
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of a water-soluble endohedrally functionalized coordination cage including polar guests†
Qingqing
Sun‡
 ab,
Luis
Escobar§
*ab,
Jorn
de Jong¶
a and
Pablo
Ballester
*ac
ab,
Luis
Escobar§
*ab,
Jorn
de Jong¶
a and
Pablo
Ballester
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: pballester@iciq.es
bUniversitat Rovira i Virgili (URV), Departament de Química Analítica i Química Orgánica, c/Marcel·lí Domingo 1, 43007 Tarragona, Spain
cICREA, Passeig Lluís Companys 23, 08010 Barcelona, Spain
First published on 13th September 2021
Abstract
Coordination cages containing endohedrally functionalized aromatic cavities are scarce in the literature. Herein, we report the self-assembly of a tetra-cationic super aryl-extended calix[4]pyrrole tetra-pyridyl ligand into a water-soluble Pd(II)-cage featuring two endohedral polar binding sites. They are defined by the four pyrrole NHs of the calix[4]pyrrole unit and the four inwardly directed α-protons of the coordinated pyridyl groups. The efficient assembly of the Pd(II)-cage requires the inclusion of mono- and ditopic pyridyl N-oxide and aliphatic formamide guests. The monotopic guests only partially fill the cage's cavity and require the co-inclusion of a water molecule that is likely hydrogen-bonded to the endohedral α-pyridyl protons. The ditopic guests are able to completely fill the cage's cavity and complement both binding sites. We observed high conformational selectivity in the inclusion of the isomers of α,ω-bis-formamides. We briefly investigate the uptake and release mechanism/kinetics of selected polar guests by the Pd(II)-cage using pair-wise competition experiments.
Introduction
Self-assembly is an effective synthetic strategy for the construction of coordination cages (CCs) in organic solvents and in water.1–3 Their molecular components are multitopic aromatic ligands (L) and metal coordination ions (M).4 The inclusion of size, shape and charge complementary guests in CCs enables not only selective molecular recognition but also controls the properties, reactivities and the confinement and release of molecular cargo.3,5–8 The inclusion of polar guests requires the functionalization of the CCs' cavities with polar groups.9,10 However, the extensive use of aromatic panels in shaping the cavity makes this endeavour synthetically difficult. Fujita and co-workers introduced a strategy to generate spherical [L24·Pd12]24+ CCs containing 24 inwardly directed functional groups.11 Subsequently, Reek and co-workers applied the strategy to develop catalyst systems.12 In addition, [L4·Pd2]4+ CCs (Fig. 1a) were assembled from bis-pyridyl ligands featuring imide,13 amide14 or urea15,16 groups as spacing units. The 4 ligands were located at the edges of the CCs' cavity and the NH hydrogen bond donors were placed in a way that they were inwardly directed. The [L4·Pd2]4+ CCs bearing endohedral amide or urea NHs bound glycosides in organic solvents owing to the establishment of intermolecular hydrogen-bonding and CH–π interactions.14,16 In the same vein, tetrahedral [L6·M4]8+ CCs (Fig. 1b) featuring inwardly directed urea NHs,17–19 hydroxyl20 or carboxylic acid21,22 groups have been reported in the literature. Martinez, Nitschke and co-workers reported the anion-templated self-assembly of an azaphosphatrane endohedrally functionalized [L4·Fe4]12+ CC (Fig. 1c) in aqueous solution.23 The [L4·Fe4]12+ CC was capable of selective anion extraction and recovery.24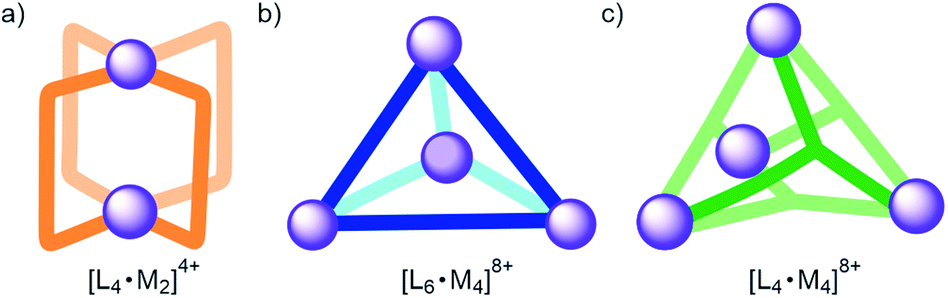 | ||
| Fig. 1 Schematic representation of different CCs previously reported in the literature: (a) [L4·M2]4+, (b) [L6·M4]8+ and (c) [L4·M4]8+. Ligands (L) contain inwardly directed functional groups, which are not shown for clarity. | ||
Recently, we described the self-assembly of the [1a·Pd]2+ CC in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CDCl3/CD3CN solution mixture25 (Fig. 2a and 3b). The tetra-pyridyl chelating ligand 1a is based on a “four-wall” super aryl-extended calix[4]pyrrole (SAE-C[4]P) scaffold. The [1a·Pd]2+ CC might also be considered as a mono-nuclear metal-coordinated cavitand. The use of coordination metals in controlling the conformations and binding properties of covalent organic hosts has also been reported by the groups of de Mendoza,60,61 Badjić26–28 and Rebek.29–33 The [1a·Pd]2+ CC features two different and converging polar binding sites34 defined by the four pyrrole NHs of the C[4]P unit and the four inwardly directed α-CH protons of the pyridyl groups coordinated to Pd(II). The assembly of the CC requires the filling of its cavity with suitable polar guests. The bound guests not only fill ca. 55% of the cavity volume35 but also satisfy the hydrogen-bonding characteristics of the two polar binding sites (Fig. 3b). We also investigated the inclusion/exchange mechanisms of the guests operating in the [1a·Pd]2+ CC.36 We proposed a “French doors” mechanism37 for the in/out exchange of planar pyridyl N-oxide derivatives. For more sterically demanding guests, we postulated an alternative process involving a partial ligand–metal dissociation mechanism.
:1 CDCl3/CD3CN solution mixture25 (Fig. 2a and 3b). The tetra-pyridyl chelating ligand 1a is based on a “four-wall” super aryl-extended calix[4]pyrrole (SAE-C[4]P) scaffold. The [1a·Pd]2+ CC might also be considered as a mono-nuclear metal-coordinated cavitand. The use of coordination metals in controlling the conformations and binding properties of covalent organic hosts has also been reported by the groups of de Mendoza,60,61 Badjić26–28 and Rebek.29–33 The [1a·Pd]2+ CC features two different and converging polar binding sites34 defined by the four pyrrole NHs of the C[4]P unit and the four inwardly directed α-CH protons of the pyridyl groups coordinated to Pd(II). The assembly of the CC requires the filling of its cavity with suitable polar guests. The bound guests not only fill ca. 55% of the cavity volume35 but also satisfy the hydrogen-bonding characteristics of the two polar binding sites (Fig. 3b). We also investigated the inclusion/exchange mechanisms of the guests operating in the [1a·Pd]2+ CC.36 We proposed a “French doors” mechanism37 for the in/out exchange of planar pyridyl N-oxide derivatives. For more sterically demanding guests, we postulated an alternative process involving a partial ligand–metal dissociation mechanism.
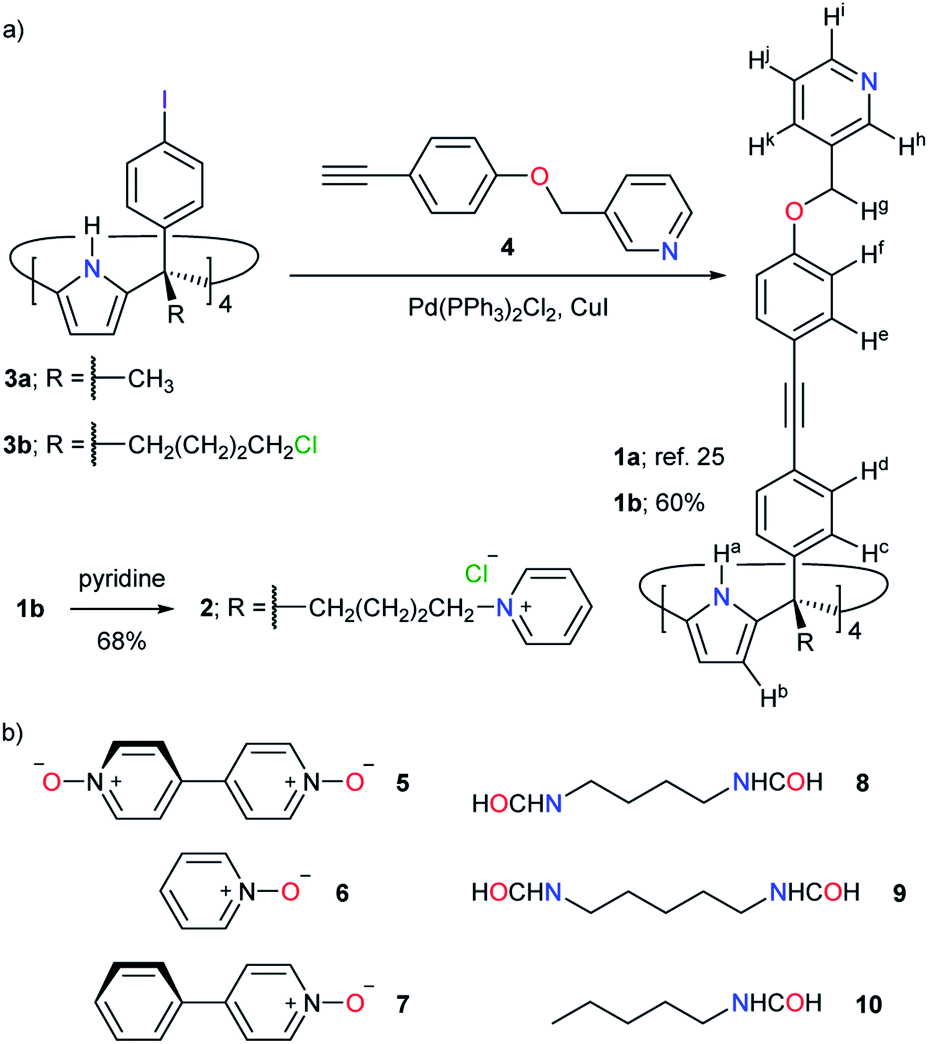 | ||
| Fig. 2 (a) Synthesis of tetra-pyridyl SAE-ligands 1a, 1b and 2. (b) Line-drawing structures of guests 5–10 used in this study. | ||
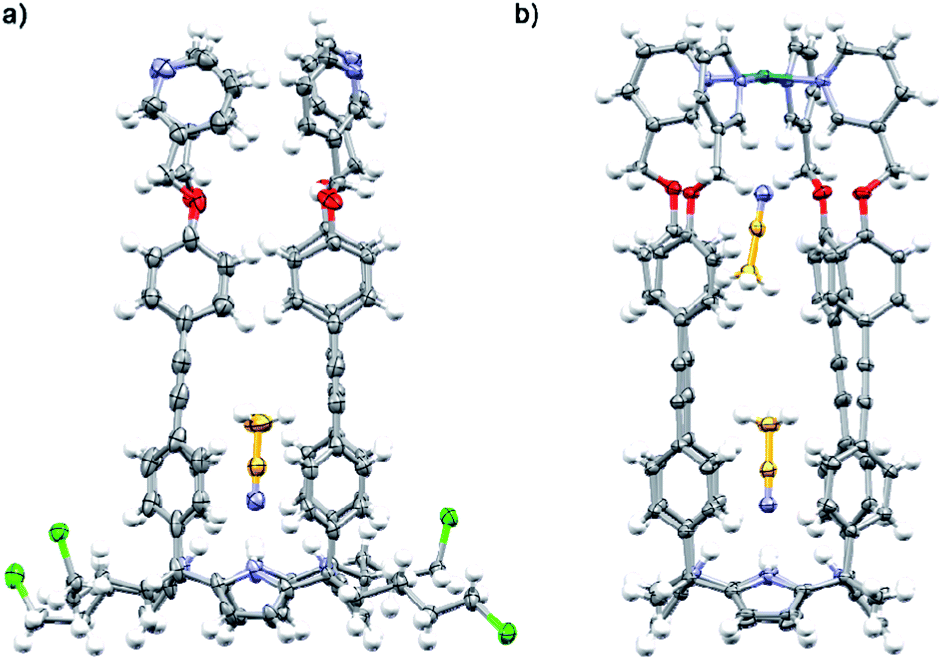 | ||
| Fig. 3 X-ray crystal structures of (a) 1b and (b) [1a·Pd]2+. The structures are shown in ORTEP view with thermal ellipsoids set at 50% probability for the non-hydrogen atoms. Hydrogen atoms are depicted as fixed-size spheres of 0.3 Å radius. | ||
Herein, we report the self-assembly of a structurally related CC, [2·Pd]6+, in water solution. We describe the synthesis of the tetra-cationic ligand precursor 24+ (Fig. 2a). The formation of the [2·Pd]6+ CC follows the assembly principles described above for the [1a·Pd]2+ counterpart. We show that the CC's assembly only occurs upon including mono- and ditopic pyridyl N-oxides, as well as aliphatic mono- and bis-formamide guests. All the polar guests used are difficult to bind in water solution with other synthetic receptors.38–40 Using 1H NMR spectroscopy, we study the conformational arrangement adopted by the end formyl groups of the α,ω-bis-formamides included in the [2·Pd]6+ CC. We disclose solid-state structures for analogous inclusion complexes with the organic-soluble [1a·Pd]2+ CC. Finally, we investigate the uptake and release mechanisms of selected polar guests in the [2·Pd]6+ CC using competitive, pair-wise binding experiments.
Results and discussion
Synthesis of tetra-pyridyl SAE-C[4]P ligands 1b and 24+
The tetra-cationic SAE-ligand 24+ was synthesized in two steps starting from the tetra-α isomer of 4-iodophenyl-4′-chlorobutyl C[4]P 3b41 (Fig. 2a). Firstly, the quadruple Sonogashira cross-coupling reaction between tetra-iodo C[4]P 3b and pyridyl mono-acetylene 425 afforded, after column chromatography purification, the tetra-chloro SAE-ligand 1b in 60% yield. Next, we treated 1b with excess of pyridine at 110 °C affording the tetra-chloride salt of 24+ in 68% yield. Both SAE-ligands, 1b and 24+, were fully characterized using a set of high-resolution spectra (ESI†).The tetra-chloro SAE-ligand 1b was also characterized by X-ray diffraction of a single crystal grown from a 1:1 CH2Cl2/CH3CN solution mixture (Fig. 3a). In the solid state, 1b adopts the cone conformation by including one molecule of CH3CN. The nitrogen atom of the included CH3CN is hydrogen-bonded to the four pyrrole NHs of the C[4]P unit (daverage (N⋯N) ∼ 3.3 Å). Moreover, the aromatic cavity of the upper section of 1b collapses owing to the establishment of intramolecular π–π interactions.42
The neutral tetra-pyridyl SAE-ligand 1b self-assembled almost quantitatively into the [1b·Pd]2+ CC in a 2:1 CDCl3/CD3CN solution mixture by addition of ca. 1 equiv. of [Pd(CH3CN)4(BF4)2] (Fig. S14†). Consequently, the meso-alkyl substituents of 1b did not have an impact on the assembly of the CC.
Attempts to self-assemble the [2·Pd]6+ CC in the absence of guests
We probed the analogous coordination of Pd(II) with the water-soluble tetra-pyridyl SAE-ligand 24+ (Fig. 2a) by means of 1H NMR spectroscopy. At 298 or 333 K, the 1H NMR spectrum of 24+ in the D2O solution showed very broad proton signals|| (Fig. S31† and 4a, respectively). However, the addition of ca. 10% DMSO to the D2O solution of 24+ at r.t. provoked the appearance of sharp proton signals (Fig. S82†). Taken together, these observations indicated that the SAE-ligand 24+ experienced significant aggregation in water solution. We cannot rule out the possibility that the aggregation process also makes the conformational exchange dynamics (cone and alternate) of 24+ become intermediate on the chemical shift time scale.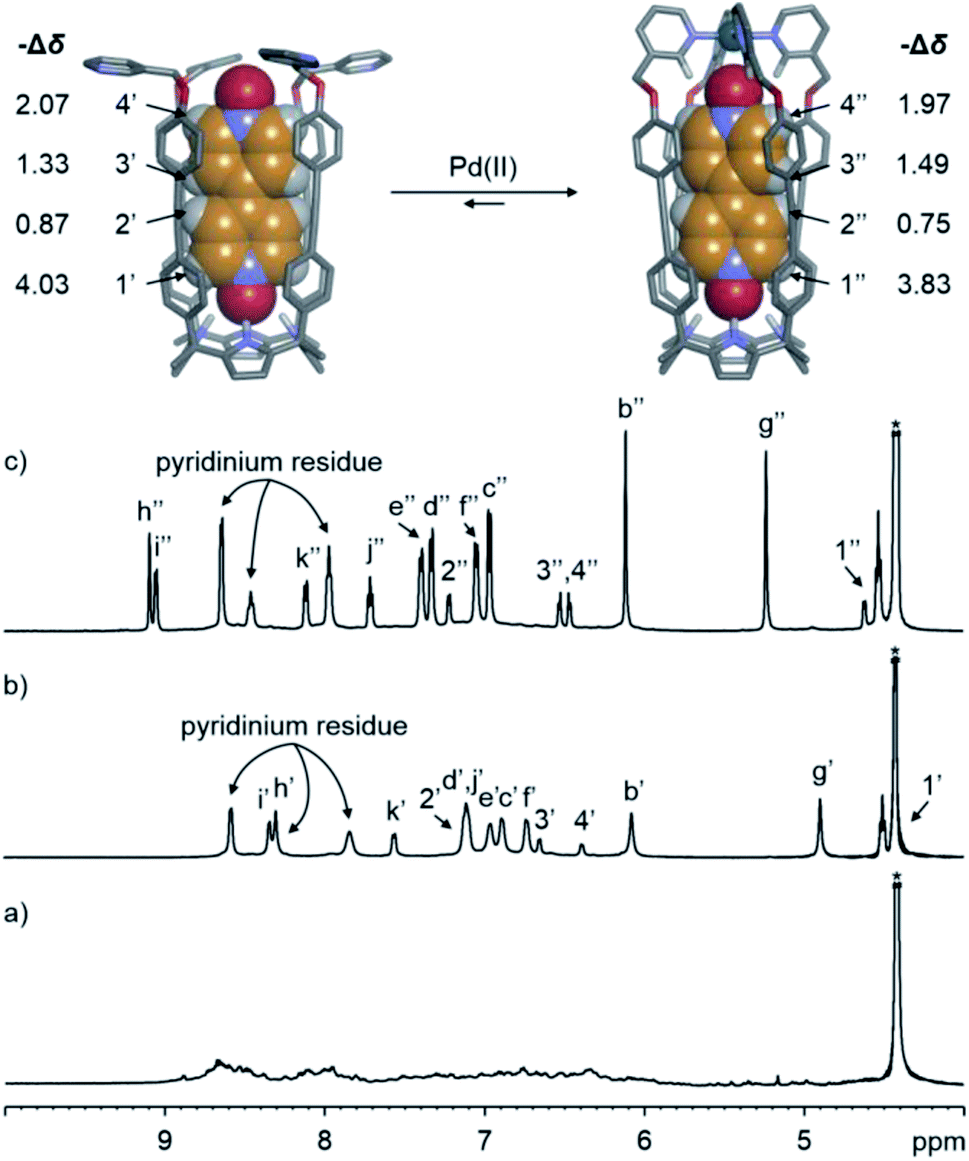 | ||
| Fig. 4 (Top) Energy minimized structures (MM3) of the simplified 5⊂24+ and 5⊂[2·Pd]6+ inclusion complexes. (Bottom) Selected regions of the 1H NMR (500 MHz with a cryoprobe, D2O, 333 K) spectra of (a) 24+, (b) 5⊂24+ and (c) 5⊂[2·Pd]6+. Primed and double primed labels correspond to the proton signals of 5⊂24+ and 5⊂[2·Pd]6+, respectively. See Fig. 2a for the proton assignment of bound 24+. *Residual solvent peak. | ||
The putative assembly of [2·Pd]6+ would assign an overall six positive charges to the CC. For this reason, we expected that the increase in the repulsive coulombic interactions will induce a reduction in the aggregation tendency of [2·Pd]6+ compared to that of the parent SAE-ligand 24+. Nevertheless, the addition of ca. 1 equiv. of Pd(NO3)2 to the D2O solution of 24+ at r.t. produced a suspension. We surmised that the insoluble material originated from the formation of metal-mediated polymeric aggregates. The analysis of the filtered solution, at 333 K, showed broad proton signals, which did not support the presence of the CC in water.
Self-assembly of the [2·Pd]6+ CC in the presence of pyridine N-oxide guests
As stated above, the emergence of the [1a·Pd]2+ CC (Fig. 3b) in organic solvents required the inclusion of suitable polar guests. One molecule of 4,4′-bis-pyridine N,N′-dioxide 5 or a combination of one pyridine N-oxide 6 (Fig. 2b) and one CH3CN were perfect fits.25,36 The N-oxides formed 1:1 inclusion complexes with the SAE-ligand 1a. The bound ligand was locked in the cone conformation and its pyridyl residues were pre-organized for tetra-coordination to Pd(II).
At 333 K, the 1H NMR spectrum of a D2O solution containing equimolar amounts of the SAE-ligand 24+ and the bis-N-oxide 5 displayed a set of well-defined proton signals in agreement with the C4v symmetry of the receptor (Fig. 4b). Specifically, the two α-pyridyl protons of 24+, Hh and Hi, resonated at 8.30 and 8.34 ppm, respectively. The four aromatic protons of the meso-biphenyl-ethynyl substituents, Hc–Hf, emerged as separate signals (Fig. 4b). The β-pyrrole protons, Hb, appeared as a singlet at 6.07 ppm. The symmetry loss and the upfield shifts experienced by the protons of 5 placed it in the polar and deep aromatic cavity of 24+ (Fig. 4, top panel).
The addition of more than 1 equiv. of 5 did not produce changes in the proton signals of 24+, but caused the emergence of those of the free guest (ESI†). All these observations supported the formation of the 5⊂24+ inclusion complex, which featured slow exchange dynamics on both the 1H NMR and the EXSY time scales (tmix = 0.3 s, Fig. S36†). We estimated a binding constant larger than 104 M−1.
The complexation-induced shifts experienced by the aromatic protons of the bound bis-N-oxide 5 are not directly related to its depth in the aromatic cavity of 24+ (see −Δδ values in Fig. 4, top panel). This result reflected the dissimilar magnetic anisotropy provided by the meso-biphenyl-ethynyl substituents of 24+.
The inclusion geometry of the 5⊂24+ complex diminishes the aromatic surface of the receptor available for water solvation. Because the binding of non-polar residues in water is mainly driven by the hydrophobic effect (HE),38,43 the reduction in the aromatic surface helps explain the lower aggregation tendency of the 5⊂24+ complex compared to that of the free receptor 24+.
The addition of ca. 1 equiv. of Pd(NO3)2 to the D2O solution of 5⊂24+ produced the complete disappearance of its proton signals and the emergence of a new set that was also in agreement with the C4v symmetry of the SAE-ligand 24+. Based on our previous findings in organic solution,25,36 the new set of signals was diagnostic of the 5⊂[2·Pd]6+ CC complex (vide infra) (Fig. 4c). Using an internal standard, we calculated that the CC complex was assembled to a 60% extent. Most likely, metal-mediated aggregates of 5⊂24+ were also formed whose proton signals broadened beyond detection.
The formation of N-Pd(II) coordination bonds in the 5⊂[2·Pd]6+ CC complex was substantiated by the downfield shifts experienced by the two α-pyridyl protons, Hh and Hi (Δδ = +0.79 and +0.71 ppm, respectively), with respect to their chemical shift values in the initial 5⊂24+ complex. The reduced downfield shift experienced by the β-pyrrole protons, Hb, of 5⊂[2·Pd]6+ indicated that the C[4]P unit maintained the cone conformation. Finally, the proton signals of 5 included in the CC suffered negligible chemical shift changes compared to those of 5⊂24+. We reported analogous observations for the structurally related 5⊂[1a·Pd]2+ CC assembled in organic solvents.25
The addition of 5 in excess did not produce chemical shift changes to the proton signals of the water-soluble 5⊂[2·Pd]6+ CC complex (ESI†). The free and bound guest 5 was involved in a slow exchange process on both the 1H NMR and the EXSY time scales (tmix = 0.3 s, Fig. S39†). The binding constant of 5⊂[2·Pd]6+ must be larger than 104 M−1.
We performed a 1H DOSY NMR experiment44,45 using the D2O solution containing the 5⊂[2·Pd]6+ CC complex (Fig. S41†). The fit of the decay of all proton signals returned the diffusion constant value (D) of 5⊂[2·Pd]6+ as logD = −9.31. Collectively, the results presented above demonstrated that the emergence of the [2·Pd]6+ CC in D2O required the filling of its polar cavity with the bis-N-oxide 5.** The assembly of the [2·Pd]6+ CC was also studied at 333 K owing to the remaining aggregation problems at lower temperatures.††
In a 2:1 CDCl3/CD3CN solution mixture, pyridine N-oxide 6 (Fig. 2b) was shown to be a good guest to induce the formation of the 6⊂[1a·Pd]2+ CC complex.25,36 X-ray crystallography revealed the co-inclusion of one CH3CN molecule in the assembled [6·CH3CN]⊂[1a·Pd]2+ CC complex. We wondered if a water molecule could replace the CH3CN for the co-inclusion of 6 in the water-soluble version of the CC.
At 333 K, the 1H NMR spectrum of an equimolar D2O solution (1 mM) of pyridine N-oxide 6 and the SAE-ligand 24+ showed a diminution in the broadening of the proton signals of the receptor/ligand (Fig. S43†). The protons of free 6 were visible as sharp signals with reduced intensity compared to those of a 1 mM solution. This result suggested that the 6⊂24+ inclusion complex was formed to some extent. The subsequent addition of ca. 1 equiv. of Pd(NO3)2 provoked the appearance of a new set of sharp signals that were diagnostic of the assembly of the 6⊂[2·Pd]6+ CC complex (Fig. S44†). We assumed that the co-inclusion of one molecule of water46 with 6 took place. The putative [6·H2O]⊂[2·Pd]6+ CC complex (Fig. S88†) was assembled only to a 30% extent.
In agreement with the double role played by the bound guests in the CC complexes of [1a·Pd]2+, 4-phenylpyridine N-oxide 7 (Fig. 2b) did not induce the emergence of the CC complex in 2:1 CDCl3/CD3CN.25 Nevertheless, 7 formed a highly stable 1:1 inclusion complex with the SAE-ligand 1a in the same solvent mixture. The addition of Pd(II) to the 7⊂1a complex resulted in the immediate appearance of metal-mediated oligomers. Owing to the reduced size of H2O compared to CH3CN, we were curious to find out if the co-inclusion of H2O with 7 could induce the emergence of the [7·H2O]⊂[2·Pd]6+ CC complex.
The 1H NMR spectrum of an equimolar mixture of 7 and 24+ in D2O solution at 333 K showed the formation of the corresponding 7⊂24+ inclusion complex. However, the extent to which the 1:1 complex was formed was lower than that in the case of the bis-N-oxide 5 (Fig. S47†). The subsequent addition of 1 equiv. of Pd(NO3)2 provoked the formation of metal-mediated aggregates of 24+, which may partially bind the N-oxide 7. In short, the system expresses an analogous behaviour to the one observed in the mixture of organic solvents.25
Self-assembly of the [2·Pd]6+ CC in the presence of aliphatic formamide guests
The successful results obtained with bis-N-oxide 5 prompted us to investigate the self-assembly of other inclusion complexes of the [2·Pd]6+ CC in water solution using ditopic guests. Based on molecular modelling studies (MM3), we selected the aliphatic α,ω-bis-formamides 8 and 9, having four and five methylene groups as spacers,47 respectively (Fig. 2b). The extended conformations of both guests are good fits to the CC's cavity. In addition, the two end formyl groups of the bis-formamides can complement the two polar binding sites of the [2·Pd]6+ CC.At 333 K, the 1H NMR spectrum of a 1:1 D2O solution mixture of the bis-formamide 8 and the SAE-ligand 24+ did not display a set of sharp signals for the receptor (Fig. S51†). Yet, the signals of free 8 were easily detected. This result indicated that the putative 8⊂24+ inclusion complex was formed to a reduced extent. Surprisingly, the addition of ca. 1 equiv. of Pd(NO3)2 to the above solution produced a set of sharp and well-defined signals corresponding to the [2·Pd]6+ CC (Fig. 5a). We also observed two sets of signals for the protons of 8 with different intensities. The low intensity signals corresponded to the protons of the free guest. The more intense set displayed upfield shifts for all protons.
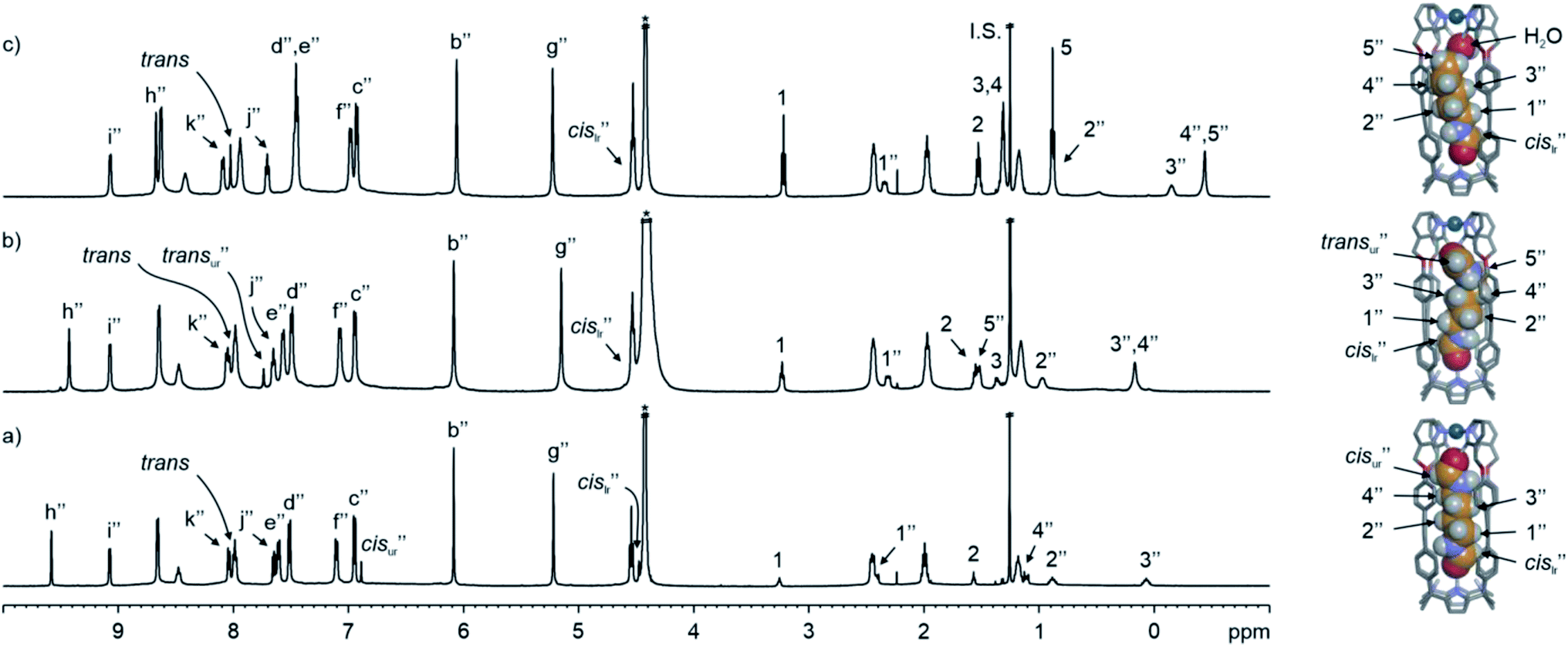 | ||
| Fig. 5 (Left) 1H NMR (500 MHz with a cryoprobe, D2O, 333 K) spectra of (a) cis,cis-8⊂[2·Pd]6+, (b) trans,cis-9⊂[2·Pd]6+ and (c) cis-10⊂[2·Pd]6+, together with free guests in solution. (Right) Energy minimized structures (MM3) of the simplified CC complexes. A putative water molecule is bound to cis-10⊂[2·Pd]6+. Double primed labels correspond to the proton signals of the CC complexes (lr = lower rim and ur = upper rim). See Fig. 2a for the proton assignment of bound 24+. I.S. = tert-butanol. *Residual solvent peak. | ||
e assigned the latter to the protons of 8 included in the [2·Pd]6+ CC. In short, the emergence of the 8⊂[2·Pd]6+ CC complex was induced by the inclusion of 8 to an extent of 60% with respect to its molecular components.
One of the formyl protons of bound 8 displayed a significant upfield shift ( , Δδ = −3.52 ppm), placing it within the four meso-aryl substituents of the C[4]P unit47,48 of the [2·Pd]6+ CC (Fig. 5a, right). The other formyl proton also experienced a reduced magnetic shielding (
, Δδ = −3.52 ppm), placing it within the four meso-aryl substituents of the C[4]P unit47,48 of the [2·Pd]6+ CC (Fig. 5a, right). The other formyl proton also experienced a reduced magnetic shielding ( , Δδ = −1.11 ppm), owing to its inclusion in the upper aromatic cavity of the CC. The intermolecular cross-peaks present in a 2D ROESY experiment also supported the location of the latter formamide in the upper aromatic cavity of the CC (Fig. S55†). Moreover, the ROESY experiment also displayed intramolecular close-contact cross-peaks between the formyl protons at the two ends of included 8 and their respective α-CH2 groups, H1 and H4 (Fig. 6). This result indicated the selective inclusion of the cis,cis-isomer of 8 in the cavity of [2·Pd]6+. Bis-formamide 8 exists in solution as a mixture of three isomers with respect to the conformation adopted by the two terminal amides: trans,trans (∼72%), trans,cis (∼25%) and cis,cis (∼3%). The [2·Pd]6+ CC selectively binds the cis,cis-isomer, most likely in a fully extended conformation (Fig. 5a, right). The binding of long-chain α,ω-bis-formamides to a water-soluble resorcin[4]arene deep cavitand was previously investigated by the Rebek group.49 The cavitand displayed a reduced binding preference for the trans,cis-isomer compared to the trans,trans-counterpart. To the best of our knowledge, the high binding selectivity of the [2·Pd]6+ CC is unprecedented.
, Δδ = −1.11 ppm), owing to its inclusion in the upper aromatic cavity of the CC. The intermolecular cross-peaks present in a 2D ROESY experiment also supported the location of the latter formamide in the upper aromatic cavity of the CC (Fig. S55†). Moreover, the ROESY experiment also displayed intramolecular close-contact cross-peaks between the formyl protons at the two ends of included 8 and their respective α-CH2 groups, H1 and H4 (Fig. 6). This result indicated the selective inclusion of the cis,cis-isomer of 8 in the cavity of [2·Pd]6+. Bis-formamide 8 exists in solution as a mixture of three isomers with respect to the conformation adopted by the two terminal amides: trans,trans (∼72%), trans,cis (∼25%) and cis,cis (∼3%). The [2·Pd]6+ CC selectively binds the cis,cis-isomer, most likely in a fully extended conformation (Fig. 5a, right). The binding of long-chain α,ω-bis-formamides to a water-soluble resorcin[4]arene deep cavitand was previously investigated by the Rebek group.49 The cavitand displayed a reduced binding preference for the trans,cis-isomer compared to the trans,trans-counterpart. To the best of our knowledge, the high binding selectivity of the [2·Pd]6+ CC is unprecedented.
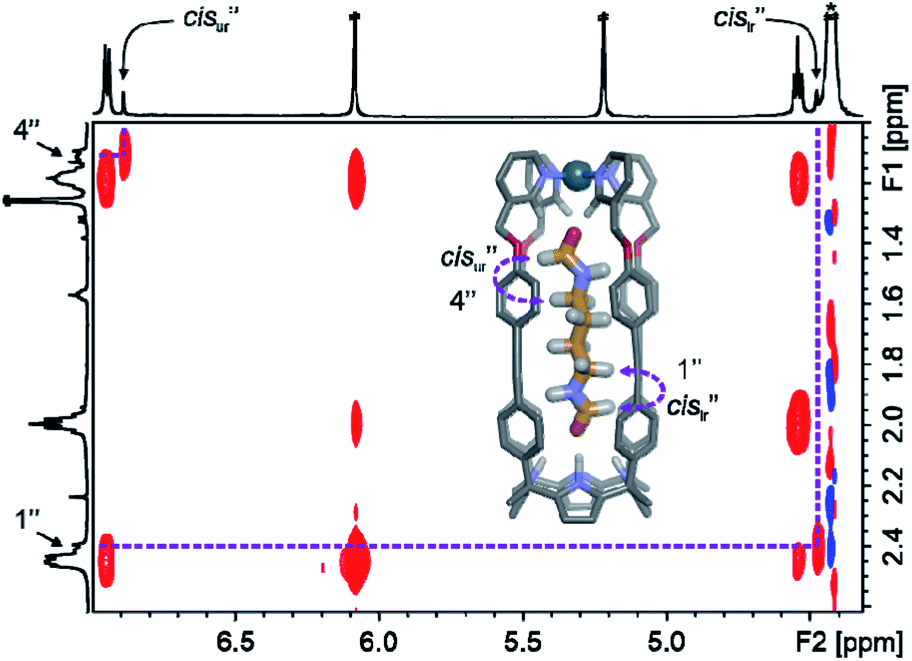 | ||
| Fig. 6 Selected region of the 2D ROESY NMR (500 MHz with a cryoprobe, D2O, 333 K, spin lock = 0.3 s) spectrum of cis,cis-8⊂[2·Pd]6+. Intramolecular close-contacts are indicated in the energy minimized structure (MM3) of the simplified CC complex. Double primed labels correspond to the proton signals of the included guest in the CC. | ||
We estimated that the stability constant for the cis,cis-8⊂[2·Pd]6+ CC complex should be larger than 105 M−1. The inwardly directed α-pyridyl protons, Hh, in cis,cis-8⊂[2·Pd]6+ were shifted further downfield than those in the 5⊂[2·Pd]6+ complex (9.58 vs. 9.09 ppm, respectively).
The homologated bis-formamide 9 (Fig. 2b), possessing an extra methylene group, produced similar results (Fig. 5b). However, the ROESY experiment of the 9⊂[2·Pd]6+ CC complex indicated that the CC showed binding selectivity for the trans,cis-isomer of 9 (Fig. 5b, right) instead of the cis,cis-counterpart observed in the case of 8.
The DOSY NMR experiments performed with cis,cis-8⊂[2·Pd]6+ and trans,cis-9⊂[2·Pd]6+ yielded similar diffusion constant values (logD = −9.32 and −9.31, respectively) to those of the two CC complexes (Fig. S58 and S67†). The calculated magnitudes are almost identical to that determined for the 5⊂[2·Pd]6+ counterpart (vide supra), supporting the structural size and shape similarity of the three diffusing species.
The binding geometries assigned in water solution to the included bis-formamides in cis,cis-8⊂[2·Pd]6+ and trans,cis-9⊂[2·Pd]6+ were in complete agreement with those obtained from X-ray diffraction analyses of single crystals grown for the structural analogues of CC complexes: cis,cis-8⊂[1a·Pd]2+ and trans,cis-9⊂[1a·Pd]2+ (Fig. 7a and b). The X-ray structure of trans,cis-9⊂[1a·Pd]2+ evidenced the bending of the larger alkyl spacer to better adapt to the dimensions of the CC's cavity. The formamide end of 9 hydrogen-bonded to the C[4]P unit adopts the cis-conformation, while the opposite end, included in the upper aromatic cavity of the CC, remains in the trans-configuration.
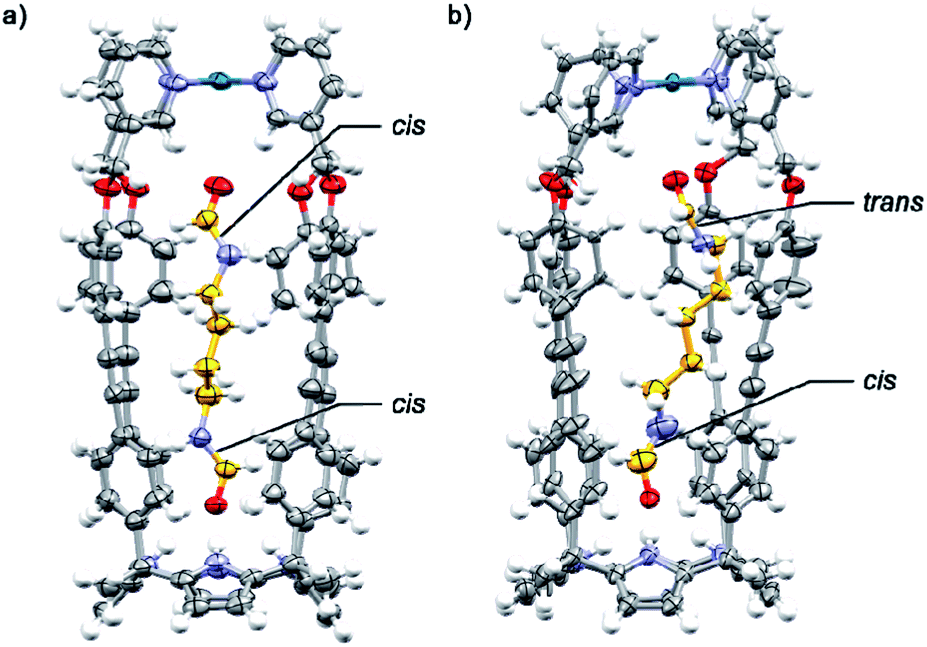 | ||
| Fig. 7 X-ray crystal structures of (a) cis,cis-8⊂[1a·Pd]2+ and (b) trans,cis-9⊂[1a·Pd]2+. The structures are shown in ORTEP view with thermal ellipsoids set at 50% probability for the non-hydrogen atoms. Hydrogen atoms are depicted as fixed-size spheres of 0.3 Å radius. | ||
The carboxyl oxygen atom of the trans-formamide is hydrogen-bonded to the inwardly directed α-pyridyl protons of the CC. The groups of Rebek and Gibb previously reported examples of unusual conformations adopted by alkyl chains of encapsulated guests in self-assembled dimeric capsules.50–52
The results with the pyridine N-oxide 6 suggested that the assembly of the CC requires the co-inclusion of a water molecule to properly fill the space. We tested the generality of this notion with the monotopic N-pentylformamide 10 (Fig. 2b). The 1H NMR spectra of an equimolar mixture of 10 and 24+ in D2O solution at 333 K showed the formation of the corresponding 10⊂24+ inclusion complex to a significant extent (Fig. S69†). The addition of Pd(II) to this partially formed complex produced a set of sharp signals indicative of the assembly of the 10⊂[2·Pd]6+ CC complex (Fig. 5c). Using an internal standard, we determined that the CC complex was assembled to a 60% extent.
The upfield shift experienced by the formyl proton (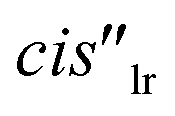 , Δδ = −3.46 ppm) of bound 10 placed it in the C[4]P unit. The conformation adopted by the formamide guest was determined by a ROESY experiment as exclusively cis (Fig. S72†). The inwardly directed α-pyridyl protons, Hh, in the cis-10 complex were less downfield shifted than those in the 6 counterpart (8.68 vs. 9.44 ppm, respectively). The co-inclusion of a water molecule in the putative [10·H2O]⊂[2·Pd]6+ CC complex satisfies the functional complementarity of the binding site induced by metal coordination46 (Fig. 5c, right). The binding dynamics of this complex are slow on the chemical shift time scale, but intermediate on the diffusion time scale of a DOSY experiment. A typical DOSY experiment (td ∼ 200 ms) assigned a diffusion constant to bound cis-10 that differed from that of [2·Pd]6+ (Fig. S76†). Likewise, the diffusion constant of free 10 did not coincide with the value determined for the guest alone in solution (Fig. S78†). The use of a shorter diffusion time (td ∼ 10 ms) showed the expected coincidence of diffusion constants for bound cis-10 and [2·Pd]6+ (Fig. S77†). This finding indicated that the [cis-10·H2O]⊂[2·Pd]6+ CC complex is kinetically less stable53 than those involving bis-formamide guests (vide supra).
, Δδ = −3.46 ppm) of bound 10 placed it in the C[4]P unit. The conformation adopted by the formamide guest was determined by a ROESY experiment as exclusively cis (Fig. S72†). The inwardly directed α-pyridyl protons, Hh, in the cis-10 complex were less downfield shifted than those in the 6 counterpart (8.68 vs. 9.44 ppm, respectively). The co-inclusion of a water molecule in the putative [10·H2O]⊂[2·Pd]6+ CC complex satisfies the functional complementarity of the binding site induced by metal coordination46 (Fig. 5c, right). The binding dynamics of this complex are slow on the chemical shift time scale, but intermediate on the diffusion time scale of a DOSY experiment. A typical DOSY experiment (td ∼ 200 ms) assigned a diffusion constant to bound cis-10 that differed from that of [2·Pd]6+ (Fig. S76†). Likewise, the diffusion constant of free 10 did not coincide with the value determined for the guest alone in solution (Fig. S78†). The use of a shorter diffusion time (td ∼ 10 ms) showed the expected coincidence of diffusion constants for bound cis-10 and [2·Pd]6+ (Fig. S77†). This finding indicated that the [cis-10·H2O]⊂[2·Pd]6+ CC complex is kinetically less stable53 than those involving bis-formamide guests (vide supra).
In water, the binding of the guests in the polar aromatic cavity of the [2·Pd]6+ CC is mainly driven by the hydrophobic effect (HE).38,43 Additional selectivity and affinity in the recognition process come from hydrogen bonds, and NH–π, π–π and CH–π interactions.42 The HE's role in the binding of the mono-formamide 10 is clear: it is efficiently included in the water-soluble [2·Pd]6+ CC, but it does not bind to the organic-soluble [1b·Pd]2+ counterpart (Fig. S30†).
Guest uptake and release in the [2·Pd]6+ CC
We briefly investigated the uptake and release mechanism/kinetics6,54,55 of [2·Pd]6+ using pair-wise competition experiments (Fig. S80 and S81†). The 1H NMR spectra of equimolar D2O solutions of 24+, Pd(NO3)2, and guests 6 (pyridine N-oxide) and 8 (butane α,ω-bis-formamide) indicated the assembly of [6·H2O]⊂[2·Pd]6+ and cis,cis-8⊂[2·Pd]6+ to 30% and 60% extent, respectively. Sharp signals for the remaining free substrates were observed. Next, we added 1 equiv. of the bis-N-oxide 5 to the two solutions and, after 30 s, we reanalysed them using 1H NMR spectroscopy. The encapsulated 6 and 8 were completely replaced by 5, and the 5⊂[2·Pd]6+ CC complex was present to a 60% extent in both solutions. The signals of the protons of the released guests increased in intensity in the bulk solution. The results indicate that the 5⊂[2·Pd]6+ CC complex is thermodynamically more stable than those of guests 6 and 8. In addition, the release of the guests triggered by the uptake of 5 is fast on the human time scale (<30 s). We propose a “French doors” mechanism for the in/out exchange process.37,56 This mechanism has a low energy barrier (∼20 kcal mol−1) and involves the rotation of the four meso-aromatic substituents of the CC. It allows the release of encapsulated 6 or 8, with the simultaneous uptake of the incoming 5 without the dissociation of any of the pyridyl N-Pd(II) bonds of the host.36Conclusions
In summary, we described the efficient self-assembly of endohedrally functionalized [2·Pd]6+ CC complexes in water by inclusion of mono- and ditopic pyridyl N-oxide and aliphatic formamide guests – 5, 6 and 8–10, respectively. The precursor 24+ shows a strong aggregation tendency that is somewhat reduced in the 1:1 inclusion complexes formed with the polar guests. The complexes of [2·Pd]6+ feature six positive charges, but are still prone to aggregation in D2O solutions at r.t. An increase in temperature (333 K) or the addition of DMSO (10%) reduces this aggregation tendency. The ditopic guests (5, 8 and 9) complement the two endohedral polar binding sites of the CC and suitably fill its cavity volume. The monotopic guests (6 and 10) are bound to the C[4]P unit of the CC and only partially fill its cavity. Accordingly, we surmise that the latter complexes require the co-inclusion of a water molecule bound to the inwardly directed α-pyridyl protons. We encountered conformational selectivity in the encapsulation of bis-formamides, 8 and 9, by the [2·Pd]6+ CC: butane bis-formamide 8 is included as the (otherwise) least abundant cis,cis-conformer in the CC. Finally, we showed that guests 6 and 8 are displaced by the bis-N-oxide 5, quantitatively and rapidly on the human time scale (<30 s). This finding suggested the existence of a “French doors” mechanism for the guest exchange process. The results are rare examples of guest-induced self-assembly/emergence of CCs having polar aromatic cavities in water solution.57 The conformational selectivity displayed by the [2·Pd]6+ CC in the encapsulation of bis-formamide 8 is unprecedented.58,59
Data availability
Experimental procedures, characterization data, NMR spectra and X-ray crystal structures are provided in the ESI.†Author contributions
LE and PB designed the research. QS, LE and JDJ performed the experiments. QS, LE, JDJ and PB analysed the data. LE and PB wrote the paper with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Gobierno de España MICIN/AEI/FEDER, UE (projects CTQ2017-84319-P and CEX2019-000925-S), the CERCA Programme/Generalitat de Catalunya, and AGAUR (2017 SGR 1123) for financial support. Q. S. thanks the Chinese Research Council for a predoctoral fellowship (2017-06870013). L. E. thanks MECD for a predoctoral fellowship (FPU14/01016). We also thank Dr Eduardo C. Escudero-Adán for X-ray crystallography data, Dr Gemma Aragay for help with mass spectrometry experiments and Miss Inmaculada Sempere for synthetic assistance.Notes and references
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480–13544 CrossRef PubMed.
- M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
- T. Y. Kim, R. A. S. Vasdev, D. Preston and J. D. Crowley, Chem.–Eur. J., 2018, 24, 14878–14890 CrossRef CAS PubMed.
- L. L. K. Taylor, I. A. Riddell and M. M. J. Smulders, Angew. Chem., Int. Ed., 2019, 58, 1280–1307 CrossRef CAS PubMed.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969–984 CrossRef CAS.
- L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261–3277 RSC.
- P. M. Bogie, T. F. Miller and R. J. Hooley, Isr. J. Chem., 2019, 59, 130–139 CrossRef CAS.
- M. Tominaga, K. Suzuki, T. Murase and M. Fujita, J. Am. Chem. Soc., 2005, 127, 11950–11951 CrossRef CAS PubMed.
- Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
- M. D. Johnstone, E. K. Schwarze, J. Ahrens, D. Schwarzer, J. J. Holstein, B. Dittrich, F. M. Pfeffer and G. H. Clever, Chem.–Eur. J., 2016, 22, 10791–10795 CrossRef CAS PubMed.
- X. Schaapkens, E. O. Bobylev, J. N. H. Reek and T. J. Mooibroek, Org. Biomol. Chem., 2020, 18, 4734–4738 RSC.
- H. Dasary, R. Jagan and D. K. Chand, Inorg. Chem., 2018, 57, 12222–12231 CrossRef CAS PubMed.
- X. Schaapkens, J. H. Holdener, J. Tolboom, E. O. Bobylev, J. N. H. Reek and T. J. Mooibroek, ChemPhysChem, 2021, 22, 1187–1192 CrossRef CAS PubMed.
- R. Custelcean, J. Bosano, P. V. Bonnesen, V. Kertesz and B. P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025–4029 CrossRef CAS PubMed.
- R. Custelcean, P. V. Bonnesen, N. C. Duncan, X. Zhang, L. A. Watson, G. Van Berkel, W. B. Parson and B. P. Hay, J. Am. Chem. Soc., 2012, 134, 8525–8534 CrossRef CAS PubMed.
- S. Yi, V. Brega, B. Captain and A. E. Kaifer, Chem. Commun., 2012, 48, 10295–10297 RSC.
- M. C. Young, L. R. Holloway, A. M. Johnson and R. J. Hooley, Angew. Chem., Int. Ed., 2014, 53, 9832–9836 CrossRef CAS PubMed.
- L. R. Holloway, P. M. Bogie, Y. Lyon, C. Ngai, T. F. Miller, R. R. Julian and R. J. Hooley, J. Am. Chem. Soc., 2018, 140, 8078–8081 CrossRef CAS PubMed.
- C. Ngai, C. M. Sanchez-Marsetti, W. H. Harman and R. J. Hooley, Angew. Chem., Int. Ed., 2020, 59, 23505–23509 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez, L. Guy and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 6574–6577 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez and J. R. Nitschke, Angew. Chem., Int. Ed., 2018, 57, 3717–3721 CrossRef CAS PubMed.
- L. Escobar, D. Villarón, E. C. Escudero-Adán and P. Ballester, Chem. Commun., 2019, 55, 604–607 RSC.
- Z. Yan, S. Xia, M. Gardlik, W. Seo, V. Maslak, J. Gallucci, C. M. Hadad and J. D. Badjić, Org. Lett., 2007, 9, 2301–2304 CrossRef CAS PubMed.
- S. Rieth, Z. Yan, S. Xia, M. Gardlik, A. Chow, G. Fraenkel, C. M. Hadad and J. D. Badjić, J. Org. Chem., 2008, 73, 5100–5109 CrossRef CAS PubMed.
- S. Stojanović, D. A. Turner, C. M. Hadad and J. D. Badjić, Chem. Sci., 2011, 2, 752–759 RSC.
- F.-U. Rahman, Y.-s. Li, I. D. Petsalakis, G. Theodorakopoulos, J. Rebek and Y. Yu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17648–17653 CrossRef CAS PubMed.
- M. Petroselli, V. Angamuthu, F.-U. Rahman, X. Zhao, Y. Yu and J. Rebek, J. Am. Chem. Soc., 2020, 142, 2396–2403 CrossRef CAS PubMed.
- F.-U. Rahman, J.-M. Yang, Y.-H. Wan, H.-B. Zhang, I. D. Petsalakis, G. Theodorakopoulos, J. Rebek and Y. Yu, Chem. Commun., 2020, 56, 6945–6948 RSC.
- Y.-H. Wan, F.-U. Rahman, J. Rebek Jr and Y. Yu, Chin. J. Chem., 2021, 39, 1498–1502 CrossRef CAS.
- H.-B. Zhang, K. Kanagaraj, J. Rebek and Y. Yu, J. Org. Chem., 2021, 86, 8873–8881 CrossRef CAS PubMed.
- S. Peng, Q. He, G. I. Vargas-Zúñiga, L. Qin, I. Hwang, S. K. Kim, N. J. Heo, C.-H. Lee, R. Dutta and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 865–907 RSC.
- S. Mecozzi and J. Rebek Jr, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- L. Escobar, E. C. Escudero-Adán and P. Ballester, Angew. Chem., Int. Ed., 2019, 58, 16105–16109 CrossRef CAS PubMed.
- K. N. Houk, K. Nakamura, C. Sheu and A. E. Keating, Science, 1996, 273, 627–629 CrossRef CAS PubMed.
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS PubMed.
- M. A. Beatty and F. Hof, Chem. Soc. Rev., 2021, 50, 4812–4832 RSC.
- J. Dong and A. P. Davis, Angew. Chem., Int. Ed., 2021, 60, 8035–8048 CrossRef CAS PubMed.
- L. Escobar and P. Ballester, Org. Chem. Front., 2019, 6, 1738–1748 RSC.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- N. E. Ernst and B. C. Gibb, in Supramolecular Chemistry in Water, ed. S. Kubik, Wiley-VCH, 2019, pp. 1–33 Search PubMed.
- L. Avram and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586–602 RSC.
- Y. Cohen, S. Slovak and L. Avram, Chem. Commun., 2021, 57, 8856–8884 RSC.
- B. Htan, D. Luo, C. Ma, J. Zhang and Q. Gan, Cryst. Growth Des., 2019, 19, 2862–2868 CrossRef.
- Q. Sun, L. Escobar and P. Ballester, Angew. Chem., Int. Ed., 2021, 60, 10359–10365 CrossRef CAS PubMed.
- L. Escobar, A. Díaz-Moscoso and P. Ballester, Chem. Sci., 2018, 9, 7186–7192 RSC.
- Y.-S. Li, L. Escobar, Y.-J. Zhu, Y. Cohen, P. Ballester, J. Rebek and Y. Yu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19815–19820 CrossRef CAS PubMed.
- J. J. Rebek, Chem. Commun., 2007, 2777–2789 RSC.
- K.-D. Zhang, D. Ajami and J. Rebek, J. Am. Chem. Soc., 2013, 135, 18064–18066 CrossRef CAS PubMed.
- S. Liu, D. H. Russell, N. F. Zinnel and B. C. Gibb, J. Am. Chem. Soc., 2013, 135, 4314–4324 CrossRef CAS PubMed.
- L. Escobar, Y.-S. Li, Y. Cohen, Y. Yu, J. Rebek Jr and P. Ballester, Chem.–Eur. J., 2020, 26, 8220–8225 CrossRef CAS PubMed.
- M. M. J. Smulders and J. R. Nitschke, Chem. Sci., 2012, 3, 785–788 RSC.
- S. Akine and Y. Sakata, Chem. Lett., 2020, 49, 428–441 CrossRef CAS.
- S. Rieth, K. Hermann, B.-Y. Wang and J. D. Badjić, Chem. Soc. Rev., 2011, 40, 1609–1622 RSC.
- D. Yang, L. K. S. von Krbek, L. Yu, T. K. Ronson, J. D. Thoburn, J. P. Carpenter, J. L. Greenfield, D. J. Howe, B. Wu and J. R. Nitschke, Angew. Chem., Int. Ed., 2021, 60, 4485–4490 CrossRef CAS PubMed.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Org. Chem., 2008, 73, 7132–7136 CrossRef CAS PubMed.
- H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574–578 CrossRef CAS PubMed.
- E. Botana, E. Da Silva, J. Benet-Buchholz, P. Ballester and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 198–201 CrossRef CAS PubMed.
- E. Botana, M. C. Lubinu, E. Da Silva, P. Espinet and J. de Mendoza, Chem. Commun., 2010, 46, 4752–4754 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 2093909–2093911. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03751j |
| ‡ Present address: Yangzhou University, School of Chemistry and Chemical Engineering, Yangzhou, 225002 Jiangsu, China. |
| § Present address: Ludwig-Maximilians-Universität München (LMU), Department of Chemistry, Butenandtstrasse 5-13, 81377 München, Germany. E-mail: E-mail: luis.escobar@cup.lmu.de |
| ¶ Present address: Leiden University, Leiden Institute of Chemistry, Einsteinweg 55, 2333 CC Leiden, The Netherlands. |
| || The tetra-pyridyl SAE-ligand 24+ became soluble in D2O at mM concentrations after heating the initial suspension at 50–60 °C for 1 h. |
| ** All our attempts to characterize the CC complexes in the gas-phase were unsuccessful. We only detected ion-peaks for the CC in different charged states. |
| †† The addition of ca. 10% DMSO to the r.t. D2O solution of the 5⊂[2·Pd]6+ CC complex provoked the appearance of sharp proton signals that coincided with those observed at 333 K. |
| This journal is © The Royal Society of Chemistry 2021 |