
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed C–H glycosylation and retro Diels–Alder tandem reaction via structurally modified norbornadienes (smNBDs)†
Yang
An‡
a,
Bo-Sheng
Zhang‡
 b,
Ya-Nan
Ding
a,
Zhe
Zhang
a,
Xue-Ya
Gou
a,
Xue-Song
Li
a,
Xiaolei
Wang
a,
Yuke
Li
*c and
Yong-Min
Liang
*a
b,
Ya-Nan
Ding
a,
Zhe
Zhang
a,
Xue-Ya
Gou
a,
Xue-Song
Li
a,
Xiaolei
Wang
a,
Yuke
Li
*c and
Yong-Min
Liang
*a
aState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: liangym@lzu.edu.cn
bCollege of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou, Gansu 730070, P. R. China
cDepartment of Chemistry, Centre for Scientific Modeling and Computation, Chinese University of Hong Kong, Shatin, Hong Kong, P. R. China. E-mail: yukeli@link.cuhk.edu.hk
First published on 31st August 2021
Abstract
This report describes palladium-catalyzed C–H glycosylation and retro Diels–Alder tandem reaction via structurally modified norbornadienes (smNBDs). smNBDs were proposed to regulate the reactivity of the aryl-norbornadiene-palladacycle (ANP), including its high chemoselectivity and regioselectivity, which were the key to constructing C2 and C3 unsubstituted C4-glycosidic indoles. The scope of this substrate is extensive; the halogenated six-membered and five-membered glycosides were applied to the reaction smoothly, and N-alkyl (primary, secondary and tertiary) C4-glycosidic indoles can also be obtained by this method. In terms of mechanism, the key ANP intermediates characterized by X-ray single-crystal diffraction and further controlled experiments proved that the migration-insertion of smNBDs with phenylpalladium intermediate endows them with high chemo- and regioselectivity. Finally, density functional theory (DFT) calculation further verified the rationality of the mechanism.
Introduction
A variety of glycoconjugates exhibit a range of essential biological functions, such as immune response, intercellular communication, and the bioactive modulation of natural products.1–3 Their unstable biochemical properties, such as glycosidic linkages undergoing hydrolysis in the living body, limit their practical development. Notably, the C-aryl glucoside skeleton, which is widely found in nature, has high biological metabolism stability, and C-glycosyl indole derivatives, which are widely used in the field of medicinal chemistry, have unique biological activities.4–14 In 1994, the Hofsteenge group first discovered α-C-mannosyltryptophan (α-C-Man-Trp) I from the Trp7 of ribonuclease.15–17 Due to their unique structure and biological activities such as cellular signaling and communication, these compounds have attracted considerable attention. Compounds II and III with 3-indolyl-C-glycoside skeleton are sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors and are considered potential drugs for the therapy of type 2 diabetes.18,19 Compounds IV and V with 2-indolyl-C-glycoside skeleton were isolated from Isatis indigotica and they show significant cytotoxic activities against human liver cancer HepG2 cells, human myeloid leukemia Mata cells and human myeloid leukemia HL-60 (Scheme 1a).20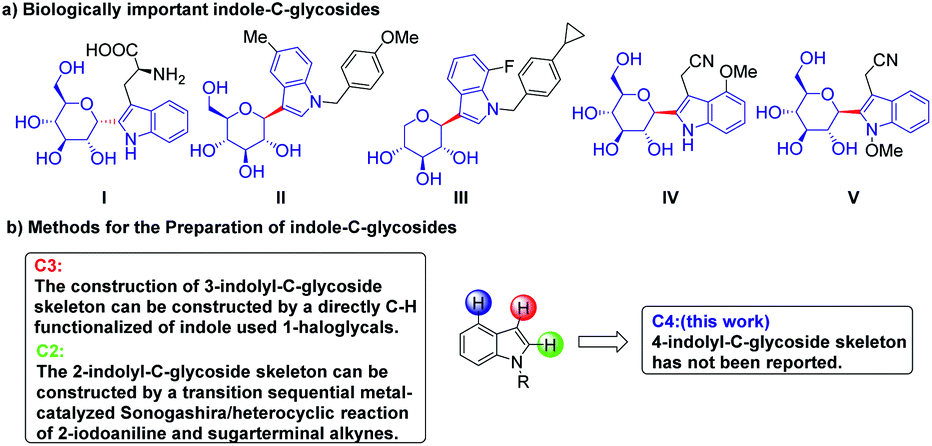 | ||
| Scheme 1 The study of glycosidic indoles was limited to C2 and C3 position substitution. | ||
Due to indolyl-C-glycosides' DNA modulatory properties and great potential in disease treatment, a variety of synthetic methods have been developed to construct these valuable scaffolds. These compounds are mainly synthesized using the following two approaches. The first method is to construct a 2-indolyl-C-glycoside skeleton by Larock indole synthesis with 2-iodoanilines and glycosyl alkyne as the substrate.21–27 The second approach is the transition-metal-catalyzed direct C–H functionalization of indole,28–35 and the most efficient is the Pd-catalyzed ortho-directed C–H glycosylation reactions to construct 2-indolyl-C-glycoside skeleton using glycosyl chloride donors (Scheme 1b).36 However, the construction of 4-indole-C-glycosides has not been reported. Therefore, the development of an efficient C–H glycosylation reaction to construct a 4-glycosylindole skeleton has important application value.
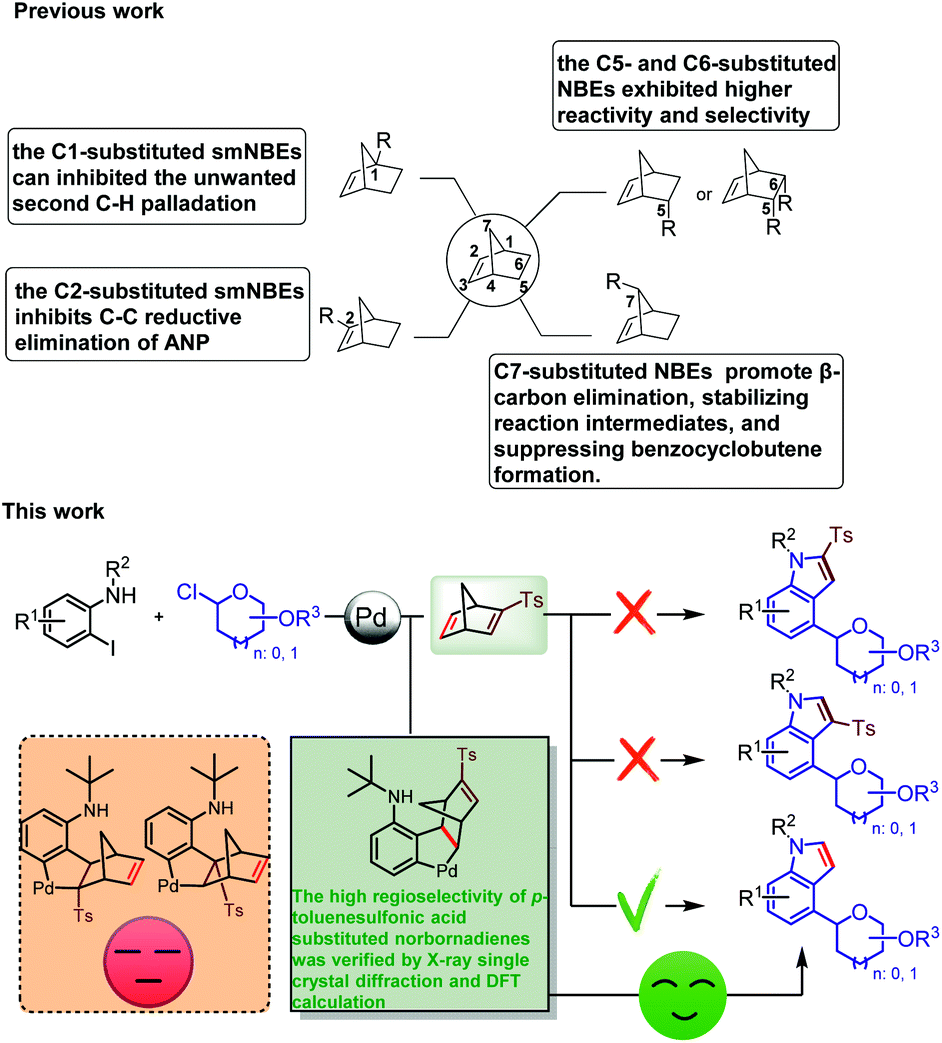
The Catellani reaction was discovered in 1997,37 then Lautens expanded the chemical compatibility of Pd/NBE chemistry by utilizing phosphine ligands in 2000.38 Later, it was named the Catellani–Lautens reaction system. The mechanism of the Catellani reaction is rather lengthy,39–49 so there are a lot of competitive reactions to produce a variety of by-products.50 For example, the reductive elimination of aryl-norbornene-pallada-cycle (ANP) intermediate may take place to form the byproduct benzocyclobutene.51,52 In recent years, structurally modified norbornene (smNBE) strategy provides a good solution to this problem.
In 2015, Dong et al. first reported the smNBE (C5-amide-substituted),53,54 and since then, Yu,55 Zhou,56–60 Gu,61 Ding,62 Cheng,63 Liang64 and others have developed numerous C2-, C5- and C6-substituted NBEs that exhibited unique reactivity and higher selectivity compared to the simple unsubstituted NBE. The amide-substituted NBE was reported by Dong et al.65–68 and the ester-substituted NBE reported by Yu et al. was the most effective type of C2-substituted smNBEs that greatly inhibit the C–C reductive elimination of ANP to form the by-product benzocyclobutanes and effectively reduce the overall activation barrier. Notably, Yu et al. made a breakthrough in using the C2-substituted smNBEs and quinoline-type ligand to realize meta-C–H activation with guiding groups. In 2018, Dong et al. developed a series of C1-substituted smNBEs that significantly inhibited the β-carbon elimination of NBE in ortho-unsubstituent and inhibited the unwanted second C–H palladation.69,70 Recently, Dong et al. discovered that C7-substituted NBE can promote β-carbon elimination, stabilizing reaction intermediates, and tetrahydrobenzo[b]azepine derivatives were efficiently synthesized from ortho-unsubstituted iodobenzene (Scheme 2).71
 | ||
| Scheme 2 Synthesis of C4-glycosidic indoles via palladium/structurally modified norbornadiene (smNBDs). | ||
Although high achievements have been made in the study of palladium/structurally modified norbornene (Pd/smNBEs) system, the palladium/structurally modified norbornadiene (Pd/NBD) system has not been reported yet. The main reason is that the migration-insertion reaction between smNBDs and phenylpalladium intermediates has four potential reaction sites, which is more challenging than our previous work to synthesize C4-aminated indoles using structurally unsubstituted NBD.72 In this reaction, we first used structurally modified norbornadiene (smNBDs) as the coupling partner of the Catellani reaction to realize the ortho-C–H glycosylation reaction and used the reverse Diels–Alder reaction of norbornadiene to realize the synthesis of C4-glycoside indole.
Results and discussion
Initially, we used N-(tert-butyl)-2-iodoaniline (1a) as a model substrate and glycosyl chloride donors (2a) as an ortho-glycosylation reagent, using palladium/norbornadiene (NBD) cooperative catalysis to investigate this reaction (Table 1). Unfortunately, the desired product (4a) was isolated in a 23% yield (entries 1) with NBD (N1, 3.5 equiv.), Pd(OAc)2 (10 mol%), tri(2-furyl)phosphane (TFP, 20 mol%), and Cs2CO3 (4.0 equiv.) in 1,4-dioxane under Ar at 90 °C (12 h)–150 °C (24 h), and most of the reaction proceeded toward the intramolecular Buchwald coupling without ortho-glycosylation. We hypothesized that a wide spectrum of NBDs may have significant effects on the reactivity of this transformation. Then, we tried a series of modified NBDs and found that the intramolecular Buchwald coupling without ortho-glycosylation by-product was inhibited. After intensive investigation, the NBD with substitution para-toluenesulfonyl groups (N8) was the optimal ortho-C–H glycosylation coupling partner for the Catellani reaction, and it gave an isolated yield of 79%.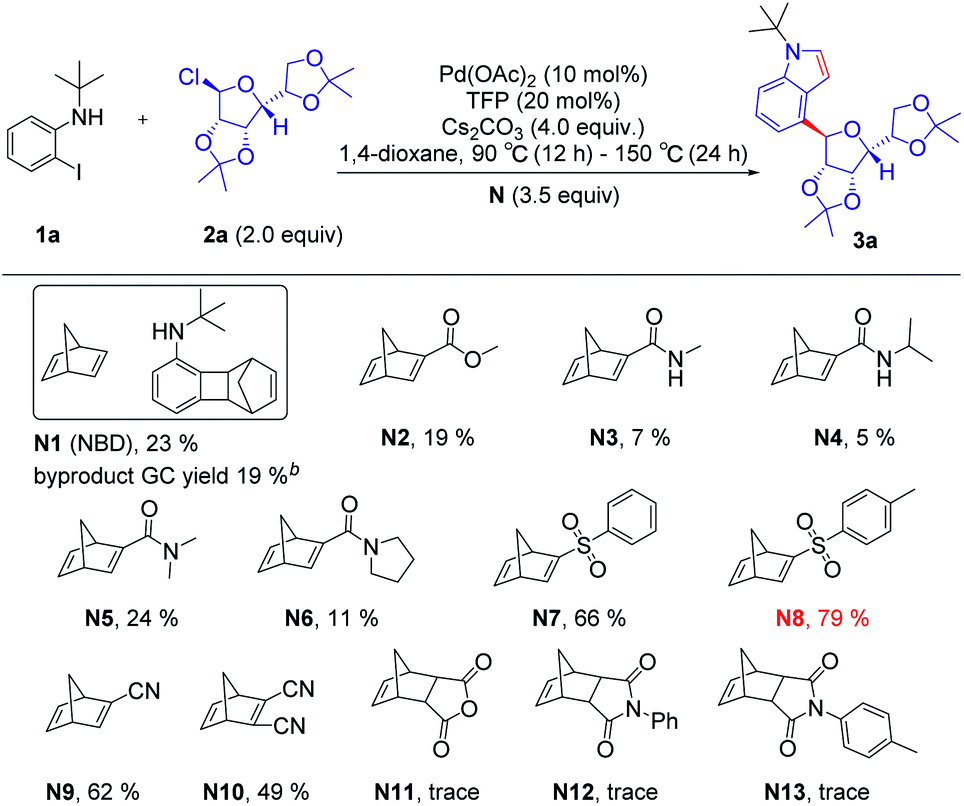
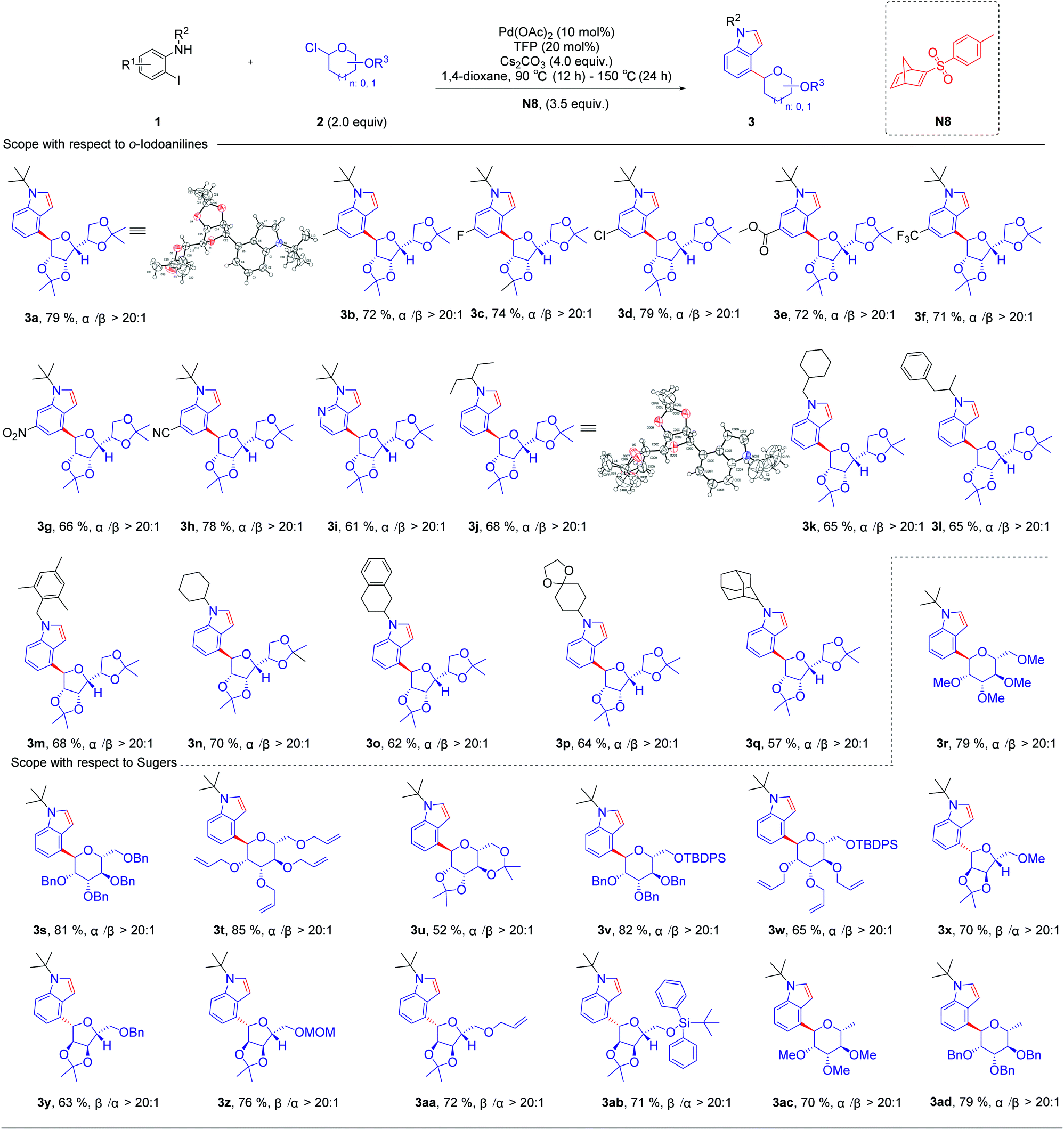
With optimized reaction conditions in hand, we first examined the substrate scope of o-iodoanilines. The reactions of o-iodoanilines, electron-donating group (–Me), halogen (–F, Cl), and strongly electron-withdrawing group (–NO2, CN, CO2Et, CF3) substituents performed smoothly, affording the desired 4-glycosylindole products (3b–3h) in 66–79% yield. It is noteworthy to mention that 3-iodopyridine-2-amine as a heteroaromatic substrate reacted favourably and 4-glycosyl-7-azaindole (3i) was obtained in 61% yield. Subsequently, a range of the groups on the nitrogen atom of o-iodoaniline was also examined under the optimal reaction conditions. Chain alkane (3j), 3-phenylbutane-2-yl (3l) containing aromatic hydrocarbons, benzyl (3m), cycloalkane (3n), tetrahydronaphthalene (3o), and adamantane (3q) with a large steric hindrance were outstandingly tolerated and afforded the desired products in excellent yields. It can be seen that this method synthesizes a variety of N-alkyl substituted indoles, which are difficult to generate by the direct coupling reaction of indole with tertiary or secondary carbon. Inspired by these encouraging results, we then studied a range of glycosyl chlorides. Tetramethyl-, tetrabenzyl-, and tetraallyl-protected α-mannosyl chlorides performed excellently and delivered the corresponding product (3r–3t) in 79–85% yield. The corresponding α-C-aryl mannoside (3u) can be obtained using 2,3,4,6-di-O-isopropylidene-α-mannosyl chloride as a substrate in 52% yield. Other mannosyl chlorides with TBDPS are methyl- and benzyl-, such as 3v and 3w, affording the desired products in 65–82% yields with exclusive α-selectivity. Delightfully, α-ribofuranosyl chlorides with different functional groups, such as methyl-, benzyl-, methoxymethyl-, allyl- and TBDPS can also deliver the corresponding products in 63–76% yield (3x–3ab). In addition, α-rhamnosyl chloride can also be used as a suitable substrate for this reaction, and the corresponding targeted product can be obtained with an excellent yield of 70–79% (Table 2).
| a Reaction conditions unless otherwise noted: 1 (0.2 mmol), 2 (0.4 mmol), N8 (3.5 equiv.), Pd(OAc)2 (10 mol%), TFP (20 mol%), and Cs2CO3 (4.0 equiv.) in 1,4-dioxane, 90 °C (12 h)–150 °C (24 h) isolated yields. |
|---|
|
It can be known from the introduction that pharmaceutical compounds and natural products usually have unprotected carbohydrate skeleton structures, so we conducted deprotection experiments on 4-glycosylindole. We tried using ethylene dithioglycol and boron trifluoride ether for deprotection. Finally, we found that by adding boron trifluoride ether as the Lewis acid at −78 °C, ethylene dithioglycol and DCM as the solvent to react at −10 °C, the yield of the deprotected product (4a–4b) was 79–83%. Urinary tract infections (UTIs) are mostly caused by Gram-negative uropathogenic Escherichia coli (UPEC) bacteria. C-Mannosides have good inhibitory activity against the type 1 pilus adhesion, FimH.73,74 After obtaining the compounds with a more stable and unprotected structure of 4-indole-C-glycosides, we tested the bioactivity of compounds 4a and 4b. The hemagglutination (HA) inhibition assay test was performed on guinea pig red blood cells using clinical E. coli strain UTI89. We found that the 4b compound has moderate activity (HAI = 33.3 μM) in the hemagglutination (HA) inhibition test. Here, we disclosed that α-D-mannofuranose provided a possible new structure for the inhibitory activity against FimH compared with traditional α-D-mannopyranose (Scheme 3).
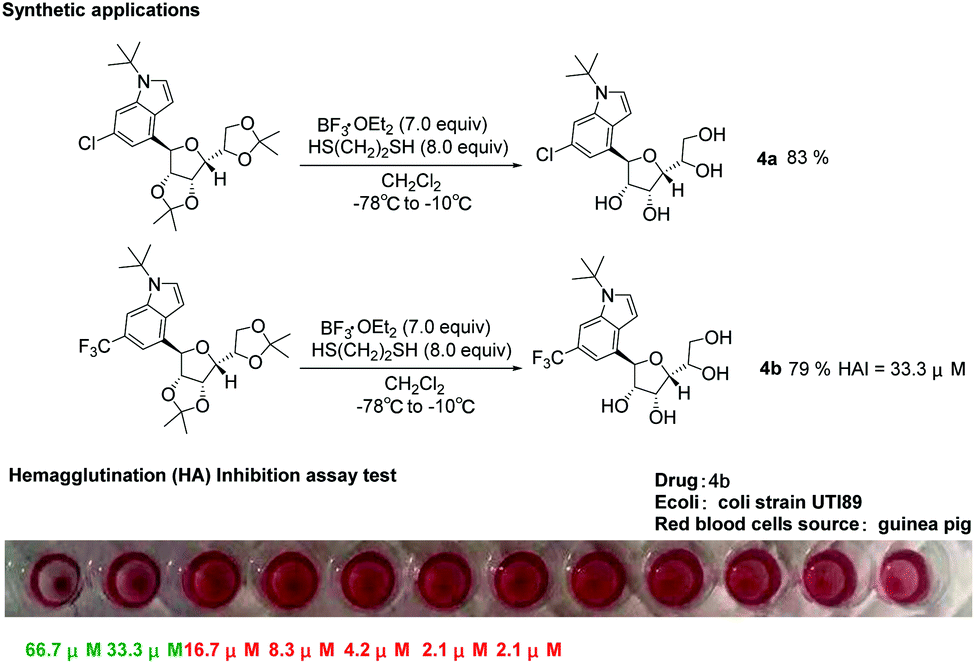 | ||
| Scheme 3 Synthetic applications and biochemical experiment. | ||
According to the above experimental results, the structure-modified norbornadienes (smNBDs) can effectively regulate the reaction activity of aryl-norbornadiene-palladacycle (ANP), thus successfully realizing the ortho C–H glycosylation. Due to the unique structural characteristics of norbornadiene, the retro-Diels–Alder reaction could further be realized to construct the indole skeleton. However, the modified norbornadiene has two olefins and four migration insertion sites. In theory, four isomers may be formed, among which J1 and J2 have migratory-insertion on the non-p-toluenesulfonyl side olefins, while J3 and J4 are migratory-insertions on the side of the p-toluene sulfonic acid. Therefore, we first carried out experimental verification. Under the system of potassium phenolate as the base, we successfully obtained the single crystal of the ANP intermediate (3j) and did not find other types of isomers through NMR spectroscopy. Then, we carried out the reaction with an equivalent or catalytic amount of intermediate (3j). Finally, the target products were obtained in good yield (Scheme 4). The experimental results directly proved that the migration-insertion of palladium compounds with p-toluenesulfonyl substituted norbornadiene was highly chemo- and regioselective, and the mechanism of promoting the activity of the ANP intermediate was inductive effect. In addition, the C2, C3 position unsubstituted C4-glycosidic indoles indirectly proved that the intramolecular Buchwald coupling step occurred on the non p-toluenesulfonyl side olefins.
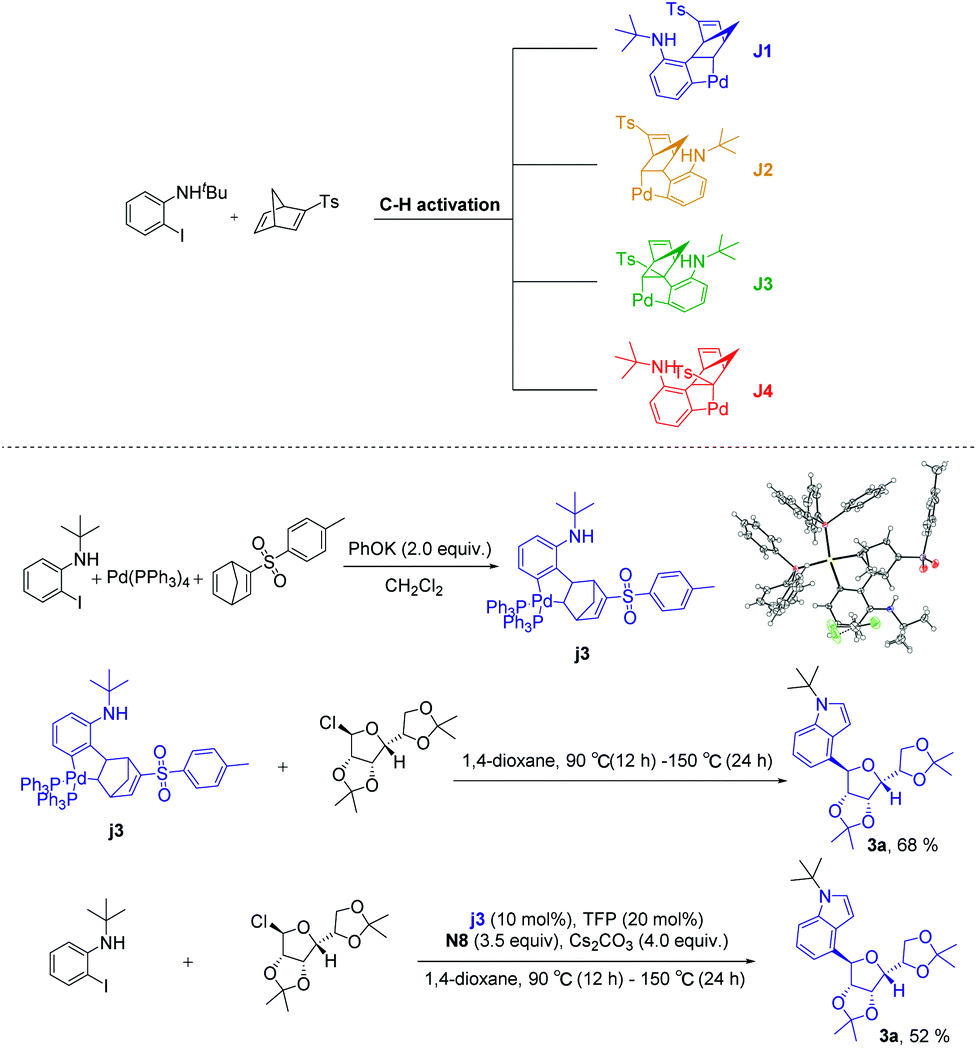 | ||
| Scheme 4 Structurally modified norbornadiene (smNBDs) has four potential reaction sites. | ||
Density functional theory (DFT) was used to study the reaction selectivity. At the initial stage, 1a attacks the Pd(0) catalyst A and one ligand leaves forming the complex B, as shown in Fig. 1. This process is roughly endothermic around 13.0 kcal mol−1. Then, oxidative addition takes place through the transition state of C-ts to form intermediate D with a barrier of 14.8 kcal mol−1 from A to C-ts. Cs2CO3 attacks the Pd center generating complex E, as suggested in Liu's work,75 releasing around 56.8 kcal mol−1. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of N8 coordinates with Pd forming four isomers F1, F2, F3 and F4, as shown in Fig. 2.
C of N8 coordinates with Pd forming four isomers F1, F2, F3 and F4, as shown in Fig. 2.
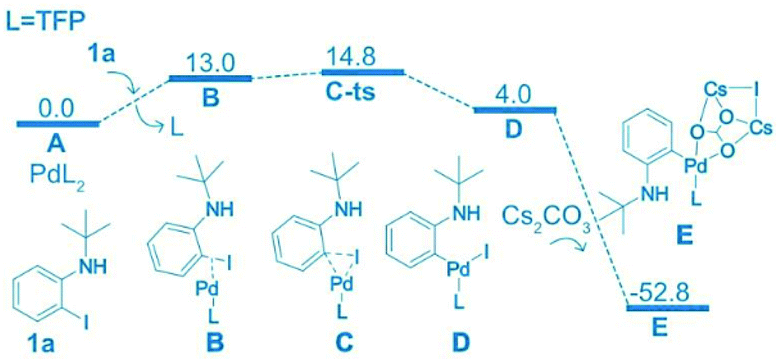 | ||
| Fig. 1 Computed free energy surface of the oxidation addition reaction. The relative free energies are presented in kcal mol−1. | ||
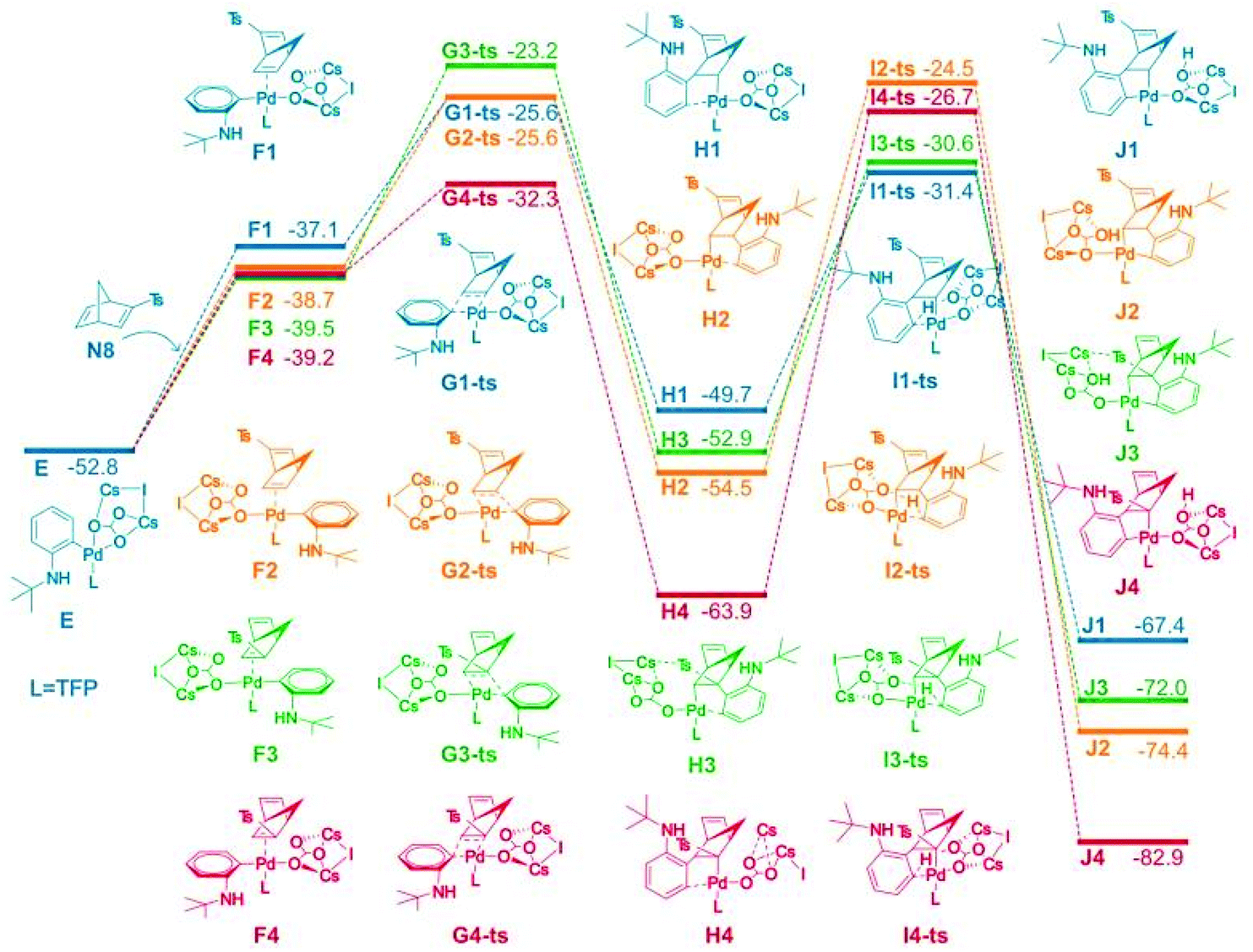 | ||
| Fig. 2 Computed free energy surface for alkene insertion and C–H activation process. The relative free energies are presented in kcal mol−1. | ||
The alkene insertion occurred with a barrier of 27.2, 27.1, 29.6, and 20.4 kcal mol−1 through G1-ts, G2-ts, G3-ts and G4-ts, respectively, while releasing 12.6, 14.2, 15.0, and 24.7 kcal mol−1 forming H1, H2, H3 and H4, respectively. The benzene ring coordinates with the Pd for H1, H2, H3 and H4. The barrier of the reverse process of the alkene insertion was 24.1, 27.3, 31.3, and 31.6 kcal mol−1 from the complex H series through the G-ts series. We found that the alkene insertion is a reversible process. C–H activation occurred with barriers of 18.3, 28.4, 23.9, and 37.2 kcal mol−1 for H1, H2, H3, and H4, respectively, generating five-membered ring complex J1, J2, J3, and J4. The C–H activation barrier for H1 was 18.3 kcal mol−1, which is the smallest barrier in the C–H activation barriers. The C–H activation occurred from H1 to I2-ts most favorably generating K1, agreeing with the experiment. The reverse C–H activation barrier is −36.0, −49.9, −41.4, and 56.2 kcal mol−1 from complex J series to complex H series.
In conclusion, we directly proved that the migratory insertion of p-toluenesulfonyl substituted norbornadiene with the phenylpalladium intermediate was highly chemo- and regioselective manner by X-ray single-crystal diffraction and density functional theory (DFT) calculation. Achieving high chemo- and regioselectivity was crucial for the subsequent C–H glycosylation, and it can be inferred that the regulation of the norbornadiene p-toluenesulfonyl group on the reaction was the induction effect. In particular, the chemo- and regioselectivity of p-toluenesulfonyl substituted norbornadiene was also very important for the formation of target products, avoiding the formation of p-toluenesulfonyl substituted indoles. Of course, the key step of C–H glycosylation, which is the transition state between ANP intermediates and halogenated glycosides, was complex and still under further exploration.
Conclusion
This report describes the palladium-catalyzed C–H glycosylation and retro Diels–Alder tandem reaction via structurally modified norbornadiene (smNBDs). A highly functionalized 4-glycosylindole was synthesized via the three-component cross-coupling of o-iodoaniline, glycosyl chloride and structurally modified norbornadiene. And the skeleton structure of unprotected glycosyl indole was obtained by the deprotection experiment. In terms of mechanism, the key ANP intermediates characterized by X-ray single-crystal diffraction and further control experiment proved that the migration-insertion reaction of smNBDs with phenyl palladium intermediate has high chemo- and regioselectivity. Finally, density functional theory (DFT) calculations were used to study the main five-membered aryl-norbornadiene-palladacycle (ANP) intermediate formation process.Data availability
All Data associated with this article are available in the ESI.†Author contributions
Y. A. and B.-S. Z. performed the methodology, synthesis, characterization, analysis, and wrote the manuscript, and these authors contributed equally. Y.-N. D., Z. Z., X.-Y. G. and X.-S. L. analyzed the data, discussed the results. X. W. performed the biological activity experiment. Y. L. provided carried out DFT calculations. Y.-M. L. designed the project and supervised the whole experiment. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (NSF 21532001 and 21772075), and the Innovation Fund Project of the Gansu Education Department (2021A-004). Computations reported in this paper were performed on the computer clusters at the Centre for Scientific Modeling and Computation, CUHK.References
- F. Berti and R. Adamo, Chem. Soc. Rev., 2018, 47, 9015–9025 RSC.
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- D. E. Levy and C. Tang, The chemistry of C-glycosides, Elsevier, 1995 Search PubMed.
- K. Krohn, A. Agocs and C. Bäuerlein, J. Carbohydr. Chem., 2003, 22, 579–592 CrossRef CAS.
- T. Bililign, B. R. Griffith and J. S. Thorson, Nat. Prod. Rep., 2005, 22, 742–760 RSC.
- J. Štambaský, M. Hocek and P. Kočovský, Chem. Rev., 2009, 109, 6729–6764 CrossRef PubMed.
- L. Nicolas, E. Izquierdo, P. Angibaud, I. Stansfield, L. Meerpoel, S. Reymond and J. Cossy, J. Org. Chem., 2013, 78, 11807–11814 CrossRef CAS PubMed.
- X. Cai, K. Ng, H. Panesar, S.-J. Moon, M. Paredes, K. Ishida, C. Hertweck and T. G. Minehan, Org. Lett., 2014, 16, 2962–2965 CrossRef CAS PubMed.
- N. Taniguchi, T. Endo, W. Hart, G. P. H. Seeberger and C. H. Wong, Glycoscience: Biology and Medicine Glycoscience: Biology and Medicine, Springer, 2015 Search PubMed.
- E. De Clercq, J. Med. Chem., 2016, 59, 2301–2311 CrossRef CAS PubMed.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef CAS PubMed.
- K. Kitamura, Y. Ando, T. Matsumoto and K. Suzuki, Chem. Rev., 2018, 118, 1495–1598 CrossRef CAS PubMed.
- Q. Sun, H. Zhang, Q. Wang, T. Qiao, G. He and G. Chen, Angew. Chem., Int. Ed., 2021, 60, 19620–19625 CrossRef CAS PubMed.
- J. Hofsteenge, D. R. Mueller, T. de Beer, A. Loeffler, W. J. Richter and J. F. G. Vliegenthart, Biochemistry, 1994, 33, 13524–13530 CrossRef CAS PubMed.
- S. Manabe and Y. Ito, J. Am. Chem. Soc., 1999, 121, 9754–9755 CrossRef CAS.
- T. Nishikawa, M. Ishikawa, K. Wada and M. Isobe, Synlett, 2001, 0945–0947 CrossRef CAS.
- C.-H. Yao, J.-S. Song, C.-T. Chen, T.-K. Yeh, T.-C. Hsieh, S.-H. Wu, C.-Y. Huang, Y.-L. Huang, M.-H. Wang, Y.-W. Liu, C.-H. Tsai, C. R. Kumar and J.-C. Lee, Eur. J. Med. Chem., 2012, 55, 32–38 CrossRef CAS PubMed.
- N. Kerru, A. Singh-Pillay, P. Awolade and P. Singh, Eur. J. Med. Chem., 2018, 152, 436–488 CrossRef CAS PubMed.
- S. Yonekubo and N. Fushimi, E. P Appl. EP1813611A1, August 1, 2007.
- T. Nishikawa, Synlett, 1999, 123–125 CrossRef CAS.
- T. Nishikawa, Y. Koide, A. Kanakubo, H. Yoshimura and M. Isobe, Org. Biomol. Chem., 2006, 4, 1268–1277 RSC.
- C. Wiebe, C. Schlemmer, S. Weck and T. Opatz, Chem. Commun., 2011, 47, 9212–9214 RSC.
- C. Wiebe, S. Fusté de la Sotilla and T. Opatz, Synthesis, 2012, 44, 1385–1397 CrossRef CAS.
- A. Yepremyan and T. G. Minehan, Org. Biomol. Chem., 2012, 10, 5194–5196 RSC.
- P. Subramanian and K. P. Kaliappan, Eur. J. Org. Chem., 2013, 595 CrossRef CAS.
- F. Zhang, D. Mu, L. Wang, P. Du, F. Han and Y. Zhao, J. Org. Chem., 2014, 79, 9490–9499 CrossRef CAS PubMed.
- E. Y. Aguilera and M. S. Sanford, Organometallics, 2019, 38, 138–142 CrossRef CAS PubMed.
- P. J. Cabrera and M. S. Sanford, J. Am. Chem. Soc., 2018, 140, 5599–5606 CrossRef CAS PubMed.
- M. R. Luzung, C. A. Lewis and P. S. Baran, Angew. Chem., Int. Ed., 2009, 48, 7025–7029 CrossRef CAS PubMed.
- Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547–2550 CrossRef CAS PubMed.
- Q. Wang, Y. Fu, W. Zhu, S. An, Q. Zhou, S.-F. Zhu, G. He, P. Liu and G. Chen, CCS Chem., 2020, 2, 1729–1736 Search PubMed.
- S. Zhang, Y.-H. Niu and X.-S. Ye, Org. Lett., 2017, 19, 3608–3611 CrossRef CAS PubMed.
- Q. Wang, S. An, Z. Deng, W. Zhu, Z. Huang, G. He and G. Chen, Nat. Catal., 2019, 2, 793–800 CrossRef CAS.
- J. Liu, X. Xiao, P. Han, H. Zhou, Q.-S. Yin and J.-S. Sun, Org. Biomol. Chem., 2020, 18, 8834 RSC.
- Q. Wang, W. Zhu, Q. Sun, G. He and G. Chen, Chin. J. Chem., 2021, 39, 571–576 CrossRef CAS.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed., 1997, 36, 119–122 CrossRef CAS.
- M. Lautens and S. Piguel, Angew. Chem., Int. Ed., 2000, 39, 1045 CrossRef CAS PubMed.
- M. Catellani, E. Motti and S. Baratta, Org. Lett., 2001, 3, 3611–3614 CrossRef CAS PubMed.
- N. D. Ca, G. Sassi and M. Catellani, Adv. Synth. Catal., 2008, 350, 2179–2182 CrossRef.
- M. Catellani, E. Motti and N. Della Ca', Acc. Chem. Res., 2008, 41, 1512–1522 CrossRef CAS PubMed.
- G. Maestri, N. Della Ca' and M. Catellani, Chem. Commun., 2009, 4892–4894 RSC.
- E. Motti, N. Della Ca', S. Deledda, E. Fava, F. Panciroli and M. Catellani, Chem. Commun., 2010, 46, 4291–4293 RSC.
- M.-H. Larraufie, G. Maestri, A. Beaume, É. Derat, C. Ollivier, L. Fensterbank, C. Courillon, E. Lacôte and M. Catellani, Angew. Chem., Int. Ed., 2011, 50, 12253–12256 CrossRef CAS PubMed.
- G. Maestri, E. Motti, N. Della Ca', M. Malacria, E. Derat and M. Catellani, J. Am. Chem. Soc., 2011, 133, 8574–8585 CrossRef CAS PubMed.
- N. Della Ca', M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed.
- H.-G. Cheng, S. Chen, R. Chen and Q. Zhou, Angew. Chem., Int. Ed., 2019, 58, 5832–5844 CrossRef CAS PubMed.
- J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS PubMed.
- K. Zhao, L. Ding and Z. Gu, Synlett, 2019, 30, 129–140 CrossRef CAS.
- R. Li and G. Dong, J. Am. Chem. Soc., 2020, 142, 17859–17875 CrossRef CAS PubMed.
- G. Bocelli, M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1985, 279, 225–232 CrossRef CAS.
- M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1985, 286, 13–16 CrossRef.
- Z. Dong, J. Wang, Z. Ren and G. Dong, Angew. Chem., Int. Ed., 2015, 54, 12664–12668 CrossRef CAS PubMed.
- Z. Wu, N. Fatuzzo and G. Dong, J. Am. Chem. Soc., 2020, 142, 2715–2720 CrossRef CAS PubMed.
- P.-X. Shen, X.-C. Wang, P. Wang, R.-Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574–11577 CrossRef CAS PubMed.
- S. Chen, Z.-S. Liu, T. Yang, Y. Hua, Z. Zhou, H.-G. Cheng and Q. Zhou, Angew. Chem., Int. Ed., 2018, 57, 7161–7165 CrossRef CAS PubMed.
- Z.-S. Liu, G. Qian, Q. Gao, P. Wang, H.-G. Cheng, Q. Wei, Q. Liu and Q. Zhou, ACS Catal., 2018, 8, 4783–4788 CrossRef CAS.
- Q. Gao, Y. Shang, F. Song, J. Ye, Z.-S. Liu, L. Li, H.-G. Cheng and Q. Zhou, J. Am. Chem. Soc., 2019, 141, 15986–15993 CrossRef CAS PubMed.
- Z.-S. Liu, G. Qian, Q. Gao, P. Wang, H.-G. Cheng, Y. Hua and Q. Zhou, Tetrahedron, 2019, 75, 1774–1780 CrossRef CAS.
- P. Wang, S. Chen, Z. Zhou, H.-G. Cheng and Q. Zhou, Org. Lett., 2019, 21, 3323–3327 CrossRef CAS PubMed.
- W. Cai and Z. Gu, Org. Lett., 2019, 21, 3204–3209 CrossRef CAS PubMed.
- J. Liu, Q. Ding, W. Fang, W. Wu, Y. Zhang and Y. Peng, J. Org. Chem., 2018, 83, 13211–13216 CrossRef CAS PubMed.
- W. Lv, Y. Chen, S. Wen, D. Ba and G. Cheng, J. Am. Chem. Soc., 2020, 142, 14864–14870 CrossRef CAS PubMed.
- Y.-N. Ding, W.-Y. Shi, C. Liu, N. Zheng, M. Li, Y. An, Z. Zhang, C.-T. Wang, B.-S. Zhang and Y.-M. Liang, J. Org. Chem., 2020, 85, 11280–11296 CrossRef CAS PubMed.
- R. Li, F. Liu and G. Dong, Org. Chem. Front., 2018, 5, 3108–3112 RSC.
- J. Wang, Z. Dong, C. Yang and G. Dong, Nat. Chem., 2019, 11, 1106–1112 Search PubMed.
- J. Wang, Y. Zhou, X. Xu, P. Liu and G. Dong, J. Am. Chem. Soc., 2020, 142, 3050–3059 Search PubMed.
- R. Li and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 1697–1701 CrossRef CAS PubMed.
- J. Wang, R. Li, Z. Dong, P. Liu and G. Dong, Nat. Chem., 2018, 10, 866–872 CrossRef CAS PubMed.
- Z. Dong and G. Dong, J. Am. Chem. Soc., 2013, 135, 18350–18353 CrossRef CAS PubMed.
- X. Liu, J. Wang and G. Dong, J. Am. Chem. Soc., 2021, 143, 9991–10004 CrossRef CAS PubMed.
- B.-S. Zhang, Y. Li, Z. Zhang, Y. An, Y.-H. Wen, X.-Y. Gou, S.-Q. Quan, X.-G. Wang and Y.-M. Liang, J. Am. Chem. Soc., 2019, 141, 9731–9738 CrossRef CAS PubMed.
- M. Scharenberg, O. Schwardt, S. Rabbani and B. Ernst, J. Med. Chem., 2012, 55, 9810–9816 CrossRef CAS PubMed.
- L. Mydock-McGrane, Z. Cusumano, Z. Han, J. Binkley, M. Kostakioti, T. Hannan, J. S. Pinkner, R. Klein, V. Kalas, J. Crowley, N. P. Rath, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2016, 59, 9390–9408 CrossRef CAS PubMed.
- B.-W. Li, M.-Y. Wang, S. Fang and J.-Y. Liu, Organometallics, 2019, 38, 2189–2198 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2047890, 2047891 and 2067951. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03569j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |