
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasticizer and catalyst co-functionalized PEDOT:PSS enables stretchable electrochemical sensing of living cells†
Jing
Yan
a,
Yu
Qin
a,
Wen-Ting
Fan
a,
Wen-Tao
Wu
a,
Song-Wei
Lv
b,
Li-Ping
Yan
a,
Yan-Ling
Liu
 *a and
Wei-Hua
Huang
a
*a and
Wei-Hua
Huang
a
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: yanlingliu@whu.edu.cn; whhuang@whu.edu.cn
bSchool of Pharmacy, Changzhou University, Changzhou 213164, China
First published on 12th October 2021
Abstract
Recently, stretchable electrochemical sensors have stood out as a powerful tool for the detection of soft cells and tissues, since they could perfectly comply with the deformation of living organisms and synchronously monitor mechanically evoked biomolecule release. However, existing strategies for the fabrication of stretchable electrochemical sensors still face with huge challenges due to scarce electrode materials, demanding processing techniques and great complexity in further functionalization. Herein, we report a novel and facile strategy for one-step preparation of stretchable electrochemical biosensors by doping ionic liquid and catalyst into a conductive polymer (poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate), PEDOT:PSS). Bis(trifluoromethane) sulfonimide lithium salt as a small-molecule plasticizer can significantly improve the stretchability and conductivity of the PEDOT:PSS film, and cobalt phthalocyanine as an electrocatalyst endows the film with excellent electrochemical sensing performance. Moreover, the functionalized PEDOT:PSS retained good cell biocompatibility with two extra dopants. These satisfactory properties allowed the real-time monitoring of stretch-induced transient hydrogen peroxide release from cells. This work presents a versatile strategy to fabricate conductive polymer-based stretchable electrodes with easy processing and excellent performance, which benefits the in-depth exploration of sophisticated life activities by electrochemical sensing.
Introduction
Cells in the body are constantly exposed to mechanical forces and could perceive and transduce them into biochemical signals.1–6 Currently, cell mechanotransduction has gained tremendous attention due to the crucial role of mechanical forces in cell function, and the accurate characterization of the ensuing biochemical signals during the mechanotransduction process is vitally important for understanding cellular mechanical signalling.2–5 Since cell mechanotransduction involves rapid mechanochemical conversion (within a second),3 soft stretchable electrochemical sensors with a fast response and high sensitivity have emerged as a powerful technique to induce the mechanical deformation of cells and simultaneously monitor the transient biochemical response in real time.7–11 Recently, stretchable electrochemical sensors have made significant progress in exploring dynamic mechanotransduction, and mechanically evoked molecules (e.g. hydrogen peroxide (H2O2), nitric oxide (NO) and serotonin) release from stretched endothelial cells and inflated intestine were successfully monitored.9,12–15 This notable advance is of great benefit to study the mechanism of force-activated signalling in mechanotransduction.As for the fabrication of stretchable electrodes, most of the reported sensors were based on well-designed zero- or one-dimensional nanomaterials. Zero-dimensional nanomaterials with high density, such as agglomerated Au nanoparticles,16 could fill stretch-evoked interspaces between the particles to retain continuous electronic pathways. One-dimensional nanomaterials, such as Au nanowires,17 Au nanotubes7,18 or carbon nanotubes (CNTs),9,19,20 could accommodate the applied strain by sliding and rotating against each other. However, the available materials that could be employed to construct stretchable electrodes are still very lacking, and the fabrication of stretchable electrodes usually involves demanding and time-consuming processes to in situ synthesize these engineered nanomaterials.7,16,17 Moreover, to monitor the very weak biochemical signals in the primary mechanotransduction, stretchable electrodes usually need further functionalization to improve their performance, such as sensitivity, selectivity or stretchability.9,13,15,19 Therefore, stretchable electrochemical sensors face with great challenges due to the limitation of both electrode materials and processing techniques, and facile and versatile fabrication strategies are urgently needed to advance their applications.
By virtue of high conductivity and good electrochemical performance, the conductive polymer poly(3,4-ethylenedioxythiophene) (PEDOT) commonly dispersed in poly(styrene sulfonate) (PSS) aqueous solution has been extensively used as an electrode material.21–24 However a low fracture strain (∼5%) hinders the direct application of PEDOT:PSS film in stretchable electrodes. Many recent efforts have been devoted to increasing the stretchability by blending PEDOT:PSS with various additives, including elastomeric polymers and small-molecule plasticizers.25–27 Generally, polymeric blends lead to worse conductivity due to the addition of insulating polymers.28,29 Although small-molecule plasticizers (e.g. Zonyl30 and Triton X-100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31) could enhance both the elastic modulus and conductivity, the leaching of these toxic additives posed a threat to biological systems. Very recently, the doping of ionic liquids into PEDOT:PSS has achieved the coexistence of high stretchability and conductivity as well as satisfactory biocompatibility.32,33 However, no impressive progress in stretchable electrodes based on PEDOT:PSS for electrochemical biosensing has been witnessed so far.
31) could enhance both the elastic modulus and conductivity, the leaching of these toxic additives posed a threat to biological systems. Very recently, the doping of ionic liquids into PEDOT:PSS has achieved the coexistence of high stretchability and conductivity as well as satisfactory biocompatibility.32,33 However, no impressive progress in stretchable electrodes based on PEDOT:PSS for electrochemical biosensing has been witnessed so far.
Herein, we report a facile method to fabricate a PEDOT-based stretchable electrochemical biosensor for the first time. The functionalized polymer was obtained by straightforwardly incorporating bis(trifluoromethane) sulfonimide lithium salt (LiTFSI) and cobalt phthalocyanine (CoPc) into PEDOT:PSS solution via simply mixing and stirring, forming the complex PEDOT:PSS–LiTFSI–CoPc (PPLC) (Scheme 1A). LiTFSI significantly improved the stretchability and conductivity of the PEDOT:PSS film, and CoPc endowed the film with excellent electrocatalytic property toward the oxidation of H2O2 (a signaling molecule in diverse biological processes34,35). As a proof-of-concept, the stretchable PPLC/PDMS sensor was used to in situ load strains and simultaneously monitor mechanically induced H2O2 release from human bronchial epithelial cells (Scheme 1B).
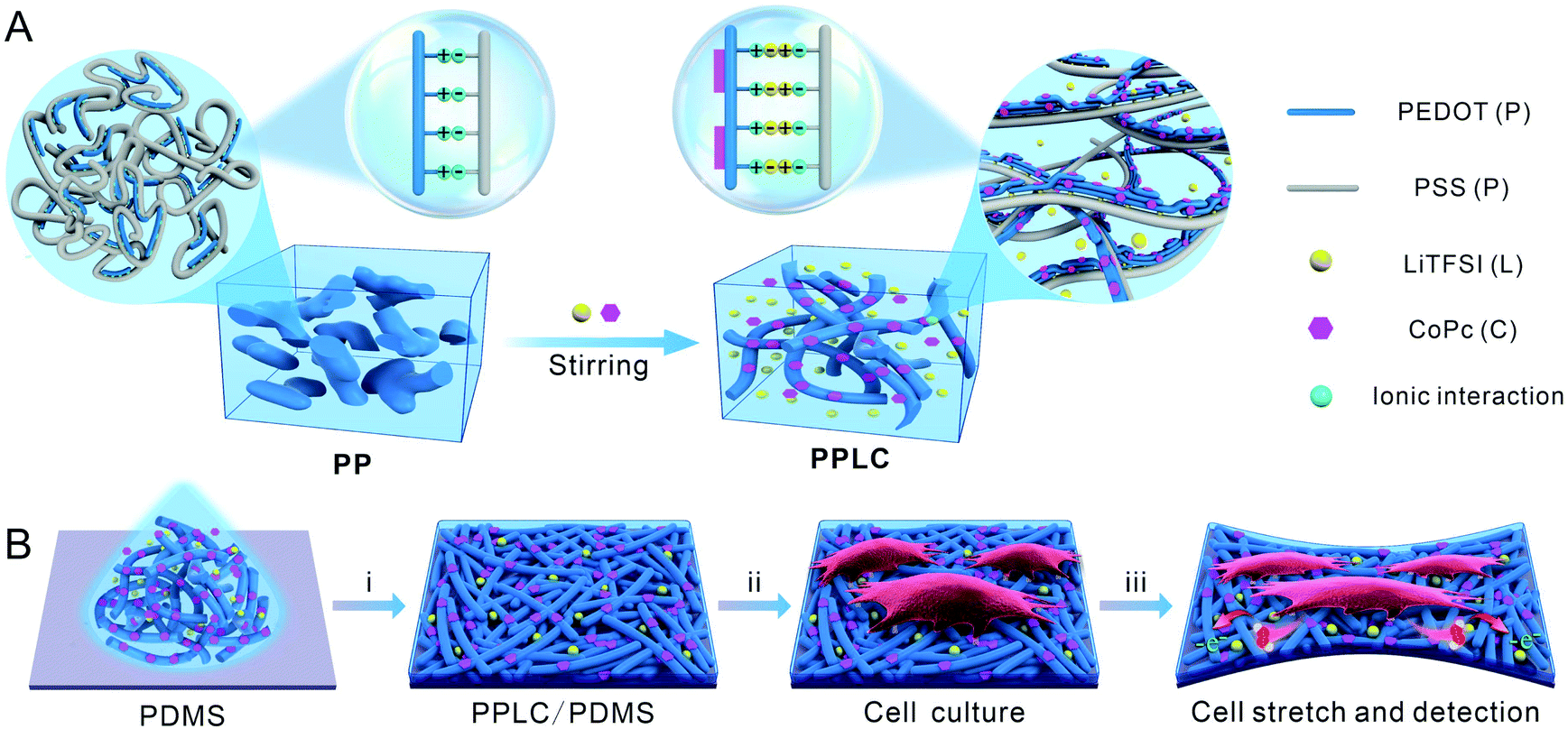 | ||
| Scheme 1 (A) Schematic diagram representing the microstructures of traditional PEDOT:PSS (PP, left) and stretchable PEDOT:PSS (PPLC, right) functionalized with LiTFSI and CoPc. (B) Fabrication of a stretchable PPLC/PDMS film for monitoring cell mechanotransduction. (i) Spin-coating PPLC on PDMS. (ii) Cell seeding and culture. (iii) Electrochemical detection of H2O2 release from stretched cells. | ||
Results and discussion
Fabrication and characterization of PPLC polymer
The functionalized PPLC polymer was obtained by simple blending of the plasticizer LiTFSI and catalyst CoPc with the solution of PEDOT:PSS. To investigate the microstructures of different polymer systems, PEDOT:PSS (PP), PEDOT:PSS–LiTFSI (PPL) and PPLC films were characterized by using an atomic force microscope (AFM). As a conductive polymer, PP dispersions are typically described as gel-like particles, which are composed of PEDOT-rich cores stabilized by PSS-rich shells in aqueous solvent36,37 (Scheme 1A, left), and thus a large number of aggregated small grains appeared in the pristine PP film (Fig. 1A). Meanwhile, after being doped with the plasticizer LiTFSI, the polymer film processed distinct nanofibrous structures with good continuity (Fig. 1B). The morphological change is attributed to that LiTFSI can easily interact with both negatively charged PSS and positively charged PEDOT, and the weakened coulombic interaction between PSS and PEDOT chains allows PEDOT to decouple from the highly coiled PSS and grow into large-scale conducting domains, forming a continuous nanofiber network33 (Scheme 1A, right). Interestingly, the subsequent incorporation of catalyst CoPc molecules had little impact on the fibrous morphology (Fig. 1C), which will be beneficial for retaining the stable conductivity of the stretchable sensor.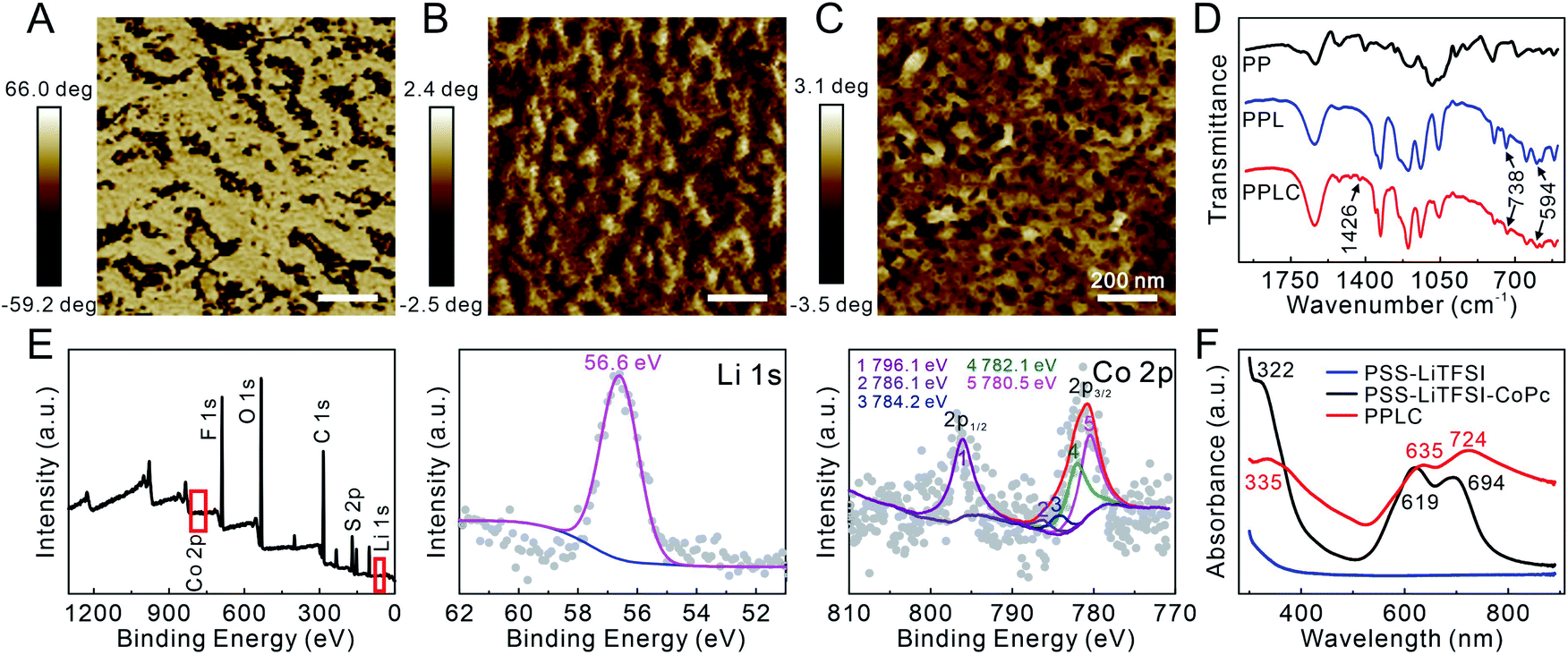 | ||
| Fig. 1 AFM phase images of the (A) PP film, (B) PPL film and (C) PPLC film. (D) FTIR spectra of different materials. (E) XPS spectra of the PPLC film. (F) UV-visible spectra of different solutions. | ||
Subsequently, Fourier transform infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS) were performed to further characterize the composition of the PPLC/PDMS film respectively. Compared with that of original PP, the FTIR spectrum of PPLC displayed characteristic absorption peaks assigned to the overlapping of the symmetric bending mode of CF3, C–S and S–N stretching (738 cm−1), asymmetric bending mode of CF3 (594 cm−1) and C–C stretching in isoindole (1426 cm−1), which were attributed to the existence of LiTFSI38 and CoPc39 in the functionalized PP, respectively (Fig. 1D). The full XPS spectrum of the PPLC film, together with the deconvolution of F 1s, Li 1s, Co 2p, C 1s, O 1s and S 2p spectra, demonstrated the presence of F, Li, O, C, S and Co elements (Fig. 1E and S1†), in which F and Li corresponded to LiTFSI and Co corresponded to CoPc, indicating the successful doping of LiTFSI and CoPc into the PPLC film.40–42
Furthermore, UV-visible spectra were recorded to investigate the existing form of the catalyst CoPc in the PPLC film (Fig. 1F). Compared with that of PSS–LiTFSI–CoPc, the spectrum of PPLC showed that the existence of PEDOT made the peak corresponding to the B band of CoPc at 322 nm shift to 335 nm, and the peaks at 619 nm and 694 nm corresponding to the Q bands of CoPc shift to 635 nm and 724 nm, respectively.43 These red shifts in the spectrum revealed that CoPc molecules were assembled on the PEDOT chains via π–π interaction.
Mechanical properties of the PPLC/PDMS electrode
To evaluate the mechanical properties of the PPLC-based sensor, a homogeneous mixture of PP, LiTFSI and CoPc was spin-coated on the surface of the PDMS film, which was pretreated with polydopamine to improve the wetting of PPLC solution. Due to the morphology of isolated grains, the fracture strain of the PP film is as low as 5%, because the grains will separate from each other and lead to a rapid decline in conductivity when strains are applied (Fig. S2A†). After introducing LiTFSI and CoPc, the electrostatic interaction between PEDOT and PSS could be greatly weakened, which resulted in the structure of conductive PEDOT-rich domains embedded in the soft PSS matrix, and this will theoretically improve the stable conductivity during stretching (Fig. 2A). Since the ionic liquid is the key to realizing the coexistence of high stretchability and conductivity, the amount of LiTFSI (1–3 wt%) in the PPL film was optimized by comparing the cyclic voltammetric (CV) characteristics in K3[Fe(CN)6] (an electrochemical probe) solution (Fig. S3A†). The CV curves of PPL films with the additive of 2 wt% and 3 wt% LiTFSI had smaller potential differences than that of the PPL film doped with 1 wt% LiTFSI, and they displayed very similar potential differences and electric capacities. Therefore, we chose 2 wt% as the optimal additive amount in PP solution in principle of less addition of LiTFSI. Though the undeformed PPL/PDMS films spin-coated at a speed of 1000 rpm, 1500 rpm, 2000 rpm and 3000 rpm had similar electrochemical performances (Fig. S3B†), the PPL/PDMS film of 1500 rpm had the least cracks after recovering from being stretched to a strain of 50% and the most stable electrochemical performance (Fig. S4 and S5†). As a result, 2 wt% LiTFSI and 1500 rpm speed for 60 s were selected as the optimal conditions for the fabrication of PPL/PDMS and PPLC/PDMS films, and the thickness of spin-coated PPLC was approximately 400 nm (Fig. S6†). Besides, we examined the electrical conductivities of PPL and PPLC, and they were 1798.2 ± 348.4 and 1036.2 ± 120.5 S cm−1, respectively. These results revealed that the doping of the ionic liquid LiTFSI dramatically improved the electrical conductivity of PP whose reported electrical conductivity is about 4 S cm−1.33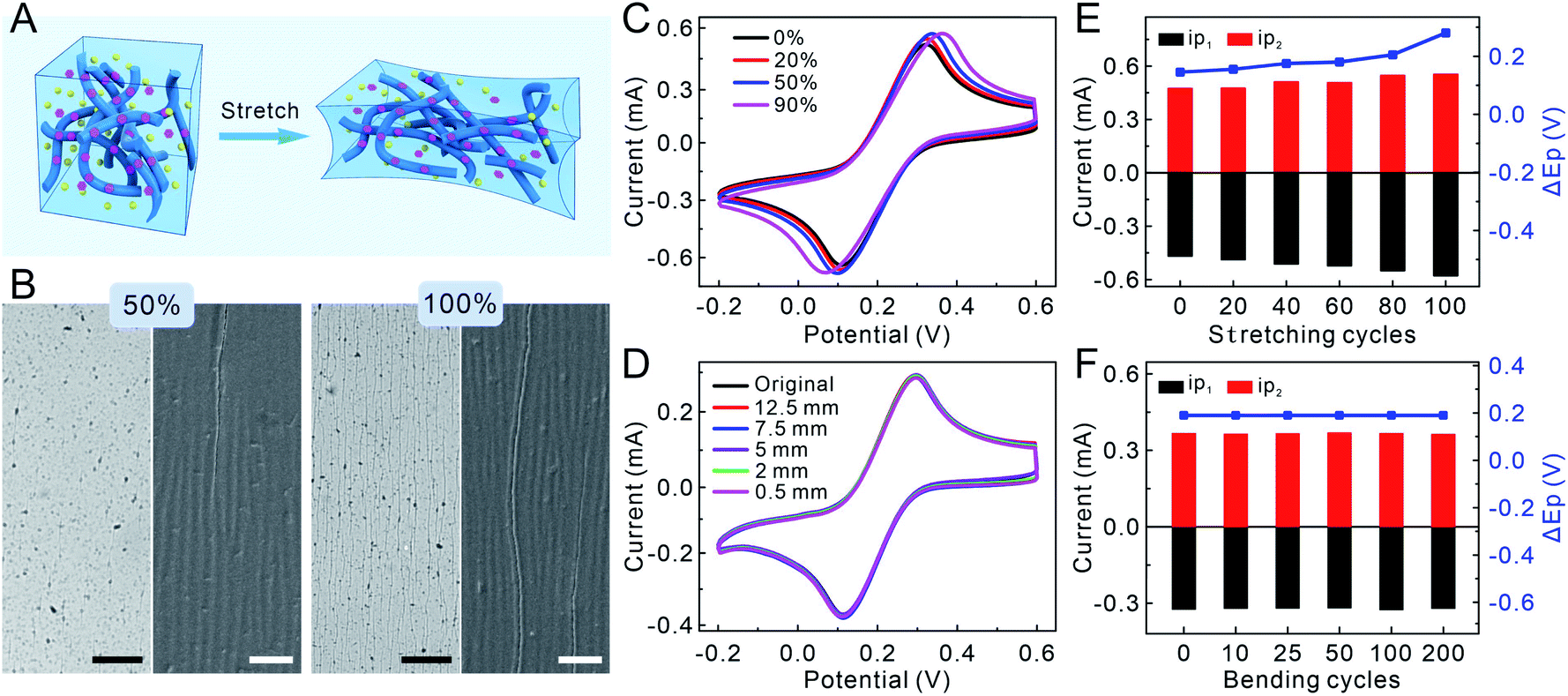 | ||
| Fig. 2 (A) Schematic illustration showing the stretchability principle of the PPLC film. (B) Optical microscope (left) and SEM (right) images of the PPLC/PDMS film recovering from being submitted to various strains. Scale bar: 100 μm (black) and 10 μm (white). CVs of the PPLC/PDMS film obtained in 10 mM K3[Fe(CN)6] (C) under different tensile strains and (D) after recovering from being bent at different radii (scan rate: 0.05 V s−1). Statistical results of the CVs of PPLC/PDMS films obtained in 10 mM K3[Fe(CN)6] after recovering from being (E) stretched to 50% and (F) bent at a radius of 3 mm for different cycles. | ||
To compare the stretchability of different films, the morphologies of PP, PPL and PPLC films were characterized after they were loaded with a series of tensile strains (10–100%) and then released to the original states. The pristine PP film obviously exhibited many wide vertical cracks (ca. 30 μm in width) after suffering from only 10% strain, and the cracks became sharply larger with the increase of mechanical strains (Fig. S2B†). In contrast, the surface of the PPL/PDMS film kept smooth even with a strain of 50%, and only few hairline cracks appeared when stretched up to 100% (Fig. S7,† top row). Scanning electron microscope (SEM) results showed that the PPL film maintained high integrality within the 50% strain, and uniform wrinkles and very few tiny cracks (ca. 500 nm in width) were observed under 100% strain due to the buckling of the PPL composite (Fig. S7,† bottom row), manifesting that it was the LiTFSI additive that greatly improved the stretchability of the PPL film. As for the PPLC/PDMS film (Fig. 2A), similar to that of the PPL film, optical microscope images displayed that few short cracks appeared until the film was stretched up to 100%, and SEM images revealed that few perceptible wrinkles started to form when stretched to a strain of 50% (Fig. 2B and S8†). Whereas, the appeared cracks did not affect the overall continuity and stability of the PPLC film even under 100% strain.
To study the electrical stability of the PPLC-based stretchable sensor, the relative resistance (ΔR/R0) of the PPLC/PDMS film was recorded in the stretching–releasing cycle (0–50–0%) and bending process, and no significant increase in ΔR/R0 occurred during the dynamic deformations, as well as stretching (50% strain) and bending (a radius of 1 mm) for 1000 cycles, respectively (Fig. S9†). The almost invariable resistance revealed the stable conductivity in the PPLC polymer. Then, to investigate the electrochemical stability, CVs of the PPLC/PDMS film in K3[Fe(CN)6] solution were collected after subjected to different mechanical deformations, including stretching to various tensile strains (0–50%), bending with different radii (0.5–12.5 mm) and repeated loading. The potentials and peak currents of ferricyanide showed high consistency even when the PPLC/PDMS film was stretched with a strain of up to 90% (Fig. 2C) or bent with a small radius of 0.5 mm (Fig. 2D). Moreover, the currents of reduction (ip1) and oxidation (ip2) peaks and their potential differences (ΔEp) of the recorded CVs were further analyzed after being repeatedly stretched to a strain of 50% and bent to a curvature radius of 3 mm for different cycles (Fig. 2E, F and S10†), and they had little change compared with the original undeformed states. The superior electrochemical stability is probably attributed to the percolation network of nanofibrous PEDOT:PSS, which could accommodate the applied strain by rotating and sliding against each other. The slight increase in ip and ΔEp in the case of higher strain (e.g. 90%) or being recovered from stretching for more cycles might be attributed to a combined result of more exposed PEDOT:PSS nanofibers (larger electrode area and ip) and some irreversible interlacement with others (higher electrical resistivity and ΔEp). Overall, these results clearly demonstrated the excellent electrochemical stability of the PPLC-based stretchable electrode against mechanical deformations.
Moreover, compared with other electrode materials of stretchable electrochemical sensors, the prominent feature of water-soluble PPLC is the high processability, and many processing techniques could be employed for convenient, controllable and high-throughput electrode fabrication. For example, the pattern and size of the PPLC-based electrode could be easily regulated with the aid of photolithography or an ink-jet printer (Fig. S11†). Convincingly, the superb processibility will make PPLC hold great potential application in various flexible and stretchable sensors.
Electrochemical sensing performance of the PPLC/PDMS film
The electrochemical performance of the PPLC/PDMS film was studied after the confirmation of its mechanical stability. Compared with the PP/PDMS electrode, the electrochemical performance of the PPL/PDMS electrode was greatly improved because of the increased conductivity caused by doping LiTFSI. The CV of the PPLC/PDMS electrode was almost the same as that of the PPL/PDMS film, which indicated that the addition of CoPc had little effect on the electron transfer in the original PPL/PDMS electrode (Fig. 3A). Meanwhile, as an electron mediator, CoPc could endow the electrode with remarkable sensing ability by catalyzing the target molecules involved in cell activities, such as the oxidation of H2O2 to O2via a redox reaction between CoIIPc and CoIIIPc (Fig. 3B).44 It was observed that the CV curve obtained in H2O2 solution of the PPLC/PDMS electrode showed an obvious oxidation peak at +0.55 V (Fig. 3C), while the response of the PPL/PDMS electrode in H2O2 was almost identical to that in phosphate buffered solution (PBS), manifesting the excellent catalysis of CoPc molecule towards H2O2 oxidation. Since the sensing performance was ascribed to the catalyst CoPc, the additive amount of CoPc (0–50 mM) in the PPLC polymer system was optimized by comparing the CV responses of different PPLC/PDMS electrodes to 1 mM H2O2 (Fig. S12†). The current responses caused by H2O2 oxidation increased with the increase of CoPc contents and reached a peak at a level of 20 mM, and the declining response at a concentration of 25 mM or even higher probably resulted from the formation of CoPc precipitates45 with low electrocatalytic efficiency towards H2O2 oxidization.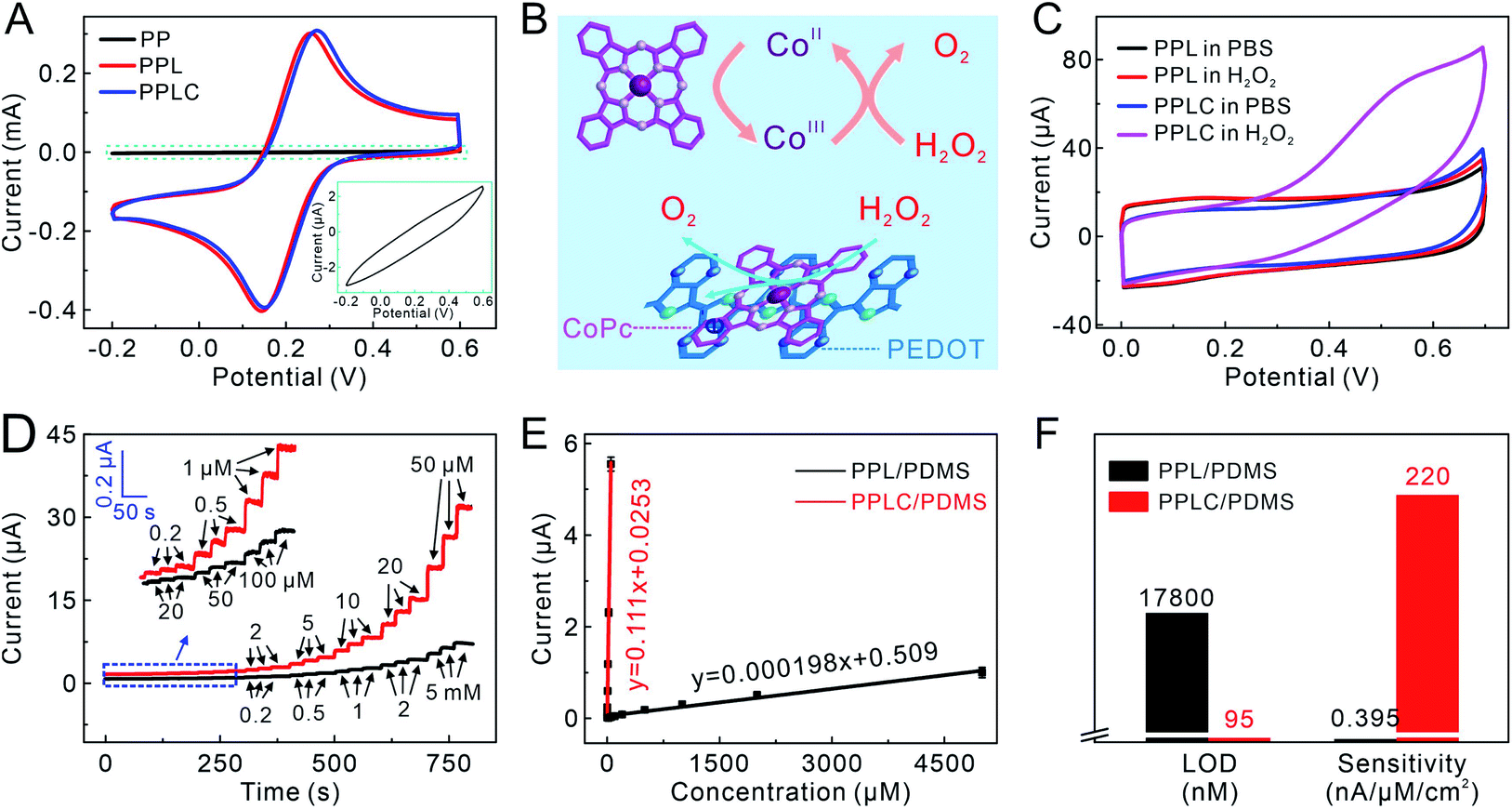 | ||
| Fig. 3 (A) CVs of different electrodes obtained in 10 mM K3[Fe(CN)6]. Inset: the enlarged view for CV of the PP electrode. (B) Schematic illustration of the electrocatalysis mechanism. (C) CVs of different electrodes with and without 1 mM H2O2. (D) Amperometric responses of PPL/PDMS (black lines) and PPLC/PDMS (red lines) electrodes to H2O2 at a potential of +0.55 V (vs. Ag/AgCl) to increasing H2O2 concentrations. Inset: the enlargements of amperometric responses framed in blue. (E) Calibration curves of PPL/PDMS and PPLC/PDMS electrodes to increasing H2O2 concentrations (data presented as mean ± standard error, n = 3). (F) Calculated LOD and sensitivity of PPL/PDMS and PPLC/PDMS electrodes to H2O2. | ||
The superiority of CoPc molecules to H2O2 electrooxidation was further demonstrated by the amperometric responses to a series of H2O2 solutions with increasing concentrations. The PPLC/PDMS electrode could respond sensitively to 200 nM H2O2 (Fig. 3D, red lines) and exhibit a good linear relationship with H2O2 in a wide concentration range from 200 nM to 50 μM (Fig. 3E). Conversely, the PPL/PDMS electrode generated a detectable signal until the concentration of H2O2 increased to as high as 20 μM (Fig. 3D, black lines). The calculated detection limit (LOD) of the PPLC/PDMS electrode was 95 nM (S/N = 3), which was approximately 200-fold lower than that of the PPL/PDMS electrode (17800 nM), and the sensitivity of the PPLC/PDMS electrode (220 nA μM−1 cm−2) was about 560-fold higher than that of the PPL/PDMS electrode (0.395 nA μM−1 cm−2) (Fig. 3F). In addition, we recorded the CV responses of the PPLC/PDMS electrode to 1 mM H2O2 and the amperometric responses of this electrode to a series of H2O2 solutions before deformation and after recovering from being repeatedly stretched to a strain of 50% (Fig. S13†), and the almost perfectly consistent curves indicated that the repeated deformations had little negative influence on the H2O2 sensing performance.
Real-time monitoring of stretch-induced H2O2 released from 16HBECs
As an important signaling molecule, H2O2 is strongly implicated in cell proliferation, aging, death and signal transduction, and the levels of H2O2 can reflect the normal or abnormal conditions of living organisms.34,35 Recently increasing studies have revealed that the production of H2O2 is also regulated by mechanical forces exposed on cells or organs via mechanotransduction, besides the inherent cell metabolism.46–48 Exemplarily, the secretions of lung cells are regulated by mechanical stretch under both physiological (breathing) and pathophysiological (ventilator-induced) conditions,49–51 and the content of exhaled H2O2 was elevated in the lungs of patients with bronchiectasis.52–54 Considering that the dynamic changes of H2O2 concentration emitted from lung cells have been poorly studied, the PPLC/PDMS electrode was employed as a platform for real-time monitoring of stretch-induced H2O2 release from human bronchial epithelial cells (16HBECs) under different strains (Fig. 4A). | ||
| Fig. 4 (A) Schematic diagram of cellular mechanotransduction in stretched 16HBECs. (B) SEM image of 16HBECs cultured on a PPLC/PDMS film for 4 h. (C) Fluorescence microscopic images of 16HBECs cultured on a PPLC/PDMS film stained with Calcein-AM (green) and PI (red) before and after being stretched. Amperometric responses (D) and the corresponding statistical results (E) detected from 16HBECs submitted to different stretch stimuli (applied potential: +0.55 V vs. Ag/AgCl, data presented as mean ± standard error, n = 6, P-values were calculated using one-way ANOVA, **P < 0.01, ***P < 0.001). (F) Amperometric responses detected from 16HBECs submitted to 30% strain with different treatments at a potential of +0.55 V (vs. Ag/AgCl). Inset: corresponding normalized statistical results of amperometric responses recorded from cells stimulated with only strain (red cylinder) and cells pretreated with L-NMMA (blue cylinder) (data presented as mean ± standard error, n = 4, P-values were calculated using one-way ANOVA, n.s.: no significance). The vertical gray dashed lines in (D) and (F) indicated the beginning of the stretch stimuli; the hauling speed was 0.85 mm s−1. | ||
Firstly, the biocompatibility should be taken into account due to the direct contact between the sensor and living cells, and this is a particularly important issue since two additives were added into the PPLC compared with the native PP form. It was expressly observed that 16HBECs could attach well onto the surface of the PPLC film with the pseudopodia outspread fully after being cultured thereon for 4 h (Fig. 4B). Thereafter, 16HBECs were cultured on the PPLC/PDMS film for 6 h, 24 h, 48 h and 96 h (Fig. S14†), until they proliferated and covered almost all over the electrode. The staining by live/dead cell markers 3′,6′-di(o-acetyl)-4′,5′-bis[N,N-bis(carboxymethyl)-aminomethyl] fluorescein, tetraacetoxymethylester (Calcein-AM) and propidium iodide (PI) revealed that all cells kept high viability after being cultured for up to 96 h (Fig. S14E†). The satisfactory biocompatibility demonstrated that the added plasticizer LiTFSI and catalyst CoPc have little effect on the original cytocompatibility of PP.
Before the monitoring of mechanotransduction, the normal metabolism activity of 16HBECs cultured on the PPLC/PDMS electrode was examined. After the cells were stimulated with phorbol 12-myristate 13-acetate (PMA),55 which could activate nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (the main enzyme for ROS production in the cells), a rapid increase in the current was detected by the PPLC/PDMS electrode (Fig. S15,† red line). When catalase (H2O2 scavenger) was added together with PMA, the current decreased significantly (Fig. S15,† blue line). The above results indicated that the PPLC/PDMS electrode had the ability of real-time monitoring of H2O2 release from 16HBECs. Subsequently, the electrode bearing 16HBECs was submitted to a tensile strain of about 35% to fully cover the physiological (5–15%) and pathological strain ranges (20–30%) of lung cells,49,56 and the fluorescence staining of Calcein-AM and PI revealed that the cells still adhered firmly to the substrate and remained perfectly alive during the stretching process (Fig. 4C).
Then, the PPLC/PDMS electrode with 16HBECs cultured thereon was loaded with different stretching stimuli to model natural (10% strain) and bronchiectasis (20% and 30% strain) conditions.56 It was observed that the ampere-currents rose upon the stretching stimuli (Fig. 4D), and the increases of ampere-currents were proportional to the incremental mechanical strains (Fig. 4D, red line). To confirm that the increased current was caused by H2O2 release from the cells triggered by mechanical stimuli, 16HBECs were submitted to the same strains after being pre-treated with catalase and diphenyleneiodonium chloride (DPI, a widely used NADPH oxidase inhibitor), respectively. The results showed that the recorded current increases from cells pre-treated with catalase (Fig. 4D, blue line) were obviously smaller than those from cells with only mechanical stimuli (Fig. 4D, red line), and pre-treating with DPI further decreased the current responses due to the inhibition of NADPH oxidase (Fig. 4D, purple line). Altogether, these results revealed that the rise of ampere-current was evoked by H2O2 release via the activation of NADPH oxidase in response to mechanical stretch. Besides, the production of H2O2 in 16HBECs caused by mechanical strain was also verified through staining with DCFH-DA (a ROS fluorescent probe) (Fig. S16†).
Considering that the sudden mechanical disturbance resulted in reasonable noise (Fig. 4D, black line), the currents recorded by the electrode under the same strains without cells cultured thereon were taken as the baseline for the statistical analysis of the above results. As clearly shown in Fig. 4E, the amount of stretch-evoked H2O2 release increased with stretching magnitudes, and the scavenging of released H2O2 by catalase made the recorded current sharply decrease, and the inhibition of NADPH oxidase activity by DPI resulted in a negligible current response. The concentrations of emitted H2O2 from 16HBECs under 10%, 20% and 30% strains were calculated to be 4.90 ± 3.00 μM (10%), 11.20 ± 3.60 μM (20%) and 22.00 ± 1.50 μM (30%), respectively, which were consistent with previous studies that much more H2O2 molecules were released under bronchiectasis than normal condition.52–54
In 16HBEC mechanotransduction, NO is another closely related signaling molecule revealed by our previous sensor at a potential of +0.8 V,9 while most of NO molecules could not be oxidized under the potential of +0.55 V for the electrooxidation of H2O2 (Fig. S17†). This viewpoint was further validated by cell experiments, in which the current response recorded from those treated with L-NMMA (a total nitric oxide synthase inhibitor) showed no significant decrease, while pre-treatment with DPI led to a dramatic fall in the current response, comparable to that of cells under only mechanical strain (Fig. 4F). These results explicitly indicated that the current increase was caused by the electrooxidation of stretch-evoked H2O2 rather than NO under the potential of +0.55 V. Taken together, the above results demonstrated that the LiTFSI and CoPc functionalized PP concurrently possessed excellent stretchability and electrochemical sensing, as well as high biocompatibility, which allowed mimicking the bronchiectasis and synchronously real-time monitoring of stretch-induced H2O2 release from 16HBECs.
Conclusions
In summary, we have developed an efficient strategy for constructing a conductive polymer-based stretchable electrochemical sensor (PPLC/PDMS) via facile blending LiTFSI and CoPc with PEDOT:PSS. The doping of LiTFSI distinctly enhanced both conductivity and stretchability, and the incorporation of CoPc enabled the sensor to sensitively detect H2O2 molecules. With excellent mechanical and electrochemical performance, as well as satisfactory biocompatibility, the PPLC/PDMS sensor has successfully achieved in situ inducing and simultaneous monitoring of H2O2 release from stretched 16HBECs. In this work, we focused on the application of stretchable PEDOT film in real-time monitoring of cell mechanotransduction. Considering the high accessibility and versatility of the preparation method, plenty of conductive polymers with specific functionality could be obtained by altering the plasticizer or catalyst. Therefore, the proposed strategy is expected to offer numerous opportunities for polymer-based wearable and stretchable sensors with practical applications in biomedical sciences and healthcare monitoring.Experimental
Materials and methods
PEDOT:PSS (Clevios PH1000) was purchased from Wuhan Zhuojia Technology Co., Ltd. (Wuhan, China). The PDMS prepolymer and cross-linker were obtained from Momentive Performance Materials (Waterford, NY, U.S.A.). Dopamine hydrochloride and bis(trifluoromethane) sulfonimide lithium salt (LiTFSI) were bought from Sigma Aldrich (St. Louis, U.S.A.). Cobalt phthalocyanine (CoPc) was purchased from Aladdin Industrial Co., Ltd. (Shanghai, China). The negative photoresist SU-8 and developer were bought from MicroChem Corp (Massachusetts, U.S.A.). 16HBECs were obtained from Shanghai Sixin Biotechnology Co., Ltd. (Shanghai, China). Fetal bovine serum (FBS), RPMI-1640 culture medium, and trypsin–EDTA (0.25%) were bought from GIBCO Corporation (U.S.A.). Calcein-AM, PI, PMA and DPI (NADPH oxidase inhibitor) were obtained from Sigma Aldrich (St. Louis, U.S.A.). DCFH-DA (ROS fluorescent probe), L-NMMA (a total nitric oxide synthase inhibitor) and catalase were bought from Beyotime Biotechnology (Shanghai, China). Ultrapure water (Millipore, 18 MΩ cm−1) was used throughout the experiments.AFM images were recorded in tapping mode using a BioScope Resolve Atomic Force Microscope (Bruker, Germany). UV-vis absorption spectra were conducted on a UV-2550 spectrophotometer (Shimadzu, Japan). XPS measurements were performed on an ESCALAB 250Xi photoelectron spectrometer (Fisher Scientific, U.S.A.) by using Al Kα X-ray radiation as the X-ray excitation source. FTIR spectra were recorded on a NICOLET FTIR5700 Fourier transform infrared spectrometer (Thermo, U.S.A.). SEM images were obtained by a Merlin compact field-emission scanning electron microscope (Zeiss, Germany). The inkjet printing was conducted by a Jetlab4 XL-A inkjet printer (MicroFab, U.S.A.). The photolithography was performed by a G17 lithography machine (Chengdu, China). All the microscopic observation and fluorescence imaging were implemented by an Axiovert 200M and Axio Observer Z1 inverted fluorescent microscope (Zeiss, Germany). All electrochemical measurements were conducted on a CHI 660A electrochemical workstation (CHI-Instruments, Shanghai) with Pt counter electrode and Ag/AgCl reference electrode at room temperature.
Fabrication of the PPLC/PDMS film
Firstly, a PDMS film was acquired by spin-coating the degassed liquid prepolymer and cross-linker (w/w = 10:1) on a Si substrate at a spin rate of 1000 rpm for 10 s and thermally cured at 80 °C for 3 h. Then, the PDMS film was immersed in dopamine hydrochloride solution (1 mg mL−1, Tris–HCl buffer solution, 10 mM, and pH ∼8.5) for 24 h to enhance its hydrophilicity. For the synthesis of PPLC polymer, LiTFSI (2 wt%) and CoPc (20 mM) were added to the PP aqueous dispersion (1 wt%), and stirred vigorously at 1000 rpm for 6 h. Then, the homogeneous mixture solution was spin-coated on the polydopamine-coated PDMS film at a spin rate of 1500 rpm for 60 s, and then annealed at 130 °C for 15 min, forming the PPLC/PDMS film.
The patterned PPLC/PDMS films were obtained by two techniques. For photolithography, firstly, the PDMS film covered with a hollowed-out mask obtained by photolithography was soaked in dopamine hydrochloride solution for 24 h to obtain the patterned polydopamine–PDMS film, and then, the homogeneous mixture solution of PPLC was spin-coated on the surface of the PDMS film, forming the patterned PPLC/PDMS film. As for inkjet printing, the patterned PPLC/PDMS was printed on the polydopamine–PDMS film by an inkjet printer with PPLC solution as the conductive ink.
Electrode fabrication for electrochemical detection
To connect with the outer workstation, the two ends of the PPLC/PDMS electrode were connected with wires, and the joints were insulated by casting PDMS prepolymer which was then thermally cured at 80 °C to ensure a chamber (1.0 × 0.5 × 0.2 cm3) for holding solution for stretch-related electrochemical measurements. As for the bending-related and other electrochemical measurements, the PPLC/PDMS electrode was connected with a copper wire via carbon paste, and the joints were insulated to ensure a 0.5 × 0.5 cm2 active area (1.0 × 0.5 cm2 active area for the amperometric i–t curve).Flexibility and stretchability test
For the flexibility test, the PPLC/PDMS film was attached to a variety of cylinders with diverse outer curvature radii (0.5–12.5 mm). As for the stretchability test, specific lengths of the film which corresponded to a set of strains were stretched by a linear sliding motor. The resistance of the film under different deformations was measured by using a multimeter.Cell culture and imaging
16HBECs were cultured using RPMI-1640 culture medium with FBS (10%) and penicillin–streptomycin (1%) in a humidified incubator (37 °C and 5% CO2). Before cell seeding, the PPLC/PDMS electrode was thoroughly sterilized with ultraviolet exposure overnight. For electrochemical detection, 16HBECs (2 × 105 cells per cm2) were seeded on the PPLC/PDMS electrode, and a chamber was built around the PPLC layer utilizing PDMS prepolymer which was then thermally cured at 80 °C to ensure a chamber (1.0 × 0.5 × 0.2 cm3) to store culture medium. After being cultured in an incubator for 20 h to allow cell adhesion, the culture medium was removed and the electrode was washed with sterile PBS for three times to remove loosely bonded cells. To assess the viability and released H2O2 of 16HBECs cultured on the PPLC/PDMS electrode, fluorescent live/dead cell markers Calcein-AM (2 μg mL−1)/PI (3 μg mL−1) and ROS fluorescent probe DCFH-DA (10 μM) were used respectively.Real-time monitoring of H2O2 release from stretched 16HBECs
To simultaneously achieve applying mechanical loading on cells and real-time monitoring of released H2O2, the PPLC/PDMS electrodes bearing 16HBECs cultured well thereon were fixed on a linear sliding device with a hauling speed of 0.85 mm s−1 and connected with the outer workstation by wires. Then, the stretchable sensors were stretched to different extents to simulate the normal state (10%) and bronchiectasis states (20% and 30%) respectively. The concentrations of added DPI, catalase and L-NMMA were 0.5 mM, 60 kU mL−1 and 1 mM, respectively. When monitoring H2O2 release from 16HBECs stimulated by PMA, the final concentrations of PMA and catalase were 50 μM and 6 kU mL−1 respectively.Data availability
The raw image data generated and analysed that support the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.Author contributions
Jing Yan and Yan-Ling Liu conceived and initiated the project. Jing Yan accomplished all the experiments and wrote the manuscript. Yu Qin, Wen-Ting Fan, Wen-Tao Wu and Li-Ping Yan helped to accomplish the cell experiments. Song-Wei Lv provided help in ink-jet printing. Yan-Ling Liu and Wei-Hua Huang supervised the project and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grants 22122408, 21804101, 21725504, and 21721005). We thank Prof. Zheng Liu from the Institute for Advanced Studies of Wuhan University for his help with the AFM characterization.References
- E. Tzima, M. Irani-Tehrani, W. B. Kiosses, E. Dejana, D. A. Schultz, B. Engelhardt, G. Cao, H. DeLisser and M. A. Schwartz, Nature, 2005, 437, 426–431 CrossRef CAS PubMed.
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265–275 CrossRef CAS.
- N. Wang, J. D. Tytell and D. E. Ingber, Nat. Rev. Mol. Cell Biol., 2009, 10, 75–82 CrossRef CAS PubMed.
- B. D. Hoffman, C. Grashoff and M. A. Schwartz, Nature, 2011, 475, 316–323 CrossRef CAS PubMed.
- J. D. Humphrey, E. R. Dufresne and M. A. Schwartz, Nat. Rev. Mol. Cell Biol., 2014, 15, 802–812 CrossRef CAS PubMed.
- A. G. Solis, P. Bielecki, H. R. Steach, L. Sharma, C. C. D. Harman, S. Yun, M. R. de Zoete, J. N. Warnock, S. D. F. To, A. G. York, M. Mack, M. A. Schwartz, C. S. Dela Cruz, N. W. Palm, R. Jackson and R. A. Flavell, Nature, 2019, 573, 69–74 CrossRef CAS PubMed.
- Y. L. Liu, Z. H. Jin, Y. H. Liu, X. B. Hu, Y. Qin, J. Q. Xu, C. F. Fan and W. H. Huang, Angew. Chem., Int. Ed., 2016, 55, 4537–4541 CrossRef CAS PubMed.
- Q. Lyu, Q. Zhai, J. Dyson, S. Gong, Y. Zhao, Y. Ling, R. Chandrasekaran, D. Dong and W. Cheng, Anal. Chem., 2019, 91, 13521–13527 CrossRef CAS PubMed.
- Y. L. Liu, Y. Qin, Z. H. Jin, X. B. Hu, M. M. Chen, R. Liu, C. Amatore and W. H. Huang, Angew. Chem., Int. Ed., 2017, 56, 9454–9458 CrossRef CAS PubMed.
- Y. L. Liu and W. H. Huang, Angew. Chem., Int. Ed., 2021, 60, 2757–2767 CrossRef CAS PubMed.
- Y. Ling, Q. Lyu, Q. Zhai, B. Zhu, S. Gong, T. Zhang, J. Dyson and W. Cheng, ACS Sens., 2020, 5, 3165–3171 CrossRef CAS PubMed.
- M. Zhou, Y. Jiang, G. Wang, W. J. Wu, W. X. Chen, P. Yu, Y. Q. Lin, J. J. Mao and L. Q. Mao, Nat. Commun., 2020, 11, 3188 CrossRef CAS PubMed.
- W. T. Fan, Y. Qin, X. B. Hu, J. Yan, W. T. Wu, Y. L. Liu and W. H. Huang, Anal. Chem., 2020, 92, 15639–15646 CrossRef CAS PubMed.
- Y. L. Liu, Y. Chen, W. T. Fan, P. Cao, J. Yan, X. Z. Zhao, W. G. Dong and W. H. Huang, Angew. Chem., Int. Ed., 2020, 59, 4075–4081 CrossRef CAS PubMed.
- Z. H. Jin, Y. L. Liu, W. T. Fan and W. H. Huang, Small, 2020, 16, 1903204 CrossRef CAS PubMed.
- X. Zhao, K. Q. Wang, B. Li, C. Wang, Y. Q. Ding, C. Q. Li, L. Q. Mao and Y. Q. Lin, Anal. Chem., 2018, 90, 7158–7163 CrossRef CAS PubMed.
- Y. Wang, S. Gong, S. J. Wang, X. Yang, Y. Ling, L. W. Yap, D. Dong, G. P. Simon and W. Cheng, ACS Nano, 2018, 12, 9742–9749 CrossRef CAS PubMed.
- M. M. Chen, S. B. Cheng, K. L. Ji, J. W. Gao, Y. L. Liu, W. Wen, X. H. Zhang, S. F. Wang and W. H. Huang, Chem. Sci., 2019, 10, 6295–6303 RSC.
- Y. Shu, Q. Lu, F. Yuan, Q. Tao, D. Jin, H. Yao, Q. Xu and X. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 49480–49488 CrossRef CAS PubMed.
- T. Sekitani, Y. Noguchi, K. Hata, T. Fukushima, T. Aida and T. Someya, Science, 2008, 321, 1468–1472 CrossRef CAS PubMed.
- H. Jiang, Y. T. Qi, W. T. Wu, M. Y. Wen, Y. L. Liu and W. H. Huang, Chem. Sci., 2020, 11, 8771–8778 RSC.
- T. Cheng, Y. Zhang, J. Zhang, W. Lai and W. Huang, J. Mater. Chem. A, 2016, 4, 10493–10499 RSC.
- Y. Liang, A. Offenhäusser, S. Ingebrandt and D. Mayer, Adv. Healthcare Mater., 2021, 10, 2100061 CrossRef CAS PubMed.
- M. Berggren, X. Crispin, S. Fabiano, M. P. Jonsson, D. T. Simon, E. Stavrinidou, K. Tybrandt and I. Zozoulenko, Adv. Mater., 2019, 31, 1805813 CrossRef PubMed.
- L. V. Kayser and D. J. Lipomi, Adv. Mater., 2019, 31, 1806133 CrossRef PubMed.
- X. Fan, W. Nie, H. Tsai, N. Wang, H. Huang, Y. Cheng, R. Wen, L. Ma, F. Yan and Y. Xia, Adv. Sci., 2019, 6, 1900813 CrossRef CAS PubMed.
- Y. Yang, H. Deng and Q. Fu, Mater. Chem. Front., 2020, 4, 3130–3152 RSC.
- P. Li, D. Du, L. Guo, Y. Guo and J. Ouyang, J. Mater. Chem. C, 2016, 4, 6525–6532 RSC.
- P. J. Taroni, G. Santagiuliana, K. Wan, P. Calado, M. Qiu, H. Zhang, N. M. Pugno, M. Palma, N. Stingelin-Stutzman, M. Heeney, O. Fenwick, M. Baxendale and E. Bilotti, Adv. Funct. Mater., 2018, 28, 1704285 CrossRef.
- D. J. Lipomi, J. A. Lee, M. Vosgueritchian, B. C. K. Tee, J. A. Bolander and Z. Bao, Chem. Mater., 2012, 24, 373–382 CrossRef CAS.
- J. Y. Oh, S. Kim, H. Baik and U. Jeong, Adv. Mater., 2016, 28, 4455–4461 CrossRef CAS PubMed.
- J. Liu, X. Zhang, Y. Liu, M. Rodrigo, P. D. Loftus, J. Aparicio-Valenzuela, J. Zheng, T. Pong, K. J. Cyr, M. Babakhanian, J. Hasi, J. Li, Y. Jiang, C. J. Kenney, P. J. Wang, A. M. Lee and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 14769–14778 CrossRef CAS PubMed.
- Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu, N. I. Rabiah, Z. Chen, J. W. Chung, C. Linder, M. F. Toney, B. Murmann and Z. Bao, Sci. Adv., 2017, 3, e1602076 CrossRef PubMed.
- H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef CAS PubMed.
- H. Sies, C. Berndt and D. P. Jones, Annu. Rev. Biochem., 2017, 86, 715–748 CrossRef CAS PubMed.
- U. Lang, E. Müller, N. Naujoks and J. Dual, Adv. Funct. Mater., 2009, 19, 1215–1220 CrossRef CAS.
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287 CrossRef PubMed.
- S. Ramesh and C. Liew, Measurement, 2013, 46, 1650–1656 CrossRef.
- R. Seoudi, G. S. El-Bahy and Z. A. El Sayed, J. Mol. Struct., 2005, 753, 119–126 CrossRef CAS.
- S. Leroy, H. Martinez, R. Dedryvère, D. Lemordant and D. Gonbeau, Appl. Surf. Sci., 2007, 253, 4895–4905 CrossRef CAS.
- K. Artyushkova, S. Levendosky, P. Atanassov and J. Fulghum, Top. Catal., 2007, 46, 263–275 CrossRef CAS.
- M. A. T. Gilmartin, R. J. Ewen, J. P. Hart and C. L. Honeybourne, Electroanalysis, 1995, 7, 547–555 CrossRef CAS.
- Y. Wang, N. Hu, Z. Zhou, D. Xu, Z. Wang, Z. Yang, H. Wei, E. S. Kong and Y. Zhang, J. Mater. Chem., 2011, 21, 3779–3787 RSC.
- P. N. Mashazi, K. I. Ozoemena and T. Nyokong, Electrochim. Acta, 2006, 52, 177–186 CrossRef CAS.
- P. M. Olmos Moya, M. Martínez Alfaro, R. Kazemi, M. A. Alpuche-Avilés, S. Griveau, F. Bedioui and S. Gutiérrez Granados, Anal. Chem., 2017, 89, 10726–10733 CrossRef CAS PubMed.
- U. Raaz, R. Toh, L. Maegdefessel, M. Adam, F. Nakagami, F. C. Emrich, J. M. Spin and P. S. Tsao, Antioxid. Redox Signaling, 2014, 20, 914–928 CrossRef CAS PubMed.
- H. J. Hsieh, C. A. Liu, B. Huang, A. H. Tseng and D. L. Wang, J. Biomed. Sci., 2014, 21, 3 CrossRef PubMed.
- K. G. Birukov, Antioxid. Redox Signaling, 2009, 11, 1651–1667 CrossRef CAS PubMed.
- A. Doryab, S. Tas, M. B. Taskin, L. Yang, A. Hilgendorff, J. Groll, D. E. Wagner and O. Schmid, Adv. Funct. Mater., 2019, 29, 1903114 CrossRef.
- N. E. Vlahakis, M. A. Schroeder, A. H. Limper and R. D. Hubmayr, Am. J. Physiol., 1999, 277, L167–L173 CAS.
- K. E. Chapman, S. E. Sinclair, D. Zhuang, A. Hassid, L. P. Desai and C. M. Waters, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2005, 289, L834–L841 CrossRef CAS PubMed.
- A. H. Gedik, E. Cakir, T. A. Vehapoglu, O. F. Ozer and S. B. Kaygusuz, Turk. J. Med. Sci., 2020, 50, 1–7 CAS.
- S. Loukides, D. Bouros, G. Papatheodorou, S. Lachanis, P. Panagou and N. M. Siafakas, Chest, 2002, 121, 81–87 CrossRef CAS PubMed.
- S. Loukides, I. Horvath, T. Wodehouse, P. J. Cole and P. J. Barnes, Am. J. Respir. Crit. Care Med., 1998, 158, 991–994 CrossRef CAS PubMed.
- M. X. Shao and J. A. Nadel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 767–772 CrossRef CAS PubMed.
- M. Schultze, M. Fiess, N. Karpowicz, J. Gagnon, M. Korbman, M. Hofstetter, S. Neppl, A. L. Cavalieri, Y. Komninos, T. Mercouris, C. A. Nicolaides, R. Pazourek, S. Nagele, J. Feist, J. Burgdorfer, A. M. Azzeer, R. Ernstorfer, R. Kienberger, U. Kleineberg, E. Goulielmakis, F. Krausz and V. S. Yakovlev, Science, 2010, 328, 1662–1668 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04138j |
| This journal is © The Royal Society of Chemistry 2021 |