
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structure and bonding nature of heavy dipnictene radical anions†
Hanns M.
Weinert
 a,
Christoph
Wölper
a,
Julia
Haak
ab,
George E.
Cutsail
III
ab and
Stephan
Schulz
*a
a,
Christoph
Wölper
a,
Julia
Haak
ab,
George E.
Cutsail
III
ab and
Stephan
Schulz
*a
aInstitute for Inorganic Chemistry, Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätsstraße 5–7, 45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de
bMax Planck Institute for Chemical Energy Conversion (CEC), Stiftstraße 34–36, 45470 Mülheim a. d. Ruhr, Germany
First published on 30th August 2021
Abstract
Cyclic voltammetry (CV) studies of two L(X)Ga-substituted dipnictenes [L(R2N)GaE]2 (E = Sb, R = Me 1; E = Bi; R = Et 2; L = HC[C(Me)NDipp]2; Dipp = 2,6-i-Pr2C6H3) showed reversible reduction events. Single electron reduction of 1 and 2 with KC8 in DME in the presence of benzo-18-crown-6 (B-18-C-6) gave the corresponding dipnictenyl radical anions (DME)[K(B-18-C-6)][L(R2N)GaE]2 (E = Sb, R = Me 3; E = Bi, R = Et 4). Radical anions 3 and 4 were characterized by EPR, UV-vis and single crystal X-ray diffraction, while quantum chemical calculations gave deeper insight into the nature of the chemical bonding.
Introduction
Heavy p-block element compounds with π-bonding contribution1 have received increasing interest after the synthesis of the first stable diphosphene [Mes*P]2 (Mes* = 2,4,6-t-Bu3-C6H2)2 and disilene [Mes2Si]2 (Mes = 2,4,6-Me3-C6H2)3 in the 1980s. Heavier dipnictenes [RE]2 (E = As, Sb, Bi) are typically kinetically stabilized by using bulky substituents, i.e. carbon-based ligands,4 amides,5 boryles,6 ferrocenyls,7 and phosphanides.8 In addition, carbene-coordinated homonuclear π-bonded diatomic p-block elements compounds9 and L(X)Ga-substituted dipnictenes [L(X)GaE]2 were structurally characterized,10 and their electronic nature was analysed by quantum chemical calculations.11 The LUMO of dipnictenes containing σ-bonded ligands is typically represented by low-lying π* orbitals, whereas the level of the molecular orbitals of the π orbitals are somewhat indistinct since their energy levels are close to those of n+ orbital or high lying σ bonds (in phase integration of lone pairs).12 The HOMO in [L(X)GaE]2 is represented by the Ga–E bond and the LUMO is ligand-centred,13 while the HOMO and LUMO in carbene-substituted dipnictenes are delocalized via the π and π* orbitals of the ligands.4aDipnictenes react in single-electron transfer reactions to the corresponding radical anions (reduction) or cations (oxidation) as was shown for (carbene-coordinated) diphosphenes,14 phosphaarsenes,15 and diarsenes.16 In contrast, distibene and dibismuthene radical cations have not been reported to date, while radical anions [(bbt)E]2˙− (E = P, Sb, Bi; bbt = 2,6-[CH(SiMe3)2]-4-[C(SiMe3)3]-C6H2) were prepared by reactions of [(bbt)E]2 with Li metal. [(bbt)Bi]2˙− was characterized in solution by UV-vis, whereas EPR measurements failed due to its quick decomposition to EPR silent bbtH.4b,12b,17 To the best of our knowledge, [(bbt)Sb]2˙−IV is the only structurally characterized heavier dipnictene radical anion (Scheme 1).17
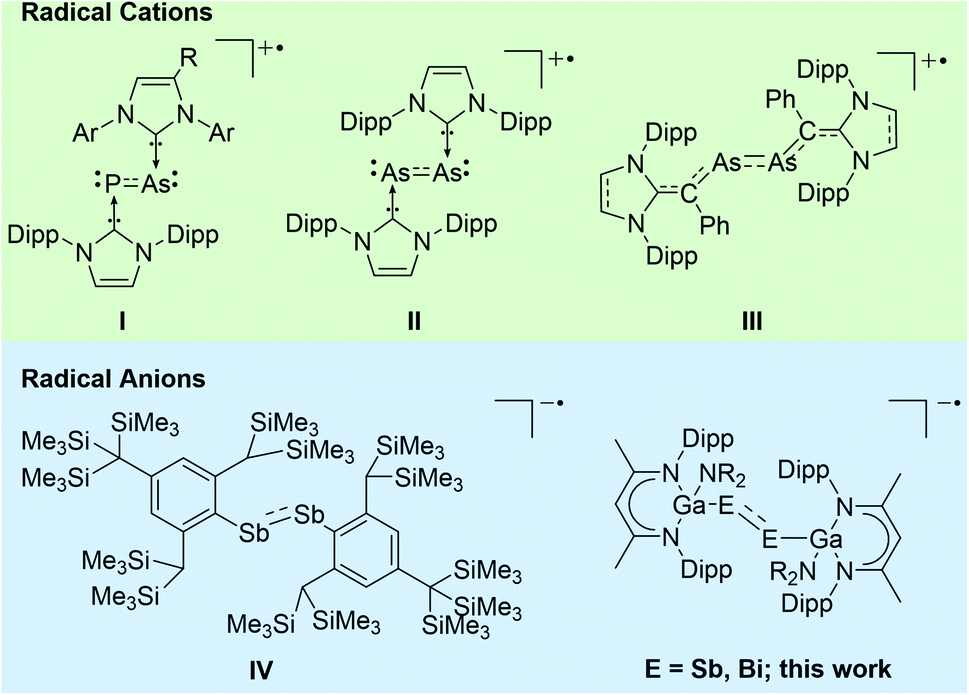 | ||
| Scheme 1 Structurally characterized heavy dipnictene radical cations and anions (E = As, Sb, Bi). | ||
Heavy main-group element-centred radicals have promising applications in organic synthesis, catalysis, and material sciences.18,19 Our interest in pnictogen-centred radicals10f,20 prompted our attention to reduction reactions of L(X)Ga-substituted dipnictenes (L = HC[C(Me)NDipp]2; Dipp = 2,6-i-Pr2C6H3), and we herein report on the synthesis and structures of two dipnictene radical anions [K(DME)(B-18-C-6)][{L(R2N)GaE}2] (E = Sb, Bi).
Results and discussion
Cyclic voltammetry
CV studies of dipnictenes [L(Me2N)GaSb]21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10b and [L(Et2N)GaBi]2210d were performed in saturated THF solution with NBu4PF6 as the electrolyte salt at 45 °C (Fig. 1).
10b and [L(Et2N)GaBi]2210d were performed in saturated THF solution with NBu4PF6 as the electrolyte salt at 45 °C (Fig. 1).
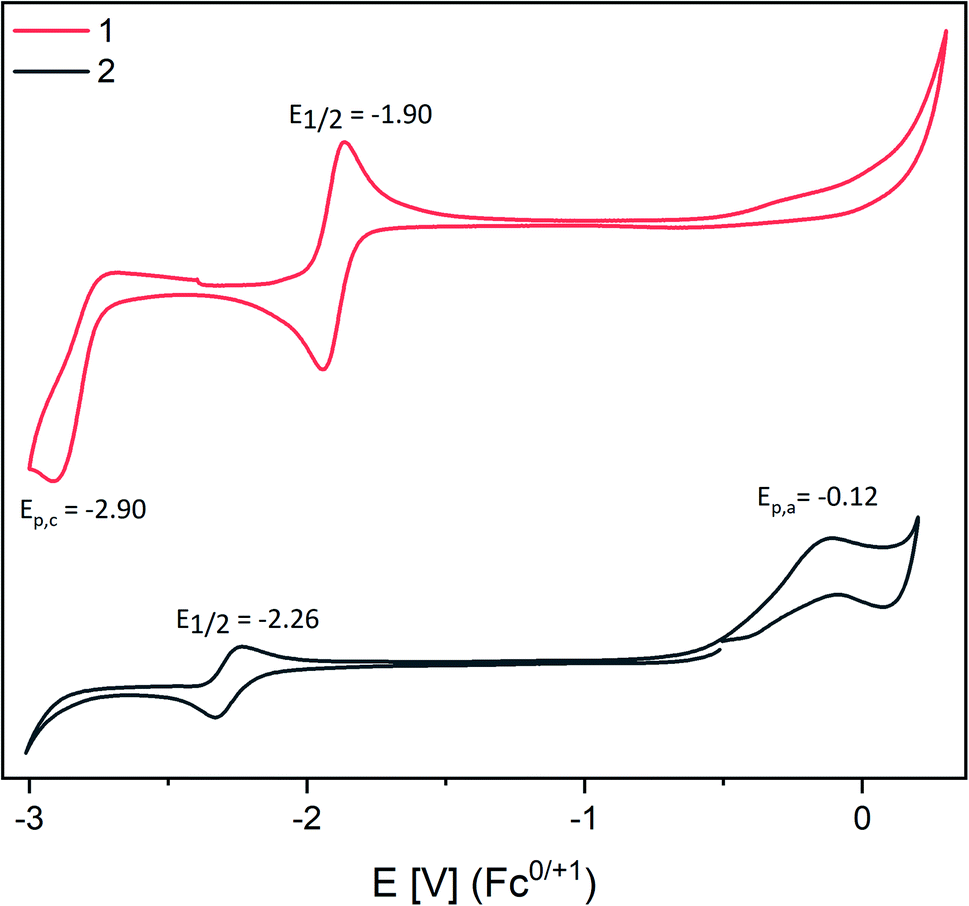 | ||
| Fig. 1 CV curves of saturated solutions of 1 and 2 in THF with [n-Bu4N][PF6] (100 mM) as electrolyte. Experiments were performed at 45 °C due to the low solubility of 1 and 2. | ||
1 and 2 showed reversible reduction events at −1.90 V (1) and −2.26 V (2) vs. the Fc0/+ couple with rather low peak-to-peak separation, which only moderately increased with increasing scan rate (Fig. S14 and S15†).21 Comparable findings were reported for dipnictenes [bbtE]2 (E = Sb −1.74 V, Bi −1.89 V12b using 0.09 V Fc vs. Ag/Ag+).22 However, [bbtE]2 showed higher reduction potentials than 1 and 2 (Bi ΔE1/2 = 0.39 V; Sb ΔE1/2 = 0.16 V). Dibismuthene 2 also showed an irreversible oxidation event at Epa = −0.12 V, whereas a second irreversible reduction reaction at −2.90 V was found for distibene 1, which indicates the reduction of initially formed radical anion to the corresponding dianion. Moreover, a pseudo reversible reduction event at E1/2 = −1.2 V (ΔEpa/c = 0.5 V, Ag/Ag+ or AgCl) was reported for [L(TfO)GaBi]2,10a which largely deviates from the potential we obtained for 2. Even assuming a high difference of 0.4 V to the Fc0/+ this is still shifted 0.7 V to lower potential.23
Radical synthesis
Dipnictenes [L(R2N)GaE]2 (E = Sb, R = Me 1; E = Bi, R = Et 2) were reacted with reducing agent, i.e. Li, Na, K, alkaline metal naphthalenides and KC8, in ether or toluene solution in the presence or absence of crown ethers and cryptands.Suspensions of 1 and 2 quickly dissolved upon addition of the reductant with formation of dark green or brown solutions, which showed broad resonances in the 1H NMR spectra (Fig. S10 and S11†), indicating the formation of radical anions. As-formed radicals typically decomposed to the corresponding dipnictene and metallic pnictogene within a short period of time, but (DME)[K(B-18-C-6)][L(R2N)GaE]2 (E = Sb, R = Me 3; E = Bi, R = Et 4) were finally isolated from reactions of dipnictenes 1 and 2 in DME with KC8 in the presence of B-18-C-6 (Scheme 2) as green (3) and brown powders (4), respectively.
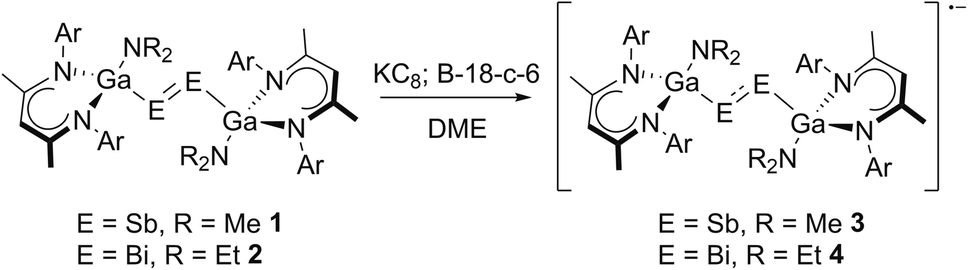 | ||
| Scheme 2 Synthesis of dipnictene radical anions 3 and 4 by reduction of dipnictenes 1 and 2; Ar = Dipp. | ||
3 showed broad resonances in the 1H NMR. The effective magnetic moment μeff of 1.85 μB (Evans method, Fig. S1†) agrees with the expected value for an unpaired electron (μeff = 1.73 μB), confirming the radical character of 3.24 Solutions of 3 were fairly stable in solution and in the solid state, whereas 4 was found to decompose in ether or toluene solutions even at low temperature (−35 °C) as well as in isolated crystalline form. Radical anion 4 was therefore only isolated in low yield (28%) by fast precipitation from a concentrated DME solution upon addition of n-hexane. The low thermal stability of radical anion 4 prevented it from purification by recrystallization, and all attempts yielded mixtures of the dibismuthene 2, radical anion 4 and elemental bismuth. The lower effective magnetic moment μeff of 1.50 μB as determined by use of the Evans method (Fig. S2†) most likely results from presence of small amounts diamagnetic impurities, i.e. dibismuthene 2 which was observed by 1H NMR spectroscopy. No paramagnetic species was detected by use of the Evans method (Fig. S13†) after storing a solution of 4 in THF for 6 h due to complete decomposition. The solution of 4 turned greenish during this time and a large amount of elemental bismuth formed within 24 h. Tokitoh et al. reported comparable findings for dibismuthene radical anion [(bbt)Bi]2˙−, which was formed as dark brown solution in the reduction of [(bbt)Bi]2 with metallic lithium,17 but EPR measurements even from freshly prepared samples failed due to its fast decomposition.
The UV-vis spectrum of [(bbt)Bi]2˙− showed a maximum at 804 nm which was assigned to the π–π* transition. UV-vis spectra of 3 (755 nm) and 4 (556, 738 nm) also showed absorption maxima in the visible region (Fig. 2), which are redshifted compared to neutral dipnictenes 1 (430 nm) and 2 (527 nm), indicating weakened π-bonds. The absorption band of 3 is in between the absorption maxima reported for [(bbt)Sb]2˙− and π–π* transitions calculated for [MesSb]2˙− (Mes = mesityl) radical anions (812, 728 nm).17
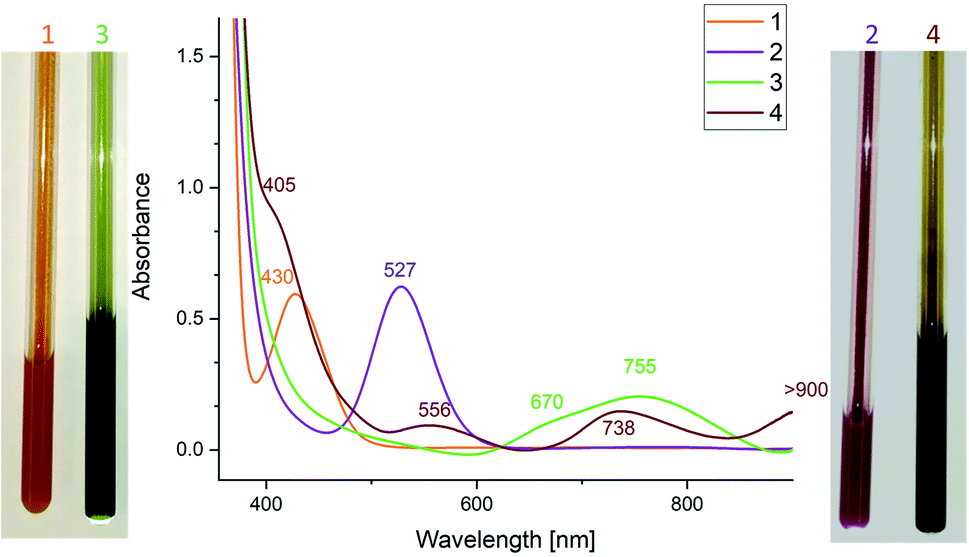 | ||
| Fig. 2 UV-vis spectra of dipnictenes 1 and 2 in benzene and dipnictene radical anions 3 and 4 in THF solution. To illustrate the colour change, pictures of solutions of 10 mg of 1 and 2 before and after addition of one equivalent of KC8 in NMR tubes are depicted. | ||
Crystallography
Single crystals were obtained from solutions in DME layered with n-hexane at −35 °C (3) and −30 °C (4). 3 and 4 (Fig. 3 and 4) crystallize in the monoclinic space group P21/c.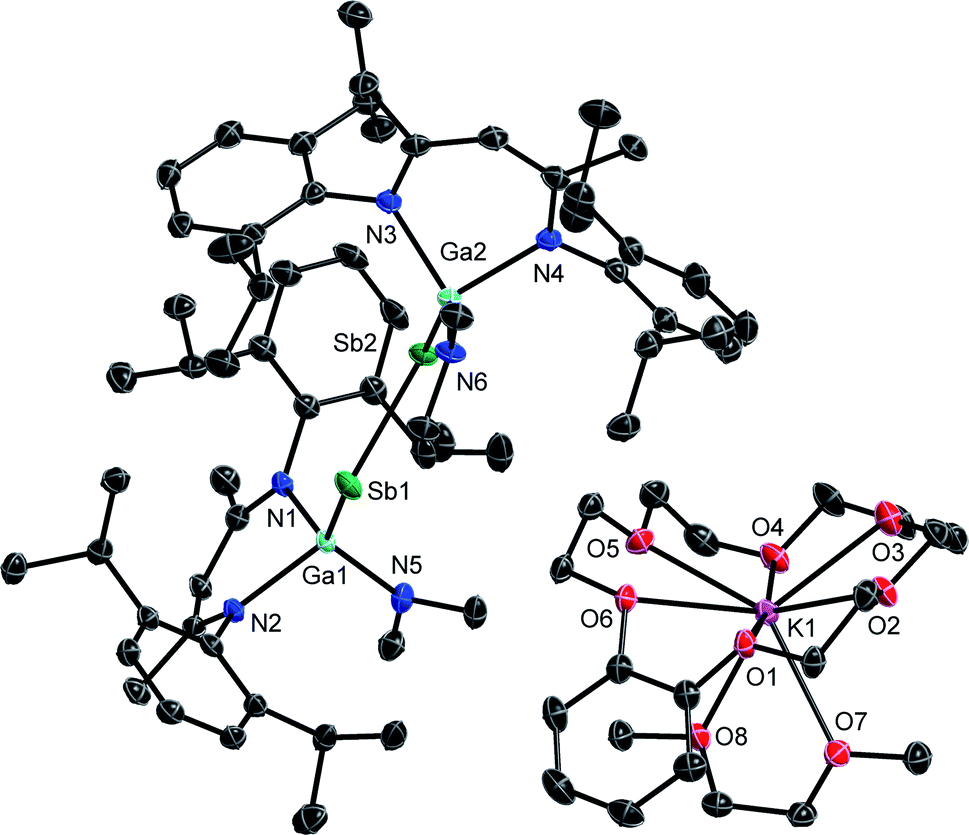 | ||
| Fig. 3 Molecular structure of 3 in the crystal. H-atoms and solvent molecules are omitted for clarity. Displacement ellipsoids are drawn at the 50% probability level. | ||
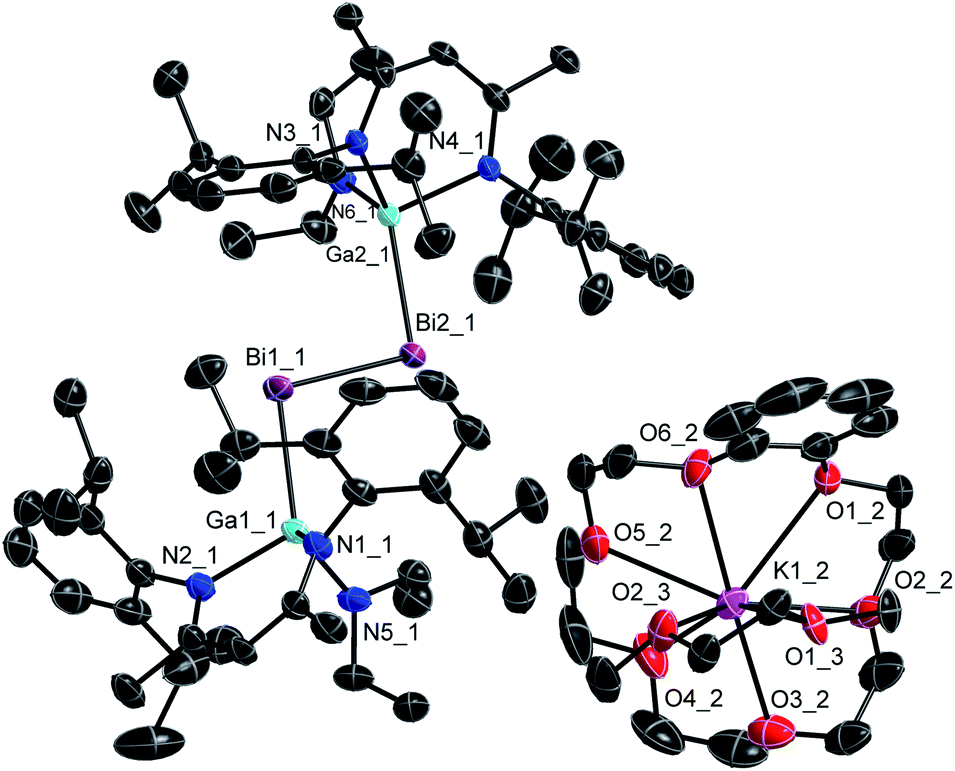 | ||
| Fig. 4 Molecular structure of 4 in the crystal. H-atoms and minor part of the disorder are omitted for clarity. Displacement ellipsoids are drawn at the 50% probability level. | ||
The Sb–Sb bond in 3 (2.7359(3) Å) is elongated compared to that in distibene [L(Me2N)GaSb]21 (2.6477(3) Å), but shorter than Sb–Sb single bonds in distibanes Sb2R4 (2.77–3.07 Å)25 and {[L(Cl)Ga](Ph)Sb}2 (2.8209(4) Å, Table 1),26 in agreement with a partially filled π*-orbital in radical anion 3. The Ga–Sb–Sb bond angle increases from distibene 1 (94.710(8)°) to radical anion 3 (100.41(1)°, 101.05(1)°), whereas neutral radicals [L(X)Ga]2Sb show slightly larger Ga–Sb–Ga bond angles (Cl: 104.89(1)°, Br: 103.47(5)°, I: 107.31(2)°).20a,c The Ga–Sb bond in radical anion 3 is slightly shortened compared to the neutral distibene 1 (2.6200(4) Å) and distibane {[L(Cl)Ga](Ph)Sb}2 (2.6255(3) Å), whereas all three Ga–N bonds in 3 are slightly elongated.
| E–E | Ga–E | Ga–Xa | Ga–N | Ga–E–Yc | N–Ga–N | X–Ga–Ea | |
|---|---|---|---|---|---|---|---|
| a X = Cl except for 1/3 (NMe2) and 2/4 (NEt2). b C–Sb–Sb. c Y = E for 1–4, any other Y = Ga. | |||||||
| 1 (ref. 10b) | 2.6477(3) | 2.6200(4) | 1.856(1) | 1.983(1), 1.989(1) | 94.710(8) | 93.16(5) | 116.37(4) |
| 3 | 2.7359(3) | 2.5826(4), 2.6052(4) | 1.886(2), 1.881(1) | 2.027(2), 2.004(2), 2.038(2), 2.038(2) | 100.41(1), 101.05(1) | 91.36(9), 90.84(9) | 120.25(7), 126.07(7) |
| [R(Ph)Sb]2 (ref. 26) | 2.8209(4) | 2.6255(3) | 2.2208(6) | 1.958(2), 1.953(2) | 102.80(1), 114.33(6)b | 95.82(7) | 116.78(2) |
| R2Sb (ref. 20a) | 2.5909(3), 2.5899(4) | 2.2028(7), 2.1623(9) | 1.956(2), 1.959(2), 1.961(2), 1.969(2) | 104.89(1) | 95.78(8), 95.69(8) | 121.46(2), 119.26(3) | |
| 2 (ref. 10d) | 2.8132(5) | 2.7061(6) | 1.884(5) | 2.006(4), 2.005(4) | 95.38(2) | 93.4(2) | 114.9(1) |
| 4 | 2.9266(3) | 2.6992(5), 2.6759(5) | 1.890(4), 1.902(3) | 2.057(3), 2.040(3), 2.036(3), 2.025(3) | 101.44(1), 98.73(1) | 91.6(1), 91.8(1) | 128.8(1), 123.6(1) |
| R2Bi˙ (ref. 10f) | 2.6485(3), 2.6619(4) | 2.2084(7), 2.2113(8) | 1.968(2), 1.955(2), 1.955(2), 1.964(2) | 105.46(1) | 95.2(1), 95.7(1) | 123.08(2), 112.92(3) | |
The Bi–Bi bond length in the dibismuthene radical anion 4 (2.9266(3) Å) is in between those of the neutral dibismuthene [L(Et2N)GaBi]22 (2.8132(3) Å) and of dibismuthanes Bi2R4 (2.98–3.18 Å).27 The Ga–Bi bond length is slightly shorter than that of the neutral dibismuthene 2, whereas all three Ga–N bond lengths in 4 are slightly elongated compared to those in 2 (Table 1). Again, the Ga–Bi–Bi bond angles substantially increased from 95.38(2)° (2) to 98.73(1)° and 101.44(1)° for the radical anion 4 as was also observed for distibene radical 3. The neutral L(X)Ga-substituted radicals [L(X)Ga]2Bi show slightly larger Ga–Bi–Ga bond angles (Cl: 105.46(1)°, I: 106.68(3)°) again.10f,20a These findings contrasts those reported for the only structurally characterized distibene radical anion [(bbt)Sb]2˙−IV, which showed a smaller C–Sb–Sb angle compared to the neutral distibene [(bbt)Sb]2.17 The origin of the increasing Ga–E–E bond angles of 3 and 4 compared to the neutral dipnictenes 1 and 2 is yet unclear. They might result from different intra- and intermolecular interactions including H⋯H and E⋯π dispersion interactions as was previously reported,13a from interactions of the radical anion with the sterically demanding cation or from packing effects.
EPR spectroscopy
The formation of the radical anion 3 was confirmed by electron paramagnetic resonance (EPR) spectroscopy. The continuous wave (CW) X-band (∼9.43 GHz) EPR spectrum of the THF solution at room temperature exhibits a highly broadened signal, centred at giso = 2.16 (Fig. 5).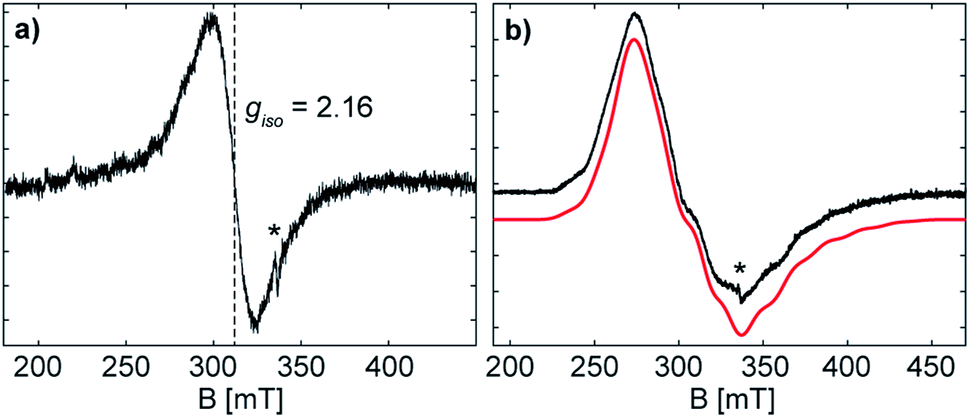 | ||
| Fig. 5 Continuous-wave EPR spectra of 3 (a) as solution in THF collected at X-band frequency (∼9.43 GHz) and (b) as frozen solution (77 K) collected at X-band frequency (∼9.45 GHz) in black with simulated spectrum in red. The asterisk indicates a small organic radical impurity. The frozen solution EPR (b) shows a broad signal with broad hyperfine features, due to the coupling of the unpaired electron with two Sb atoms, each of which possess NMR-active nucleotides (121Sb 57.21%, I = 5/2; 123Sb 42.79%, I = 7/2). EPR simulation parameters: g = [2.401, 2.051, 2.000], 2 × A(121Sb) = [120,200, 560] MHz, lw (linewidth, peak-to-peak) = 13 mT; spectrometer conditions are described in the Experimental section. | ||
The large g-shift observed from ge is the result of a large spin–orbit contribution (SOC) of the unpaired electron, supporting that the radical is metal-centred at the antimony atom(s).28 The CW X-band EPR spectrum of the frozen solution (Fig. 5) shows a broad signal with broad hyperfine features, due to the coupling of the unpaired electron with two Sb atoms. The frozen solution EPR of 3 may be simulated with a slightly rhombic g-tensor, g = [2.401, 2.051, 2.000], that has a giso value of 2.15, in good agreement with that measured at room temperature. The Sb hyperfine of the simulation, A(121Sb) = [120, 200, 560] MHz, is approximately axial with a maximum coupling of 560 MHz. The large line broadening observed is possibly due to additional unresolved hyperfine features (i.e. the 69/71Ga nuclei). The maximum Sb coupling of 3 is nearly half of that of the maximum Sb couplings observed in mononuclear Sb radical centres with similar Ga coordinating ligands,20a,28 and the estimated EPR parameters of 3 correspond well with the reported values for the distibene radical anion [(bbt)Sb]2˙−IV.17 The Sb hyperfine coupling observed for 3 and the agreement with a previously characterized distibene radical both support the assignment of the unpaired electron of 3 as delocalized in a Sb–Sb π* orbital.
The CW X-band EPR spectrum of the frozen solution of 4, (Fig. S16†), shows a highly broadened signal, that expands over a range from 100 mT to 550 mT. A multiline pattern is observed with approximately equal splitting of 20 mT (∼500 MHz), originating from the hyperfine interaction of the unpaired electron with the Bi nuclei (209Bi 100%, I = 9/2). This observed splitting is not assignable to any conical hyperfine value. The X-band EPR spectrum of 4 is significantly narrower than the spectrum of the mononuclear analogue [L(I)Ga]2Bi˙, whose X-band EPR spectrum expands over 800 mT.20a Assuming the same degree of g-anisotropy for both, a mono- and a dinuclear bismuth radical, this reduction in width of the EPR spectrum would be primarily due to a decrease in the Bi hyperfine interaction, supporting the delocalization over two metal centres as assigned by the crystal structure of 4.
The EPR spectrum of 4 at W-band frequency (94.01 GHz) (Fig. S16†) exhibits well separated g-values, but lacks resolved hyperfine features, probably due to additional strain at higher frequencies.29 Two EPR spectral components are observed with g1-values of 3.12 and 2.52 (Fig. S17†), whose origin is currently unknown; EPR measurements of multiple samples of both frozen solution and solid suspensions exhibit both components. Nonetheless, the significant shifts from ge are in line with increased SOC for heavier elements and supporting of a bismuth-centred radical.28 Simulations (Fig. S17†) allow for estimates of a maximum Bi hyperfine coupling of 700 MHz. This value is significantly smaller than the minimum coupling of ∼2800 MHz resolved by Schwamm et al.30 With respect to the weak Bi–Bi bond in 4, this radical anion may be prone to decomposition even at low temperatures, making the formation of another Bi radical possible. As discussed earlier, the significantly narrower EPR spectrum of 4 at X-band compared to [L(I)Ga]2Bi˙ eliminates the possible formation of a mononuclear Bi radical. In conclusion, the small hyperfine estimates, inferred from both the X- and W-band EPR spectra, is suggestive of a radical species delocalized over two bismuth atoms.
Quantum chemical calculations
The bonding nature and electronic structure of radical anions 3 and 4 were investigated by quantum chemical calculations at the PBE0 level of theory, and the results were compared to those obtained for neutral dipnictenes 1 and 2 to reveal electronic differences within the M2E2 unit.31 Calculated bond lengths within the M2E2 skeleton (Table 2) are in good agreement with the experimental values (Δr = 0.5–6 pm), even though one Ga atom in 4 is tilted out of the M2E2 plane.32 The spin density is concentrated within the E2 unit (Fig. 6 and 7), occupying the π* orbital. The SOMOs of 3 and 4 are similar to the LUMOs of 1 and 2 in agreement with the observed reversible reduction reactions. The calculated natural (Becke)33 spin densities show 92% (87%) location at the Sb centres and 3% (5%) at the Ga centres for 3, as well as 92% (89%)34 location at the Bi centres and 3% (4%) at the Ga centres for 4. In contrast, a contribution of 32% and 24% were reported for the As2 unit of divinyldiarsene III radical cations, with a higher density at the proximal carbon atoms (30% and 40%).16a| X–Y | r(X–Y) | q(X) | q(Y) | WBI | ONa | |
|---|---|---|---|---|---|---|
| a Squared polarization coefficients cX (|cX|2) of the σXY bond NBOs. | ||||||
| 1 | Ga1–Sb1 | 2.6040 (2.6200(4)) | 1.36 (1.37) | −0.16 (−0.16) | 0.97 | 1.96 (0.392/0.608) |
| Ga2–Sb2 | 2.6049 (2.6200(4)) | 1.38 (1.34) | −0.16 (−0.16) | 0.96 | 1.96 (0.390/0.601) | |
| Sb1–Sb2 | 2.6229 (2.6477(3)) | 1.82 | σ 1.95(0.502/0.498) | |||
| π 1.91 (0.501/0.499) | ||||||
| Sb1/2 lone-pair | 1.94/1.93 | |||||
| 2 | Ga1–Bi1 | 2.6737 (2.7061(6)) | 1.33 (1.33) | −0.10 (−0.11) | 0.98 | 1.96 (0.411/0.589) |
| Ga2–Bi2 | 2.6737 (2.7061(6)) | 1.33 (1.36) | −0.10 (−0.20) | 0.98 | 1.96 (0.412/0.588) | |
| Bi1–Bi2 | 2.7712 (2.8132(5)) | 1.81 | σ 1.94 (0.499/0.501) | |||
| π 1.90 (0.500/0.500) | ||||||
| Bi1/2 lone-pair | 1.95/1.95 | |||||
| X–Y | r(X–Y) | q(X) | q(Y) | WBI | ON(α) | ON(β)a | |
|---|---|---|---|---|---|---|---|
| 3 | Ga1–Sb1 | 2.5633 (2.5826(4)) | 1.35 (1.27) | −0.49 (−0.39) | 1.08 | 0.95 (0.376/0.624) | 0.95(0.386/0.614) |
| Ga2–Sb2 | 2.5595 (2.6052(4)) | 1.37 (1.24) | −0.51 (−0.39) | 1.08 | 0.95 (0.377/0.623) | 0.95 (0.383/0.617) | |
| Sb1–Sb2 | 2.7312 (2.7359(3)) | 1.37 | σ 0.97 (0.500/0.500) | σ 0.97 (0.499/0.501) | |||
| π 0.93 (0.494/0.506) | |||||||
| Sb1/2 lone-pair | 0.96/0.92 | 0.96/0.95 | |||||
| 4 | Ga3–Bi1 | 2.6407(2.6992(5)) | 1.34 (1.24) | −0.44 (−0.33) | 1.09 | 0.95 (0.339/0.605) | 0.94 (0.406/0.594) |
| Ga4–Bi2 | 2.6386 (2.6759(5)) | 1.34 (1.24) | −0.47 (−0.35) | 1.09 | 0.94 (0.391/0.609) | 0.94 (0.400/0.600) | |
| Bi1–Bi2 | 2.8895 (2.9266(3)) | 1.35 | σ 0.97 (0.503/0.407) | σ 0.97 (0.501/0.499) | |||
| π 0.92 (0.476/0.524) | |||||||
| Bi1/2 lone-pair | 0.96/0.92 | 0.96/0.95 |
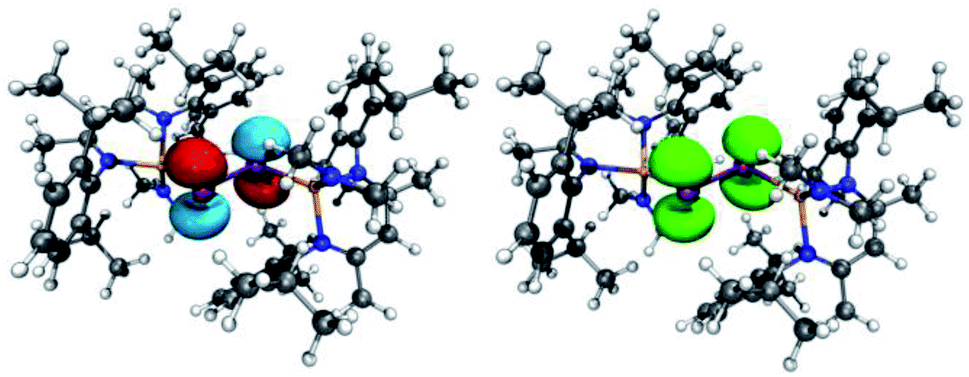 | ||
| Fig. 6 (left) LUMO of [L(Me2N)GaSb]21 (isovalue 0.05). (right) Spin density of (DME)[K(B-18-C-6)][L(Me2N)GaSb]23. | ||
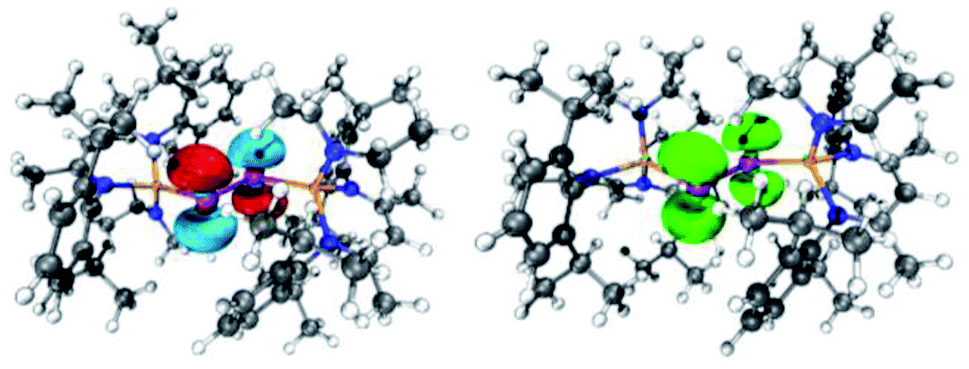 | ||
| Fig. 7 (left) LUMO of [L(Et2N)GaBi]22 (isovalue 0.05). (right) Spin density of (DME)[K(B-18-C-6)][L(Et2N)GaBi]24. | ||
Atoms in molecules (AIM), electron localization function (ELF), and natural bond orbital (NBO) analyses were performed to study the bonding situation of 1 to 4 (Table 2, Fig. S19 and Tables S2–S5†).35 NBO analyses revealed the formation of two-centre-two-electron σE–E and πE–E bonds with occupation numbers (ON) of 1.94, 0.93e for 3 and 1.94, 0.92e for 4, respectively. Compared to neutral dipnictenes (ON 1.95, 1.91e1; 1.94, 1.90e2) there is less electron density in the π-bonding orbital reflected by the reduced Wiberg bond indices (WBI) [1.82 (1) vs. 1.37 (3) and 1.81 (2) vs. 1.35 (4)].
ELF distribution reveals two disynaptic V(E,E) basins and two monosynaptic V(E) basins, and the occupation shifts towards monosynaptic basins for radical anion (![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) [V(Sb)] = 2.7 to 3.7e and [V(Sb–Sb)] = 1.4 to 0.9e and [V(Bi)] = 2.9 to 4.0e and [V(Bi–Bi)] = 1.3 to 0.7e). The increase in natural charge from −0.16 (1) to −0.50e (3) and −0.10 (2) to −0.46e (4) indicates a localization of the negative charge with in the E2 unit, whereas the natural charge on the Ga centres is not affected. However, the WBI for the Ga–E bond moderately increases from 0.97 (1) to 1.08 (3) and 0.98 (2) to 1.09 (4), which clearly reflects the shorter Ga–E bond as observed in the solid state. The number of electrons in the ELF basin remains the same (V(Ga,Sb) = 2.2e, V(Ga,Bi) = 2.2e), but the contribution of the electrons of gallium according to ELF/AIM intersection procedure ([V(Ga,Sb)|Ga]) increases from 1.00e (1,2) to 1.15e (3) and 1.20e (4).36 The ON of 1.9e and |V(rb)|/G(rb) = 2.4 (3) 2.2 (4) indicate a covalent Ga–Sb and Ga–Bi interaction for 3. In addition, weak orbital interaction between the π*E–E and the σ*Ga–N orbital were observed (NBO), and elongated Ga–N bonds found in the solid state.
[V(Sb)] = 2.7 to 3.7e and [V(Sb–Sb)] = 1.4 to 0.9e and [V(Bi)] = 2.9 to 4.0e and [V(Bi–Bi)] = 1.3 to 0.7e). The increase in natural charge from −0.16 (1) to −0.50e (3) and −0.10 (2) to −0.46e (4) indicates a localization of the negative charge with in the E2 unit, whereas the natural charge on the Ga centres is not affected. However, the WBI for the Ga–E bond moderately increases from 0.97 (1) to 1.08 (3) and 0.98 (2) to 1.09 (4), which clearly reflects the shorter Ga–E bond as observed in the solid state. The number of electrons in the ELF basin remains the same (V(Ga,Sb) = 2.2e, V(Ga,Bi) = 2.2e), but the contribution of the electrons of gallium according to ELF/AIM intersection procedure ([V(Ga,Sb)|Ga]) increases from 1.00e (1,2) to 1.15e (3) and 1.20e (4).36 The ON of 1.9e and |V(rb)|/G(rb) = 2.4 (3) 2.2 (4) indicate a covalent Ga–Sb and Ga–Bi interaction for 3. In addition, weak orbital interaction between the π*E–E and the σ*Ga–N orbital were observed (NBO), and elongated Ga–N bonds found in the solid state.
TDDFT calculation of 1–4 were performed and compared to experimental UV-vis spectra (Table S6 and Fig. S20–S23†).31 The transitions of 1 and 3 agree well with the experimental values and known π → π* transitions (Table 3). The transitions of the radical anions are redshifted compared to those of the neutral species as was previously reported for the aryl-substituted species [(bbt)Sb]2˙− and [(bbt)Bi]2˙−,4b,12b,17 respectively.
The calculated absorptions of 2 and 4 deviate slightly more from the experimental values than those of 1 and 3. Moreover, 4 shows a larger redshift than the corresponding bbt substituted radical anion [(bbt)Bi]2˙−, indicating a lower lying SOMO of 4. The gallium-based ligands seem to bind stronger to the bismuth centre compared to the bbt substituents, which also explains the higher thermal stability of 4 in solution when compared to [(bbt)Bi]2˙−. These findings might result from a better orbital overlap due to the comparable size of gallium and bismuth as well as the higher electropositive nature of gallium compared to carbon, which supports the stabilization of the negative charge.
Conclusions
L(R2N)Ga-substituted dipnictenes [L(R2N)GaE]2 (E = Sb, R = Me 1; E = Bi, R = Et 2) showed reversible reduction events in cyclic voltammetry studies. Reduction of distibene 1 and dibismuthene 2 with KC8 in the presence of B-18-C-6 yielded rare dipnictene radical anions (DME)[K(B-18-C-6)][L(R2N)GaE]2 (E = Sb, R = Me 3; E = Bi, R = Et 4), which were characterized by single-crystal X-ray diffraction. 4 represents the first structurally characterized dibismuthene radical anion, and the radical nature of 3 and 4 was confirmed by EPR spectroscopy. Quantum chemical calculations revealed the different bonding nature in compounds 1–4 and are in accordance with the formation of pnictogen-centred radicals 3 and 4 due to a high localization of spin density and charge within the central E2 unit. The elongated E–E and shorter Ga–E bond lengths of radicals 3 and 4 compared to the neutral dipnictenes 1 and 2 result from the population of the π*E–E orbital with one electron, leading to a formal bond order of 1.5.The capability of the L(X)Ga substituent for the stabilization of unusual main group metal compounds most likely results from its rather electropositive nature, which reduces orbital energies as was recently reported for the silylene [L(Br)Ga]2Si, which reacted with carbon monoxide with formation of the first room temperature-stable silylene–carbonyl complex.37 We assume that the introduction of L(X)Ga substituents to the E2 unit of the dipnictenes 1 and 2 lowers the SOMO energies of the radical anions compared to aryl-substituted species, hence allowing its population with one electron in radical anions 3 and 4. In addition, the Ga–E bonds in the radical anions 3 and 4 are slightly stronger compared to those in the neural dipnictenes 1 and 2 (WBI increase of 0.11, Table 2), thus reducing the tendency of radical anions to undergo bond homolysis reaction with formation of elemental pnictogenes.
Experimental
General procedures
All manipulations were carried out using standard Schlenk and glove-box techniques under argon, which was dried by passing through pre-heated Cu2O pellets and molecular sieves columns. n-Hexane and toluene were dried with a MBraun Solvent Purification System, benzene, THF, DME as well as deuterated THF and benzene were distilled from Na/K alloy. All solvents were degassed and stored over activated molecular sieves (4 Å). [L(Me2N)GaSb]210b and [L(Et2N)GaBi]210d were prepared according to literature methods. 1H (300 MHz) spectra were recorded using a Bruker Avance DPX-300 spectrometer and referenced to internal C6D5H (1H: δ = 7.16 or THF-d8 (1H: δ = 1.72). Solution-state magnetic susceptibilities χM and effective magnetic moments μeff were determined by 1H NMR spectroscopy using Evans method24 with pure solvent as internal reference and neglecting diamagnetic contributions.38 IR spectra were recorded in a glovebox using a Bruker ALPHAT FT-IR spectrometer equipped with a single reflection ATR sampling module. Cyclic voltammetry studies were performed in a glovebox using a Metrohm Autolab PGSTAT 204 potentiostat with a three electrodes setup consisting of a Pt disc (d = 1 mm) working electrode, Pt wire counter electrode, and Ag wire pseudo-reference electrode, and ferrocene as internal standard. Positive feedback compensation was utilized to reduce solvent resistance effects. UV-vis-spectra were recorded on a Shimadzu UV-2600i spectrophotometer in closed glass cuvette (10 mm) under an argon atmosphere.The samples of 3 in THF for the EPR measurements were prepared in a glovebox in either 50 μL capillaries (Hirschmann), sealed with Critoseal, or frozen in custom 4 mm (OD) quartz EPR tubes. Continuous-wave (CW) X-band EPR spectra at room temperature (∼9.43 GHz) and at 77 K (∼9.45 GHz) were collected with a Bruker MS 5000 spectrometer. The spectra were obtained with 100 kHz field modulation frequency, 8 G modulation amplitude, 30 mW microwave power and a scan time of 360 s. An effective time constant of 0.05 s was applied digitally to the ∼60 k point spectrum. For the spectra at room temperature and at 77 K, six and three scans were acquired, respectively. EPR samples of 4 were freshly prepared in DME and then filled in custom 2.8 mm (OD, X-band) and 0.9 mm (OD, W-band) quartz EPR tubes and frozen immediately. CW X-band EPR spectra of 4 were collected at ∼6 K on a Bruker Elexsys E500 equipped with an Oxford helium flow cryostat and temperature controller. The X-band spectra of 4 were collected with the following parameters: modulation amplitude: 6 G; modulation frequency: 100 kHz; time constant: 81.92 ms; scan time: 336 s; single scan. W-band pulsed EPR measurements of 4 were collected on a Bruker Elexsys E680 spectrometer at ∼7 K equipped with a closed cycle helium cryostat system. The spectra were collected with a two-pulse ‘Hahn’ sequence (π/2–τ–π–τ–echo) with the following parameters: π/2 = 20 ns; τ = 400 ns; repetition rate = 60 μs; effective shots per point: 11060; effective number of points: 4000; 2 scans. The magnet was swept both up and down at the same sweep rate and the offsets averaged to account for sweep delays. The EPR data was processed and analysed in Matlab R2019b and simulated using the EasySpin package (v. 6.0.0-dev.30).39
Synthesis of (DME)[K(B-18-C-6)][L(Me2N)GaSb]2 (3)
[L(Me2N)GaSb]21 (110 mg, 84 μmol), KC8 (11.4 mg, 84 μmol) and B-18-C-6 (26.3 mg, 84 μmol) were suspended in 4 ml of DME at 0 °C and the suspension was stirred for 1 h, upon which a dark green solution formed, which was concentrated to 1 ml and filtered. The filtrate was further concentrated to almost dryness and layered with n-hexane and stored at ambient temperature, yielding a green crystalline powder which was isolated by filtration. Single crystals were obtained upon storage of a solution of 3 in DME that was layered with n-hexane at −35 °C.Yield: 144 mg (80 μmol, 95%). Anal. calcd for C82H128Ga2KN6O8Sb2: C, 56.34; H, 7.38; N, 4.81. Found: C, 56.0; H, 7.49, N 4.61% ATR-IR: ν 3058, 2955, 2859, 2740, 1438, 1399, 1317, 1248, 1175, 1109, 1092, 969, 793, 741, 523, 439 cm−1. 1H NMR (300 MHz THF-d8, 25 °C): μeff = 1.85 μB. No coalescence was observed at −100 °C.
Synthesis of (DME)[K(B-18-C-6)][L(Et2N)GaBi]2 (4)
[L(Et2N)GaBi]22 (80 mg, 52 μmol), KC8 (7.0 mg, 52 μmol) and B-18-C-6 (16.3 mg, 52 μmol) were suspended in 0.3 ml of DME in a centrifuge vial and shaken for 5 minutes. Solids were separated by centrifugation and the resulting dark brown solution was mixed with 10 ml of n-hexane and stored at −30 °C for 30 min. The resulting cloudy supernatant was removed, and 4 was isolated as a dark brown crystalline solid, which washed with n-hexane twice. Storage of a freshly prepared solution of 4 in DME that was layered with n-hexane at −30 °C yielded few crystals suitable for SC-XRD.Yield: 28 mg (14 μmol, 28%). Anal. calcd for C86H136Bi2Ga2KN6O8 + one n-hexane molecule from unit cell: C, 53.52; H, 7.32; N, 4.07. Found: C, 53.5; H, 7.10, N 4.30%. ATR-IR: ν 3048, 2946, 2911, 2851, 1501, 1447, 1433, 1390, 1350, 1311, 1245, 1105, 1085, 1051, 950, 932, 789, 753, 736 cm−1. 1H NMR (300 MHz, THF-d8, 25 °C): μeff = 1.50 μB.
Data availability
All additional data is part of the ESI file.†Author contributions
M. W. performed the experiments including quantum chemical calculations, C. W. the single crystal X-ray diffraction and J. H. the EPR measurements. The work was supervised by G. C. and St. S. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the Max-Planck-Gesellschaft (GEC) and the Deutsche Forschungsgemeinschaft DFG (St. S., SCHU 1069/23-1) for generous financial support.Notes and references
- (a) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed; (b) S. Yadav, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 486–501 CrossRef CAS; (c) S. Schulz, Chem.–Eur. J., 2010, 16, 6416–6428 CrossRef CAS PubMed.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- (a) M. K. Sharma, S. Blomeyer, B. Neumann, H. G. Stammler and R. S. Ghadwal, Chem.–Eur. J., 2019, 25, 8249–8253 CrossRef CAS PubMed; (b) P. K. Majhi, H. Ikeda, T. Sasamori, H. Tsurugi, K. Mashima and N. Tokitoh, Organometallics, 2017, 36, 1224–1226 CrossRef CAS; (c) T. Sasamori and N. Tokitoh, Dalton Trans., 2008, 1395–1408 RSC; (d) T. Sasamori, Y. Arai, N. Takeda, R. Okazaki, Y. Furukawa, M. Kimura, S. Nagase and N. Tokitoh, Bull. Chem. Soc. Jpn., 2002, 75, 661–675 CrossRef CAS; (e) B. Twamley, C. D. Sofield, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1999, 121, 3357–3367 CrossRef CAS; (f) N. Tokitoh, Y. Arai, T. Sasamori, R. Okazaki, S. Nagase, H. Uekusa and Y. Ohashi, J. Am. Chem. Soc., 1998, 120, 433–434 CrossRef CAS; (g) N. Tokitoh, Y. Arai, R. Okazaki and S. Nagase, Science, 1997, 277, 78–80 CrossRef CAS.
- R. Schwamm and M. P. Coles, Chem.–Eur. J., 2019, 25, 14183–14191 CrossRef CAS PubMed.
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128–7131 RSC.
- M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, Chem.–Asian J., 2013, 8, 690–693 CrossRef CAS PubMed.
- C. von Hänisch and D. Nikolova, Eur. J. Inorg. Chem., 2006, 4770–4773 CrossRef.
- (a) L. P. Ho and M. Tamm, Dalton Trans., 2021, 50, 1202–1205 RSC; (b) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815–11832 CrossRef CAS PubMed.
- (a) G. Prabusankar, C. Gemel, P. Parameswaran, C. Flener, G. Frenking and R. A. Fischer, Angew. Chem., Int. Ed., 2009, 48, 5526–5529 CrossRef CAS PubMed; (b) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657–10661 CrossRef CAS PubMed; (c) L. Tuscher, C. Helling, C. Ganesamoorthy, J. Krüger, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2017, 23, 12297–12304 CrossRef CAS PubMed; (d) L. Tuscher, C. Helling, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2018, 24, 3241–3250 CrossRef CAS PubMed; (e) J. Schoening, L. John, C. Wölper and S. Schulz, Dalton Trans., 2019, 48, 17729–17734 RSC; (f) J. Krüger, C. Wölper and S. Schulz, Inorg. Chem., 2020, 59, 11142–11151 CrossRef PubMed.
- L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chem. Rev., 2019, 119, 8781–8845 CrossRef CAS PubMed.
- (a) P. Vilarrubias, Mol. Phys., 2017, 115, 2597–2604 CrossRef CAS; (b) T. Sasamori, E. Mieda, N. Nagahora, N. Takeda, N. Takagi, S. Nagase and N. Tokitoh, Chem. Lett., 2005, 34, 166–167 CrossRef CAS.
- (a) L. Song, J. Schoening, C. Wölper, S. Schulz and P. R. Schreiner, Organometallics, 2019, 38, 1640–1647 CrossRef CAS; (b) J. Krüger, J. Schoening, C. Ganesamoorthy, L. John, C. Wölper and S. Schulz, Z. Anorg. Allg. Chem., 2018, 644, 1028–1033 CrossRef.
- (a) K. Schwedtmann, M. H. Holthausen, C. H. Sala, F. Hennersdorf, R. Fröhlich and J. J. Weigand, Chem. Commun., 2016, 52, 1409–1412 RSC; (b) A. Doddi, D. Bockfeld, M.-K. Zaretzke, C. Kleeberg, T. Bannenberg and M. Tamm, Dalton Trans., 2017, 46, 15859–15864 RSC; (c) A. Beil, R. J. Gilliard and H. Grützmacher, Dalton Trans., 2016, 45, 2044–2052 RSC; (d) S.-S. Asami, S. Ishida, T. Iwamoto, K. Suzuki and M. Yamashita, Angew. Chem., Int. Ed., 2017, 56, 1658–1662 CrossRef CAS PubMed; (e) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 CrossRef CAS PubMed; (f) M. K. Sharma, D. Rottschäfer, S. Blomeyer, B. Neumann, H.-G. Stammler, M. van Gastel, A. Hinz and R. S. Ghadwal, Chem. Commun., 2019, 55, 10408–10411 RSC; (g) O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262–10263 CrossRef CAS PubMed; (h) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 CrossRef CAS PubMed; (i) X. Pan, Y. Su, X. Chen, Y. Zhao, Y. Li, J. Zuo and X. Wang, J. Am. Chem. Soc., 2013, 135, 5561–5564 CrossRef CAS PubMed.
- (a) A. Doddi, D. Bockfeld, M.-K. Zaretzke, T. Bannenberg and M. Tamm, Chem.–Eur. J., 2019, 25, 13119–13123 CrossRef CAS PubMed; (b) K. Tsuji, Y. Fujii, S. Sasaki and M. Yoshifuji, Chem. Lett., 1997, 9, 855–856 CrossRef.
- (a) M. K. Sharma, S. Blomeyer, B. Neumann, H.-G. Stammler, M. van Gastel, A. Hinz and R. S. Ghadwal, Angew. Chem., Int. Ed., 2019, 58, 17599–17603 CrossRef CAS PubMed; (b) M. Y. Abraham, Y. Wang, Y. R. Xie, J. Gilliard, P. Wei, B. J. Vaccaro, M. K. Johnson, H. F. Schaefer, P. V. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2013, 135, 2486–2488 CrossRef CAS PubMed.
- T. Sasamori, E. Mieda, N. Nagahora, K. Sato, D. Shiomi, T. Takui, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 12582–12588 CrossRef CAS PubMed.
- C. Helling and S. Schulz, Eur. J. Inorg. Chem., 2020, 3209–3221 CrossRef CAS.
- (a) G. Tan and X. Wang, Chin. J. Chem., 2018, 36, 573–586 CrossRef CAS; (b) S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727–4741 RSC; (c) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed; (d) C. Lichtenberg, Chem.–Eur. J., 2020, 26, 9674–9687 CrossRef CAS PubMed.
- (a) C. Ganesamoorthy, C. Helling, C. Wölper, W. Frank, E. Bill, G. E. Cutsail III and S. Schulz, Nat. Commun., 2018, 9, 87 CrossRef PubMed; (b) C. Helling, C. Wölper, Y. Schulte, G. E Cutsail III. and S. Schulz, Inorg. Chem., 2019, 58, 10323–10332 CrossRef CAS PubMed; (c) C. Helling, C. Wölper, G. E. Cutsail III, G. Haberhauer and S. Schulz, Chem.–Eur. J., 2020, 26, 13390–13399 CrossRef CAS PubMed; (d) C. Helling, G. E. Cutsail III, H. Weinert, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2020, 59, 7561–7568 CrossRef CAS PubMed.
- P. Zanello, Inorganic Electrochemistry: Theory, Practice and Application, Royal Society of Chemistry, Cambridge, 2003 Search PubMed.
- G. Inzelt, A. Lewenstam and F. Scholz, Handbook of Reference Electrodes, Springer, Berlin, Heidelberg, 2013 Search PubMed.
- (a) V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS; (b) J. R. Aranzaes, M.-C. Daniel and D. Astruc, Can. J. Chem., 2006, 84, 288–299 CrossRef.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- Cambridge Structural Database, version 5.42, see also: F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed , 55 hits containing an Sb–Sb bond classified as a single were found of the type R2Sb–SbR2 (R defined as non-antimony, c.n. (Sb) = 3). The Sb–Sb bond lengths range from 2.77–3.07 Å (mean of 2.86(5) Å excluding a single highly ring strain structure 2.64 Å)..
- C. Helling, C. Wölper and S. Schulz, Eur. J. Inorg. Chem., 2020, 4225–4235 CrossRef CAS.
- Cambridge Structural Database, version 5.42, see also: F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed , 17 unique hits containing an Bi–Bi bond classified as a single were found of the type R2Bi–BiR2 (R defined as non-bismuth, c.n. (Bi) = 3). The Bi–Bi bond lengths ranging from 2.98–3.18 Å with a mean of 3.03(4) Å..
- G. E. Cutsail III, Dalton Trans., 2020, 49, 12128–12135 RSC.
- W. Hagen, Coord. Chem. Rev., 1999, 190–192, 209–229 CrossRef CAS.
- R. Schwamm, J. Harmer, M. Lein, C. Fitchett, S. Granville and M. Coles, Angew. Chem., Int. Ed., 2015, 54, 10630–10633 CrossRef CAS PubMed.
- The ORCA quantum chemistry package was used and details regarding the quantum chemical calculations are given in the ESI† (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–6 Search PubMed; (b) F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- In all cases the geometry optimization was started from the geometry obtained by SC-XRD. In case of the dipnictenes radical anion the presence of counter ion disturbs the symmetry, with a different conformation of LX(Ga), as is reflected by the difference of the N-Ga-E-E torsion angle (Sb: 83.3(9) and 20.8(1) Bi: −23.2(1) and −89.6(1)). As no symmetry and other constraints were invoked the minimum on the energy surface exhibited an asymmetry as well and shift the Ga centre to reduce the repulsion of the ligand. This is only reflected in a small difference of spin density and charge of the Bi centres.
- A. D. Becke, J. Chem. Phys., 1998, 88, 2547–2553 CrossRef.
- Spin density: Bi1 49%, Bi2 43%. Natural charge: Bi1 −0.44e−, Bi2 −0.47e−.
- (a) C. Kalaiarasi, M. S. Pavan and v. P. Kumaradhas, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 775–786 CrossRef CAS PubMed; (b) P. Kumar, V. Shayam, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 128, 1527–1536 CrossRef CAS; (c) R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS; (d) B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS; (e) A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS; (f) L. U. Tian and C. Fei-Wu, Acta Phys.-Chim. Sin., 2011, 27, 2786–2792 Search PubMed.
- S. Raub and G. Jansen, Theor. Chem. Acc., 2001, 106, 223–232 Search PubMed.
- C. Ganesamoorthy, J. Schoening, C. Wölper, L. Song, P. R. Schreiner and S. Schulz, Nat. Chem., 2020, 12, 608–614 CrossRef CAS PubMed.
- G. J. P. Britovsek, V. C. Gibson, S. K. Spitzmesser, K. P. Tellmann, A. J. P. White and D. J. Williams, Dalton Trans., 2002, 1159–1171 RSC.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and analytical data, NMR, IR, EPR, and UV-vis spectra, computational details and cif files. CCDC 2077932 (3) and 2080779 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04230k |
| This journal is © The Royal Society of Chemistry 2021 |