. Han, M. Sandy, B. Fishman, A. E. Trindade-Silva, C. A. Soares, D. L. Distel, A. Butler and M. G. Haygood, PLoS One, 2013, 8, e76151 CrossRef CAS.
- T. B. Karpishin, T. D. P. Stack and K. N. Raymond, J. Am. Chem. Soc., 1993, 115, 6115–6125 CrossRef CAS.
- R. J. A. Ramirez, L. Karamanukyan, S. Ortiz and C. G. Gutierrez, Tetrahedron Lett., 1997, 38, 749–752 CrossRef CAS.
- S. S. Isied, G. Kuo and K. N. Raymond, J. Am. Chem. Soc., 1976, 98, 1763–1767 CrossRef CAS.
- T. B. Karpishin, T. M. Dewey and K. N. Raymond, J. Am. Chem. Soc., 1993, 115, 1842–1851 CrossRef CAS.
- W. H. Rastetter, T. J. Erickson and M. C. Venuti, J. Org. Chem., 1981, 46, 3579–3590 CrossRef CAS.
- R. C. Scarrow, D. J. Ecker, C. Ng, S. Liu and K. N. Raymond, Inorg. Chem., 1991, 30, 900–906 CrossRef CAS.
- G. N. Ramachandran, C. Ramakrishnan and V. Sasisekharan, J. Mol. Biol., 1963, 7, 95–99 CrossRef CAS.
- L. Rauscher, D. Expert, B. F. Matzanke and A. X. Trautwein, J. Biol. Chem., 2002, 277, 2385–2395 CrossRef CAS PubMed.
- Z. L. Reitz, M. Sandy and A. Butler, Metallomics, 2017, 9, 824–839 CrossRef CAS.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- K. Brillet, C. Reimmann, G. L. A. Mislin, S. Noel, D. Rognan, I. J. Schalk and D. Cobessi, J. Am. Chem. Soc., 2011, 133, 16503–16509 CrossRef CAS.
- Z. A. Youard, G. L. A. Mislin, P. A. Majcherczyk, I. J. Schalk and C. Reimmann, J. Biol. Chem., 2007, 282, 35546–35553 CrossRef CAS.
- F. Hoegy, X. Y. Lee, S. Noel, D. Rognan, G. L. A. Mislin, C. Reimmann and I. J. Schalk, J. Biol. Chem., 2009, 284, 14949–14957 CrossRef CAS.
- A. Muller, A. J. Wilkinson, K. S. Wilson and A. K. Duhme-Klair, Angew. Chem., Int. Ed., 2006, 45, 5132–5136 CrossRef PubMed.
- D. J. Raines, O. V. Moroz, E. V. Blagova, J. P. Turkenburg, K. S. Wilson and A.-K. Duhme-Klair, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5850–5855 CrossRef CAS PubMed.
- D. J. Raines, O. Moroz, E. V. Blagova, J. P. Turkenburg, K. S. Wilson and A. K. Duhme-Klair, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5850–5855 CrossRef CAS.
- T. P. Tufano and K. N. Raymond, J. Am. Chem. Soc., 1981, 103, 6617–6624 CrossRef CAS.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS.
- J. S. Davies and A. K. Mohammed, J. Chem. Soc., Perkin Trans. 1, 1981, 2982–2990 RSC.
- M. Meyer, J. R. Telford, S. M. Cohen, D. J. White, J. Xu and K. N. Raymond, J. Am. Chem. Soc., 1997, 119, 10093–10103 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- M. Baym, S. Kryazhimskiy, T. D. Lieberman, H. Chung, M. M. Desai and R. Kishony, PLoS One, 2015, 10, e0131262 CrossRef PubMed.
- A. Bankevich, S. Nurk, D. Antipov, A. A. Gurevich, M. Dvorkin, A. S. Kulikov, V. M. Lesin, S. I. Nikolenko, S. Pham, A. D. Prjibelski, A. V. Pyshkin, A. V. Sirotkin, N. Vyahhi, G. Tesler, M. A. Alekseyev and P. A. Pevzner, J. Comput. Biol., 2012, 19, 455–477 CrossRef CAS.
- A. Mikheenko, A. Prjibelski, V. Saveliev, D. Antipov and A. Gurevich, Bioinformatics, 2018, 34, 142–150 CrossRef.
- T. Tatusova, M. DiCuccio, A. Badretdin, V. Chetvernin, E. P. Nawrocki, L. Zaslavsky, A. Lomsadze, K. Pruitt, M. Borodovsky and J. Ostell, Nucleic Acids Res., 2016, 44, 6614–6624 CrossRef CAS.
- S. H. Yoon, S. M. Ha, J. Lim, S. Kwon and J. Chun, Anton Leeuw. Int. J. G., 2017, 110, 1281–1286 CrossRef CAS.
- T. B. Karpishin, M. S. Gebhard, E. I. Solomon and K. N. Raymond, J. Am. Chem. Soc., 1991, 113, 2977–2984 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on the synthesis and characterization of the cyclic diastereomeric (DHBL/DLysL/DSer)3 compounds; biosynthetic gene clusters for FSB and CTC; genome sequence statistics of D. chrysanthemi EC16; isolation and characterization of FSB; coordinates of computationally optimized structures. See DOI: 10.1039/d1sc03541j |
| ‡ Two prominent low-energy CD bands at 435 nm and 545 nm arise from characteristic LMCT transitions and are therefore sensitive to the chirality at the Fe(III) center. The CD bands at 270 nm, 310 nm, and 360 nm are assigned to ligand-based transitions corresponding to the amide (270 nm) and ester (310 and 360 nm) carbonyls, in analogy to Fe(III)–BB3−.39 |
| This journal is © The Royal Society of Chemistry 2021 |