
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvothermal depolymerization and recrystallization of imine-linked two-dimensional covalent organic frameworks†
Woojung
Ji
 a,
Leslie S.
Hamachi
ac,
Anusree
Natraj
a,
Nathan C.
Flanders
a,
Rebecca L.
Li
a,
Lin X.
Chen
ab and
William R.
Dichtel
*a
a,
Leslie S.
Hamachi
ac,
Anusree
Natraj
a,
Nathan C.
Flanders
a,
Rebecca L.
Li
a,
Lin X.
Chen
ab and
William R.
Dichtel
*a
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: wdichtel@northwestern.edu
bChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA
cDepartment of Chemistry and Biochemistry, California Polytechnic State University, San Luis Obispo, California 93407, USA
First published on 16th November 2021
Abstract
Mechanistic understanding into the formation and growth of imine-linked two-dimensional (2D) covalent organic frameworks (COFs) is needed to improve their materials quality and access larger crystallite sizes, both of which limit the promise of 2D COFs and 2D polymerization techniques. Here we report a previously unknown temperature-dependent depolymerization of colloidal 2D imine-linked COFs, which offers a new means to improve their crystallinity. 2D COF colloids form at room temperature but then depolymerize when their reaction mixtures are heated to 90 °C. As the solutions are cooled back to room temperature, the 2D COFs repolymerize and crystallize with improved crystallinity and porosity, as characterized by X-ray diffraction, infrared spectroscopy and N2 porosimetry. The evolution of COF crystallinity during the solvothermal depolymerization and repolymerization processes was characterized by in situ wide angle X-ray scattering, and the concentrations of free COF monomers as a function of temperature were quantified by variable temperature 1H NMR spectroscopy. The ability of a 2D COF to depolymerize under these conditions depends on both the identity of the COF and its initial materials quality. For one network formed at room temperature (TAPB-PDA COF), a first depolymerization process is nearly complete, and the repolymerization yields materials with dramatically enhanced crystallinity and surface area. Already recrystallized materials partially depolymerize upon heating their reaction mixtures a second time. A related 2D COF (TAPB-DMTA COF) forms initially with improved crystallinity compared to TAPB-PDA COF and then partially depolymerizes upon heating. These results suggest that both high materials quality and network-dependent properties, such as interlayer interaction strength, influence the extent to which 2D COFs resist depolymerization. These findings offer a new means to recrystallize or solvent anneal 2D COFs and may ultimately inform crystallization conditions for obtaining large-area imine-linked two-dimensional polymers from solution.
Introduction
Two-dimensional (2D) covalent organic frameworks (COFs) are crystalline and permanently porous polymer networks that polymerize and crystallize into layered structures.1–4 COFs offer designed topology, pore sizes, composition and functionality,5,6 which produce materials with properties of interest for gas storage and separation,7–9 catalysis,10–13 sensing,14–17 membranes,18–21 adsorbents22,23 and organic electronic devices.24–26 Despite their promise, many of these potential applications for COFs remain limited by the quality of synthetically accessible crystals.27 To date, the isolation of large, single-crystalline COF crystallites remains rare, with reports of boron-linked 2D COFs,28 imine-linked 3D COFs,29 and a pioneering report by Wuest and coworkers of 3D COF single crystals based on the dimerization of polyfunctional nitroso compounds.30 Improving the quality of 2D imine-linked COFs remains one of the most important frontiers in 2D polymerization. Conventionally, imine-linked COFs are synthesized solvothermally at elevated temperatures (90–120 °C) in sealed reaction vials.31,32 During synthesis, heterogeneous COF mixtures are produced, which precipitate as polycrystalline particles whose average crystallite sizes are typically on the order of tens of nanometers. To address this shortcoming and access COF materials of higher quality, a deeper mechanistic understanding of 2D polymerization is needed.Previous mechanistic studies on the synthesis and activation of imine-linked 2D COFs produced notable advances in COF materials quality.31–33 Feriante et al. demonstrated that imine-linked COFs form more rapidly than previously described, and that the previously common practice of isolating and desolvating the COF product via vacuum activation often negatively impacts its bulk crystallinity and porosity.32,34 Improved imine-linked 2D COF polymerizations remain an important frontier, as nucleation and growth processes remain difficult to control and are poorly understood. Under typical solvothermal reaction conditions, imine-linked COFs precipitate rapidly,31 and this fast rate of formation presumably yields COFs with numerous defects. Indeed, in situ mechanistic studies on imine-linked COF formation showed that the imine-linked COFs first proceed through an amorphous polymer intermediate instead of a direct crystallization process noted for boron-based 2D COFs.31,35 It remains an open question whether as-yet undiscovered reaction conditions might produce high quality imine-linked COFs via nucleation–elongation processes that have been characterized for their boron-linked counterparts.28 Other strategies that rely on slowing COF polymerization rates or improving defect correction have improved COF synthesis, such as the slow addition of monomers,28 the use of nitrile solvents,35,36 and the inclusion of monofunctional modulators, such as aniline for imine-linked COFs.29,37,38 Upon exploring the role of modulators and nitrile solvents in the synthesis of 2D COF colloids, we unexpectedly found that weakly-crystalline COFs underwent temperature-dependent depolymerization upon heating. We hypothesized that amorphous polymer regions, defects, and small crystalline domains would be preferentially depolymerized over large area crystallites with perfect sheet stacking. Thus, repeated depolymerization/repolymerization cycles would preferentially eliminate defects, resulting in higher quality materials. This temperature-dependent equilibrium of imines under the reaction conditions enables this recrystallization/annealing process of COF colloids. The hydrolysis of isolated COF powders and polymerization onto a graphene-coated substrate was reported by Wei and coworkers as a means to convert COF powders into COF films.39 These results led us to explore solvothermal depolymerization and repolymerization of COFs as a potential means to better control COF formation and improve materials quality.
Here we report a process in which defective 2D imine COFs solvothermally depolymerize and recrystallize into COF materials of higher crystallinity and porosity. We employed colloidal COF conditions, which are typically about 5 or 6 times more dilute than conventional solvothermal conditions, to separate the dissolution and reformation processes of COFs during the synthesis. Analogous to the temperature-dependent hydrolysis of an imine model compound (1) under similar conditions, colloidal TAPB-PDA COF prepared at room temperature depolymerizes when heated to 90 °C. Upon cooling the solution, soluble monomers and oligomers repolymerize to provide the COF material with improved crystallinity and increased surface area, both benchmarks of COF quality. These solvothermal depolymerization and recrystallization processes result in the formation of larger and highly ordered COF crystallites, a process that is reminiscent of molecular recrystallization. The COF depolymerization and repolymerization/recrystallization processes are characterized by in situ wide-angle X-ray scattering (WAXS) and variable temperature (VT) NMR experiments, and the COF identity and materials quality were assessed by Fourier-transform infrared spectroscopy (FTIR), powder X-ray diffraction (PXRD), and N2 porosimetry. These findings give insight into imine-linked 2D COF formation, error correction, and kinetic stability, and offer a fundamentally new means to improve COF crystallinity that is analogous to molecular recrystallization.
Results and discussion
Model compound studies demonstrate the temperature-dependent equilibrium that interconverts the diimine 1 and H2O with terephthaldehyde (PDA) and two equivalents of aniline. A pure sample of 1 was dissolved in a mixture of deuterated solvents (benzonitrile-d5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O; 16:1 v/v) along with aniline-d5 (1 equiv. with respect to 1) and benzoic acid-d5 (76 equiv. with respect to 1) and heated to 90 °C in an NMR spectrometer for 17 h and then cooled to room temperature, with spectra obtained at various reaction times and temperatures (Fig. 1). In the NMR spectra, aldehyde functional groups appear as singlets around 9.8 ppm and imines as an apparent singlet at 8.25 ppm (Fig. 1b). The overall concentrations of aldehydes (from PDA and its single condensation with aniline 2) and imines (from 1 and 2) were quantified as a function of temperature and reaction time (Fig. 1a). Prior to heating, 42% of the imines of 1 dissociated to provide a 1:1.2 ratio of aldehydes (10 μM) and imines (12 μM). Upon heating to 90 °C, the aldehyde concentration increased to 16.5 μM and the imine concentration decreased to 7.5 μM, a 2.2:1 ratio (Fig. 1a). This new equilibrium was established rapidly relative to the rate of thermal equilibration in the spectrometer, and the concentrations of imine and aldehydes did not change significantly when held at 90 °C for 17 hours. Subsequently, upon cooling back to 25 °C, the equilibrium shifts towards imine formation, as the aldehyde concentration decreased to 14 μM and the imine concentration increased to 10 μM, which are close to those established at the beginning of the experiment. These findings demonstrate that the equilibrium of imine condensation and hydrolysis is temperature dependent under conditions similar to those used for 2D COF formation (see below). It is also important to note that equilibration is rapid at all temperatures studied, as evidenced by the rapid partial hydrolysis of 1 prior to heating. These studies on PDA, a monomer used often in 2D COFs, suggest that the driving force for COF formation might be similarly temperature dependent.
:D2O; 16:1 v/v) along with aniline-d5 (1 equiv. with respect to 1) and benzoic acid-d5 (76 equiv. with respect to 1) and heated to 90 °C in an NMR spectrometer for 17 h and then cooled to room temperature, with spectra obtained at various reaction times and temperatures (Fig. 1). In the NMR spectra, aldehyde functional groups appear as singlets around 9.8 ppm and imines as an apparent singlet at 8.25 ppm (Fig. 1b). The overall concentrations of aldehydes (from PDA and its single condensation with aniline 2) and imines (from 1 and 2) were quantified as a function of temperature and reaction time (Fig. 1a). Prior to heating, 42% of the imines of 1 dissociated to provide a 1:1.2 ratio of aldehydes (10 μM) and imines (12 μM). Upon heating to 90 °C, the aldehyde concentration increased to 16.5 μM and the imine concentration decreased to 7.5 μM, a 2.2:1 ratio (Fig. 1a). This new equilibrium was established rapidly relative to the rate of thermal equilibration in the spectrometer, and the concentrations of imine and aldehydes did not change significantly when held at 90 °C for 17 hours. Subsequently, upon cooling back to 25 °C, the equilibrium shifts towards imine formation, as the aldehyde concentration decreased to 14 μM and the imine concentration increased to 10 μM, which are close to those established at the beginning of the experiment. These findings demonstrate that the equilibrium of imine condensation and hydrolysis is temperature dependent under conditions similar to those used for 2D COF formation (see below). It is also important to note that equilibration is rapid at all temperatures studied, as evidenced by the rapid partial hydrolysis of 1 prior to heating. These studies on PDA, a monomer used often in 2D COFs, suggest that the driving force for COF formation might be similarly temperature dependent.
 | ||
| Fig. 1 (a) Concentration of free aldehydes (green diamonds) and combined concentrations of imines 1 and 2 (orange triangles) present in solution when 1 was subjected to a heating and cooling cycle under similar conditions used for COF formation (12 mM in [benzonitrile-d5:D2O; 16:1 v/v]; 76 equiv. benzoic acid-d5; 1 equiv. aniline-d5). (b) Representative VT-NMR spectra and the equilibrium between the monomers (PDA and aniline) and 1. The concentration of each species was calculated by integrating relevant signals against those of the internal standard, 1,4-dinitrobenzene. | ||
To investigate how the temperature-dependent nature of imine formation under these conditions might influence the 2D polymerization process, it was first necessary to polymerize TAPB-PDA COF at room temperature, where the driving force for imine-formation is relatively high. TAPB-PDA COF was synthesized at room temperature by condensing 1,3,5-tris(4-aminophenyl)benzene (TAPB) and PDA in the presence of aniline and benzoic acid in a mixture of solvents (benzonitrile:H2O; 16:1 v/v) (Fig. 2 and S8; see ESI† for detailed procedures). Within 3 hours, a yellow colloidal suspension formed, which was comprised of 2 μM-sized spheres, as determined by extraction using methanol, and activated by supercritical CO2 drying. The resulting powder product was characterized as a crystalline sample of the imine-linked TAPB-PDA COF, as confirmed by PXRD, N2 porosimetry and FT-IR spectroscopy of the isolated colloids (Fig. 2 and 5). The PXRD pattern of the COF exhibited sharp (100), (110), (200), and (210) Bragg diffraction peaks, consistent with an eclipsed model of the COF and reported powder patterns.33 FT-IR spectroscopy showed the characteristic imine stretch at 1624 cm−1, along with the absence of both the PDA aldehyde stretch at 1687 cm−1 and the TAPB amine stretches at the range of 3300–3500 cm−1 (Fig. S1†). Analysis of the N2 adsorption isotherm provided a Brunauer–Emmett–Teller (BET) surface area of 1740 m2 g−1, from which nonlocal density functional theory analysis provided a narrow pore width distribution centered at 3.4 nm (Fig. 5b and S2†). In considering the BET surface area as a metric of the TAPB-PDA COF materials quality, this value is sufficiently high to describe the material as a successfully activated 2D COF, yet lower than the theoretical value and other literature reports.33,34,36 These bulk characterization techniques collectively indicate that polymerization of TAPB-PDA COF under these conditions at room temperature provides the target material in good but not superior materials quality.
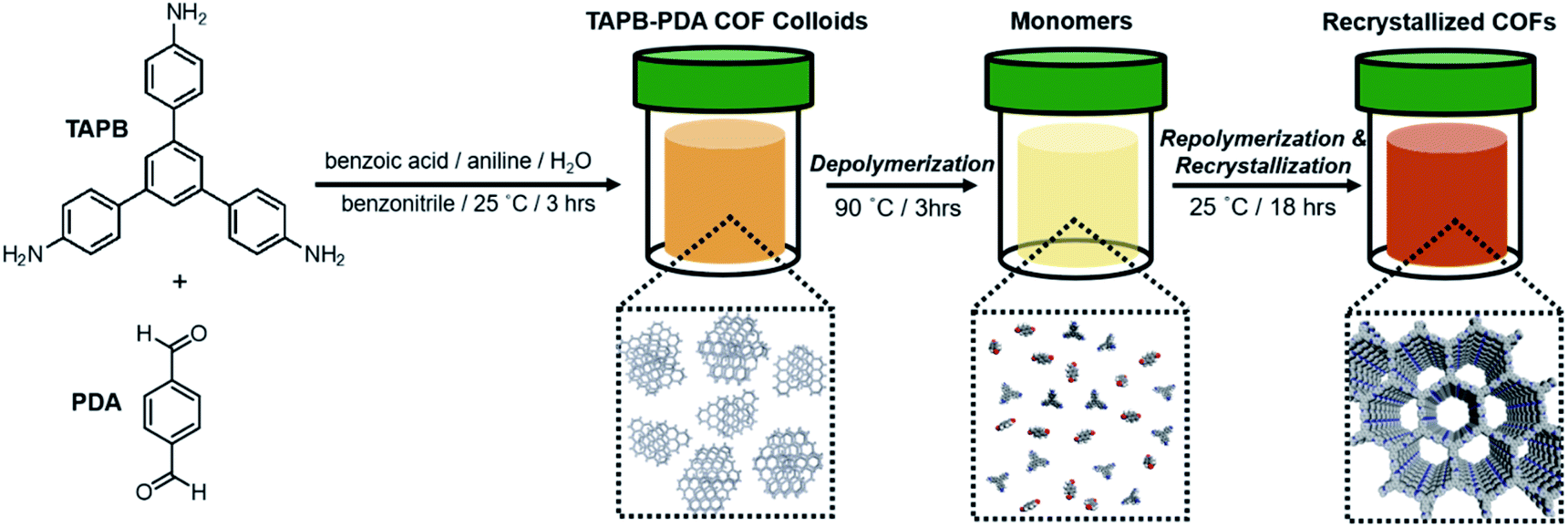 | ||
| Fig. 2 Schematic of TAPB-PDA COF recrystallization. | ||
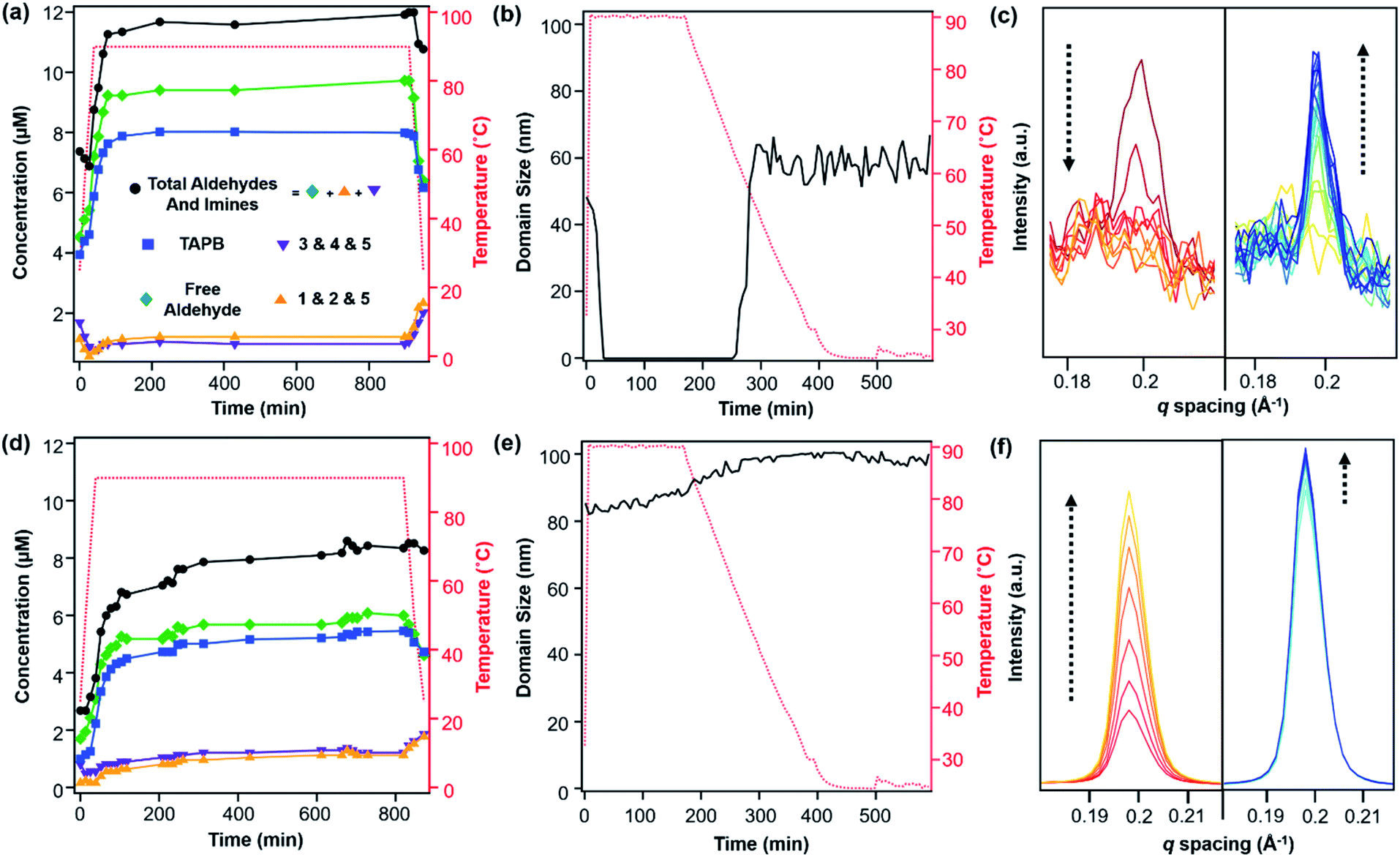
Similar to the model compound study described above, imine-linked 2D COFs show a temperature-dependent equilibrium that enables their depolymerization and repolymerization in a manner reminiscent of molecular recrystallization. The depolymerization of TAPB-PDA COF colloids prepared at room temperature was characterized by variable-temperature NMR spectroscopy, in situ wide-angle X-ray scattering, as well as product analysis following repolymerization. For the VT-NMR spectroscopy experiments,40TAPB-PDA COF was prepared as described above in the presence of deuterated solvents and reagents. The monomers were combined with aniline-d5 and benzoic acid-d5 in a mixture of deuterated solvents (benzonitrile-d5:D2O; 16:1 v/v) and heated to 90 °C in an NMR spectrometer as the temperature was varied. The overall concentrations of aldehyde functional groups (from PDA and its single condensation with aniline (2), TAPB (4), and other oligomers), as well as imine functional groups (from 1, 2, 3, 4, 5 and other oligomers, Fig. 3b) were quantified as a function of temperature and reaction time (Fig. 4a). Imines 1, 2, and 5 from the condensation between PDA and aniline appear as a single peak centered at 8.25 ppm, and imines 3, 4 and 5 from a condensation between PDA and TAPB show up as an apparent singlet at 8.4 ppm (Fig. 3a). Prior to heating, the COF reaction mixture contained approximately 61% of the free COF monomers of aldehydes (4.5 μM), TAPB (3.9 μM), and imines (2.8 μM). The other 39% was consumed in preparing the COF colloids, a form in which peaks for their protons are not observed. Within a few minutes of heating to 90 °C, the initial milky suspension turned into a clear yellow solution and remained homogeneous (Fig. 2 and S8†). Dynamic light scattering (DLS) experiments indicated the decrease in the average particle size of TAPB-PDA COF from 2.5 μM to unmeasurably small as the temperature was raised to 90 °C (Fig. S7†). At this temperature, the equilibrium shifted rapidly towards the monomers, as the aldehyde and TAPB concentrations quickly increased to 7.2 μM and 5.9 μM, respectively, and the concentrations of imine functional groups decreased to 1.5 μM. When held at 90 °C for 14.5 hours, there was an absence of a Tyndall effect associated with the presence of colloids in solution and the concentrations of both aldehydes and imines, and TAPB further increased to 12 μM and 8 μM, respectively, corresponding to depolymerization of COFs into monomers and oligomers (Fig. 4a). Upon cooling back to 25 °C, the equilibrium shifted back towards imine and COF formation, as the aldehyde and TAPB concentrations rapidly decreased to 6.4 μM and 6.2 μM, respectively, and the total imine concentrations increased to 4.4 μM. After 18 hours at room temperature, the concentrations of aldehyde, TAPB and imines all decreased to 1.5 μM, 0.9 μM, and 0.9 μM, respectively, which suggest further repolymerization overnight (Fig. 4d). Overall, TAPB-PDA COF colloids were observed to quantitatively depolymerize at 90 °C and slowly repolymerize at 25 °C. Taken together, these findings demonstrate that the driving force for COF formation is highly temperature dependent, similar to the established equilibrium between diamine 1 and PDA.
 | ||
| Fig. 3 (a) Representative VT-NMR spectra during the solvothermal depolymerization of TAPB-PDA COF. (b) Equilibrium between the TAPB-PDA COF, imines 1, 2, 3, 4 and 5, and monomers (TAPB and PDA) during the solvothermal depolymerization and recrystallization processes. | ||
 | ||
| Fig. 4 Concentration of TAPB (blue squares) and free aldehyde (green diamonds), and combined concentrations of imines 1, 2 and 5 (orange triangles), 3, 4 and 5 (purple triangles), and aldehyde and imines (black circles) present in solution during the (a) first and (d) second cycle of TAPB-PDA COF recrystallization. Change in average domain sizes of TAPB-PDA COF during the (b) first and (e) second cycle of COF recrystallization. Domain sizes are determined from the Scherrer analysis of the (100) Bragg diffraction peak. Representative WAXS patterns of TAPB-PDA COF, centered at its (100) diffraction peak, upon heating (red) and cooling (blue) during the (c) first and (f) second cycle of recrystallization. | ||
In situ WAXS of TAPB-PDA COF obtained during the solvothermal depolymerization and repolymerization processes were consistent with depolymerization at an elevated temperature, followed by the formation of a more crystalline material upon repolymerization (Fig. 4b). A colloidal suspension of TAPB-PDA COF formed at room temperature was heated to 90 °C in a capillary for 3 hours and cooled to room temperature for 7 hours, with WAXS spectra obtained at various reaction times. The average crystalline domain size of the COF was determined by applying the Scherrer equation to the full width at half max (FWHM) of the (100) Bragg diffraction peak at q = 0.20 Å−1 (Fig. 4c).36 Prior to heating, the COF has an initial domain size of 44 nm. As the sample was held at 90 °C for 3 hours, the (100) diffraction peak decreases in intensity and eventually disappears completely, consistent with depolymerization. Upon cooling to room temperature over 7 h, the domain size increases to 64 nm, approximately 20 nm higher than the initial COF domain size. After sitting overnight at room temperature, the COF domain size increased to 85 nm, indicating further crystallization at room temperature (Fig. 4e). Overall, the changes in the crystallinity and domain sizes of TAPB-PDA COF suggest that the COFs initially formed at room temperature depolymerize at 90 °C and repolymerize/recrystallize into higher quality COFs upon cooling (Fig. 5).
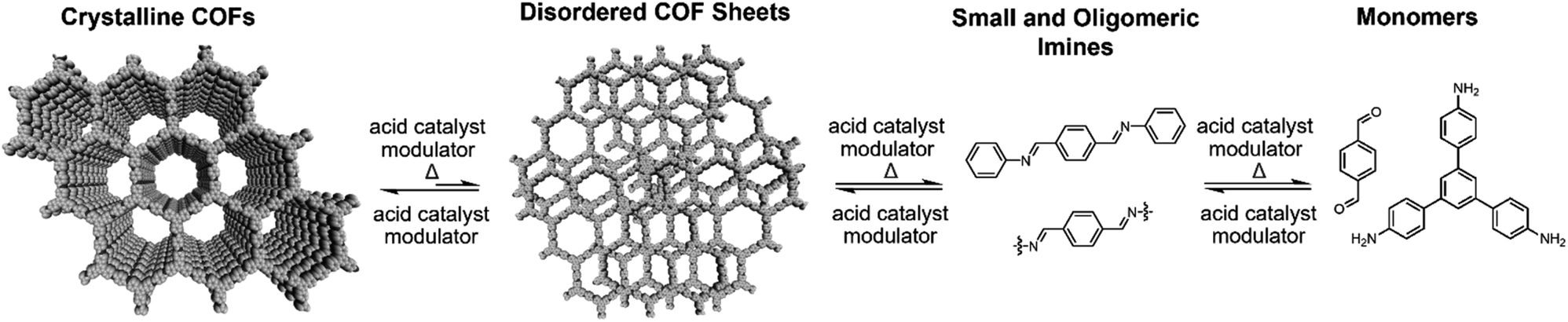 | ||
| Fig. 5 Schematic representations of proposed COF recrystallization mechanisms. | ||
Recrystallized TAPB-PDA COF exhibited enhanced crystallinity and porosity compared to the COF colloids prepared at room temperature, as characterized by PXRD and N2 adsorption isotherms (Fig. 6a and b). Recrystallized TAPB-PDA COF after isolation exhibited much sharper and more well-defined (100), (110), (200), (210) Bragg diffraction peaks than the initial COF colloids, accompanied by the appearance of higher order diffraction peaks corresponding to the (220), (320) and (001) crystalline planes. These recrystallized materials also exhibited improved porosity, as BET surface area increased from 1740 m2 g−1 to 2790 m2 g−1 (Fig. 6b). The IR spectrum of the recrystallized COF was identical to that of the initial COF colloids, with the presence of imine stretch at 1624 cm−1, and the absence of both the aldehyde stretch at 1687 cm−1 and the amine stretches at the range of 3300–3500 cm−1 (Fig. S1†). Similar observations and materials quality were obtained by cooling the solution more slowly (∼4 °C hour−1). In addition, when separate solutions of the two monomers were heated to 90 °C, combined, and cooled, the resulting COF products were of similar crystallinity and porosity as those obtained by recrystallizing the COFs themselves (Fig. S9†). Overall, the crystallinity and porosity of TAPB-PDA COF were improved after recrystallization, consistent with the increased domain sizes observed from in situ WAXS analysis (Fig. 4b and e).
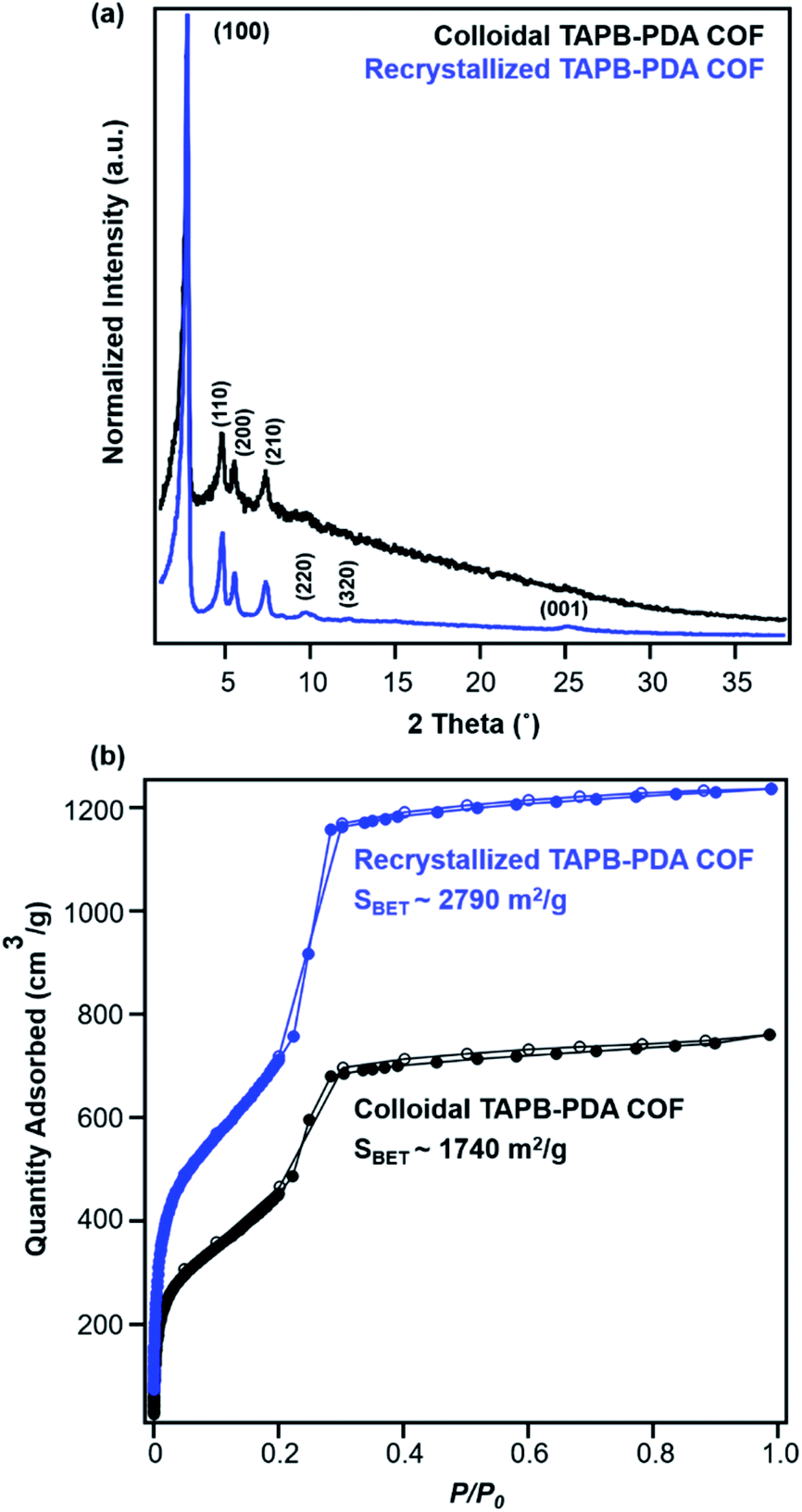 | ||
| Fig. 6 (a) PXRD patterns and (b) N2 adsorption isotherms of TAPB-PDA COF before (black) and after (blue) the first cycle of COF recrystallization. | ||
After one depolymerization/repolymerization cycle, TAPB-PDA COF samples will partially depolymerize again upon reheating to 90 °C, but a smaller percentage of the imine bonds are hydrolyzed (Fig. 4d–f and 5). After repolymerization, the reaction solution contains 22% of molecular species in the forms of aldehydes (1.7 μM), TAPB (1.0 μM), and imines (1.0 μM). Upon reheating to 90 °C, the aldehyde and TAPB concentrations increase to 3.1 μM and 2.2 μM, respectively, and the imine concentrations decrease to 0.8 μM, all of which are approximately 13% lower than those observed during the first cycle of COF recrystallization. When held at 90 °C for 10.6 hours, the concentrations of both aldehydes and imines, and TAPB increased to 8.6 μM and 5.4 μM, respectively, corresponding to 72% of the available monomers being present in solution rather than incorporated into the COF (Fig. 4d). Simultaneously, in situ WAXS analysis of this solution indicated that the average domain size of the COF network that remained had increased from 82 nm to 104 nm when held at 90 °C (Fig. 4e and f). These observations suggest that the 2D COF's initial crystallinity make it more or less susceptible to this solvothermal depolymerization process. Defect-rich COFs, such as the initially synthesized TAPB-PDA COF colloids, are more susceptible to depolymerization; whereas more crystalline samples of the same material, including the recrystallized TAPB-PDA COF, are less susceptible (Fig. 5). These observations may form the basis of procedures to solvent-anneal or otherwise upgrade the materials quality of COFs. However, it is important to note that further explorations of these conditions are needed, as the bulk crystallinity and porosity of the twice-recrystallized TAPB-PDA COF were similar to those of the samples isolated after one recrystallization cycle. These observations suggest that the repolymerization process may proceed through the nucleation of new COFs instead of the monomers adding exclusively to the residual COF crystallites. Together, these results suggest that lower quality or smaller COF crystallites are more susceptible to depolymerization than larger, more well ordered 2D COF domains.
We sought to extend this observed depolymerization/repolymerization method to a related imine-linked 2D COF, derived from 2,5-dimethoxyterephthaldehyde (DMTA), and observed different degrees of depolymerization but a similar dependence on materials quality (Fig. 7). The initial formation of TAPB-DMTA COF colloids provides more highly crystalline samples that were isolated and activated, providing a higher BET surface area (2250 m2 g−1) as compared to the initially isolated TAPB-PDA COF colloids (1740 m2 g−1) prepared under the same conditions. The TAPB-DMTA COF colloids also exhibited superior crystallinity compared to the TAPB-PDA COF colloids, as determined by their sharper Bragg diffraction peaks and through Scherer analysis (Fig. 7e and f). The TAPB-DMTA COF colloids underwent partial depolymerization upon heating to 90 °C in the presence of benzoic acid, aniline, and water. Heating the TAPB-DMTA COF colloids in its reaction mixture to 90 °C for 3 hours and cooling to room temperature yielded recrystallized TAPB-DMTA COF, which exhibited enhanced crystallinity and porosity with a modest increase in BET surface area from 2250 m2 g−1 to 2770 m2 g−1 (Fig. 7e and f). Prior to heating, the colloidal TAPB-DMTA COF reaction mixture contained free COF monomers of DMTA (0.12 μM) and TAPB (1.0 μM) (Fig. 7a). Upon heating to 90 °C, approximately 36% of the COF depolymerized to aldehydes (2.9 μM), TAPB (1.9 μM) and imines (1.5 μM), which is much lower compared to 94% depolymerization for the room-temperature synthesized TAPB-PDA COF colloids. When held at 90 °C for 16.5 hours, however, the concentrations of both aldehydes and imines, and TAPB slowly decreased to 2.3 μM and 1.6 μM, respectively, indicating repolymerization of TAPB-DMTA COF even at this elevated temperature. Simultaneously, in situ WAXS analysis of this solution clearly showed TAPB-DMTA COF colloids are highly crystalline at 90 °C with domain sizes exceeding 80 nm (Fig. 7b and c). These results are in stark contrast to the continuous solvothermal depolymerization of TAPB-PDA COF observed during the first and second cycles of COF recrystallization (Fig. 4b and d). We hypothesize that the larger crystalline domains and/or stronger interlayer interactions of TAPB-DMTA COF10,41 compared to TAPB-PDA COF are responsible for this material's increased ability to resist depolymerization. Moreover, for both networks, we speculate that less ordered regions of the materials are more susceptible to hydrolysis under these conditions, which explains why materials of improved crystallinity depolymerize to lesser extents. Going forward, we anticipate that this type of molecular approach to probe COF formation and growth mechanisms will shed new insight on developing better reaction conditions and improving the qualities of COFs. Specifically, studying the temperature dependent reversibility equilibria for various monomer pairs may be able to provide better understanding as to why some COF structures are better at crystallizing than others.
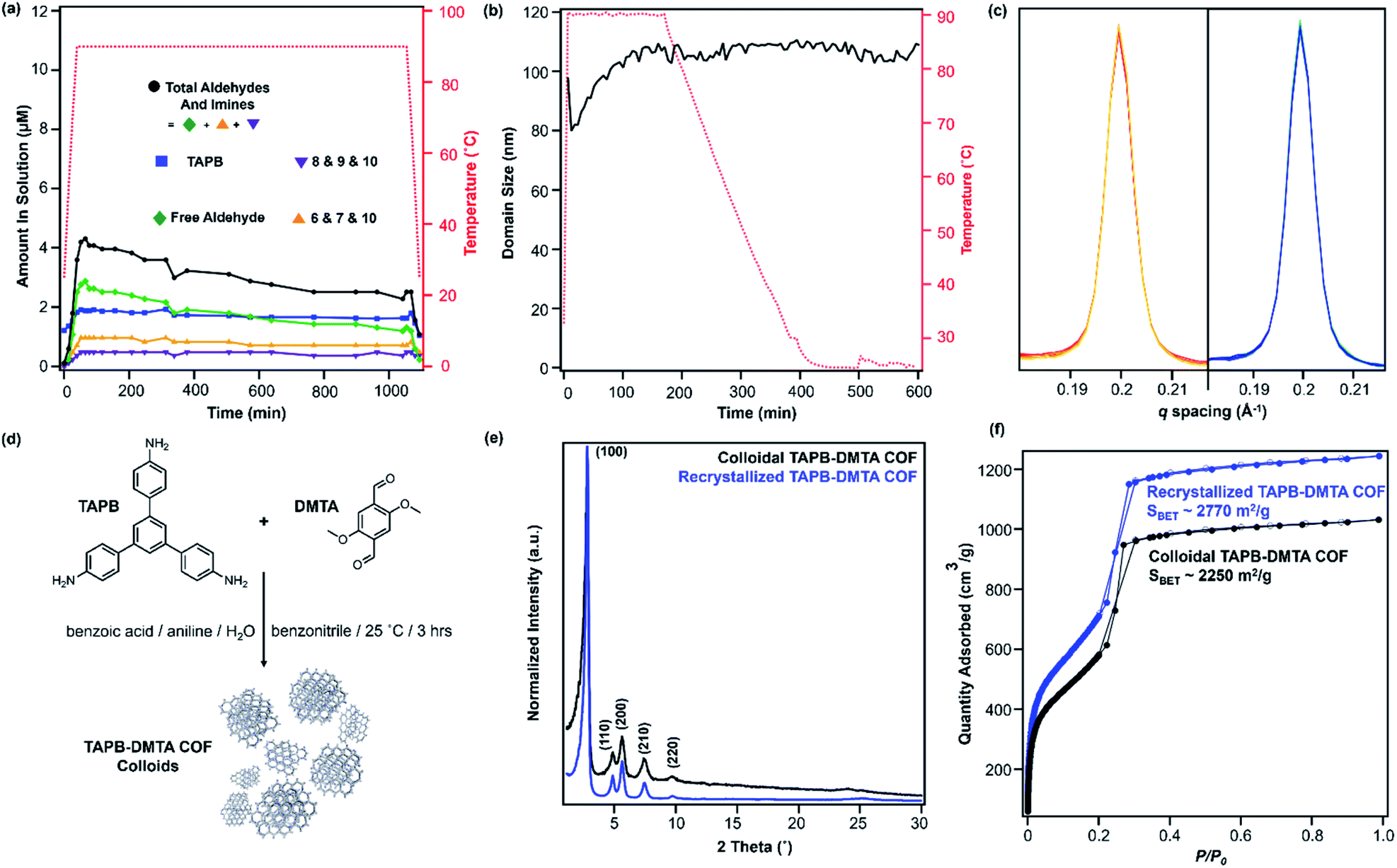 | ||
| Fig. 7 (a) Concentration of TAPB (blue squares) and free aldehyde (green diamonds), and combined concentrations of imines 6, 7 and 10 (orange triangles), 8, 9 and 10 (purple triangles), and aldehyde and imines (black circles) present in solution during the first cycle of TAPB-DMTA COF recrystallization. (b) Change in average domain sizes of TAPB-DMTA COF during the first cycle of COF recrystallization. Average domain sizes are determined from the Scherrer analysis of the (100) Bragg diffraction peak. (c) Representative WAXS patterns of TAPB-DMTA COF, centered at its (100) diffraction peak, upon heating (red) and cooling (blue) during the first cycle of recrystallization. (d) Synthesis of TAPB-DMTA COF colloids (e) PXRD patterns and (f) N2 adsorption isotherms of TAPB-DMTA COF before (black) and after (blue) the first cycle of COF recrystallization. | ||
Conclusions
We identified a previously unknown, temperature-based solvothermal equilibrium of imine condensation and repolymerization/recrystallization process for 2D imine-linked COFs. Mirroring the temperature-dependent hydrolysis of the soluble imine-containing model compound 1, the 2D COFs depolymerize in their polymerization solutions and recrystallize into higher quality COF materials upon cooling. In situ WAXS and VT-NMR experiments reported on the crystallinity and extent of depolymerization of the COFs in solution at various temperatures. Both are powerful, quantitative techniques, used here to probe COF formation mechanisms and improve the materials quality. PXRD, FTIR and N2 adsorption isotherms of the isolated materials were used to assess their structure and porosity before and after recrystallization. While TAPB-PDA COF colloids first prepared at room temperature fully depolymerize to soluble monomers and oligomers at 90 °C, recrystallized TAPB-PDA COF colloids with larger domain sizes (>80 nm) only partially depolymerize under the same conditions. Likewise, TAPB-DMTA COF is initially formed as a more crystalline network, and it also depolymerizes to a lesser extent. These observations suggest that larger and more ordered COF crystallites are more resistant to solvothermal depolymerization. Taken together, this study reports a series of novel phenomena in which 2D imine COFs undergo temperature-dependent depolymerization and recrystallization. These findings provide a new means to improve 2D COF materials quality from as-synthesized materials, as well as intriguing insight into their formation that may inform improved 2D polymerizations in the future.Data availability
Data are available upon request from the authors.Author contributions
W. J., L. S. H., A. N., and W. R. D. performed and interpreted the COF growth and characterizations. W. J., L. S. H., and W. R. D. performed and interpreted the VT NMR experiments. W. J., L. S. H., N. C. F., R. L. L., L. X. C., and W. R. D. performed and interpreted the in situ X-ray diffraction experiments. All authors wrote and revised the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the Army Research Office for a Multidisciplinary University Research Initiatives (MURI) award under grant number W911NF-15-1-0447. Parts of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by Northwestern University, E.I. DuPont de Nemours & Co., and the Dow Chemical Company. This research used resources of the Advanced Photon Source and Center for Nanoscale Materials, both U.S. Department of Energy (DOE) Office of Science User Facilities operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC0206CH11357. This work has also made use of the IMSERC, EPIC, and Keck II facility of NUANCE Center at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS1542205), the MRSEC program (NSF DMR-1720139) at the Materials Research Center, the Keck Foundation, the State of Illinois and International Institute for Nanotechnology (IIN). N. C. F. and L. X. C. are partially supported by Basic Energy Science, CBG Division, US Department of Energy through Argonne National Laboratory under Contract No. DE-AC0206CH11357.Notes and references
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS
.
- S. B. Alahakoon, S. D. Diwakara, C. M. Thompson and R. A. Smaldone, Chem. Soc. Rev., 2020, 49, 1344–1356 RSC
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 RSC
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed
- H. Ma, H. Ren, S. Meng, Z. Yan, H. Zhao, F. Sun and G. Zhu, Chem. Commun., 2013, 49, 9773–9775 RSC
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed
- R. Chen, J.-L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed
- Y. Wang, H. Liu, Q. Pan, C. Wu, W. Hao, J. Xu, R. Chen, J. Liu, Z. Li and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 5958–5963 CrossRef CAS PubMed
- Z. Li, N. Huang, K. H. Lee, Y. Feng, S. Tao, Q. Jiang, Y. Nagao, S. Irle and D. Jiang, J. Am. Chem. Soc., 2018, 140, 12374–12377 CrossRef CAS PubMed
- X. Wu, X. Han, Q. Xu, Y. Liu, C. Yuan, S. Yang, Y. Liu, J. Jiang and Y. Cui, J. Am. Chem. Soc., 2019, 141, 7081–7089 CrossRef CAS PubMed
- C. Yuan, X. Wu, R. Gao, X. Han, Y. Liu, Y. Long and Y. Cui, J. Am. Chem. Soc., 2019, 141, 20187–20197 CrossRef CAS PubMed
- S. Jhulki, A. M. Evans, X.-L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam, S. Barlow, J.-L. Brédas, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 783–791 CrossRef CAS PubMed
- D. W. Burke, C. Sun, I. Castano, N. C. Flanders, A. M. Evans, E. Vitaku, D. C. McLeod, R. H. Lambeth, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Angew. Chem., Int. Ed., 2020, 59, 5165–5171 CrossRef CAS PubMed
- A. R. Corcos, G. A. Levato, Z. Jiang, A. M. Evans, A. G. Livingston, B. J. Mariñas and W. R. Dichtel, ACS Mater. Lett., 2019, 1, 440–446 CrossRef CAS
- H. Fan, A. Mundstock, A. Feldhoff, A. Knebel, J. Gu, H. Meng and J. Caro, J. Am. Chem. Soc., 2018, 140, 10094–10098 CrossRef CAS PubMed
- H. S. Sasmal, H. B. Aiyappa, S. N. Bhange, S. Karak, A. Halder, S. Kurungot and R. Banerjee, Angew. Chem., Int. Ed., 2018, 57, 10894–10898 CrossRef CAS PubMed
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed
- W. Ji, L. Xiao, Y. Ling, C. Ching, M. Matsumoto, R. P. Bisbey, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2018, 140, 12677–12681 CrossRef CAS PubMed
- M. Wang, M. Ballabio, M. Wang, H.-H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Cánovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2019, 141, 16810–16816 CrossRef CAS PubMed
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed
- S. Jhulki, J. Kim, I.-C. Hwang, G. Haider, J. Park, J. Y. Park, Y. Lee, W. Hwang, A. A. Dar, B. Dhara, S. H. Lee, J. Kim, J. Y. Koo, M. H. Jo, C.-C. Hwang, Y. H. Jung, Y. Park, M. Kataria, Y.-F. Chen, S.-H. Jhi, M.-H. Baik, K. Baek and K. Kim, Chem, 2020, 6, 2035–2045 CAS
- R. W. Day, D. K. Bediako, M. Rezaee, L. R. Parent, G. Skorupskii, M. Q. Arguilla, C. H. Hendon, I. Stassen, N. C. Gianneschi, P. Kim and M. Dincă, ACS Cent. Sci., 2019, 5, 1959–1964 CrossRef CAS
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52 CrossRef CAS PubMed
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48 CrossRef CAS
- D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830–834 CrossRef CAS PubMed
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Chem. Commun., 2016, 52, 3690–3693 RSC
- C. Feriante, A. M. Evans, S. Jhulki, I. Castano, M. J. Strauss, S. Barlow, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 18637–18644 CrossRef CAS
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS
- C. H. Feriante, S. Jhulki, A. M. Evans, R. R. Dasari, K. Slicker, W. R. Dichtel and S. R. Marder, Adv. Mater., 2020, 32, 1905776 CrossRef CAS PubMed
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 58–65 CrossRef CAS PubMed
- R. L. Li, N. C. Flanders, A. M. Evans, W. Ji, I. Castano, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Chem. Sci., 2019, 10, 3796–3801 RSC
- I. Castano, A. M. Evans, H. Li, E. Vitaku, M. J. Strauss, J.-L. Brédas, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2019, 5, 1892–1899 CrossRef CAS
- S. Wang, Z. Zhang, H. Zhang, A. G. Rajan, N. Xu, Y. Yang, Y. Zeng, P. Liu, X. Zhang, Q. Mao, Y. He, J. Zhao, B.-G. Li, M. S. Strano and W.-J. Wang, Matter, 2019, 1, 1592–1605 CrossRef
- L. Yang, Q. Guo, H. Kang, R. Chen, Y. Liu and D. Wei, Chem. Mater., 2020, 32, 5634–5640 CrossRef CAS
- L. S. Hamachi, I. Jen-La Plante, A. C. Coryell, J. De Roo and J. S. Owen, Chem. Mater., 2017, 29, 8711–8719 CrossRef CAS
- S. Wang, A. D. Chavez, S. Thomas, H. Li, N. C. Flanders, C. Sun, M. J. Strauss, L. X. Chen, A. J. Markvoort, J.-L. Bredas and W. R. Dichtel, Chem. Mater., 2019, 31, 7104–7111 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional characterization. See DOI: 10.1039/d1sc03963f |
| This journal is © The Royal Society of Chemistry 2021 |