
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetic resolution of sulfur-stereogenic sulfoximines by Pd(II)–MPAA catalyzed C–H arylation and olefination†
Kallol
Mukherjee
a,
Nicolas
Grimblat‡
 b,
Somratan
Sau‡
a,
Koushik
Ghosh
a,
Majji
Shankar
a,
Vincent
Gandon
*bc and
Akhila K.
Sahoo
*a
b,
Somratan
Sau‡
a,
Koushik
Ghosh
a,
Majji
Shankar
a,
Vincent
Gandon
*bc and
Akhila K.
Sahoo
*a
aSchool of Chemistry, University of Hyderabad, Hyderabad, 500046, India. E-mail: akhilchemistry12@gmail.com; akssc@uohyd.ac.in
bNicolas Grimblat and Prof. Vincent Gandon, Institut de Chimie Moléculaire et des Matériaux d’Orsay, CNRS UMR 8182, Université Paris-Saclay, Bâtiment 420, 91405 Orsay Cedex, France. E-mail: vincent.gandon@universite-paris-saclay.fr
cLaboratoire de Chimie Moléculaire (LCM), CNRS UMR 9168, Ecole Polytechnique, Institut Polytechnique de Paris, Route de Saclay, 91128 Palaiseau Cedex, France
First published on 20th October 2021
Abstract
A direct Pd(II)-catalyzed kinetic resolution of heteroaryl-enabled sulfoximines through an ortho-C–H alkenylation/arylation of arenes has been developed. The coordination of the sulfoximine pyridyl-motif and the chiral amino acid MPAA ligand to the Pd(II)-catalyst controls the enantio-discriminating C(aryl)–H activation. This method provides access to a wide range of enantiomerically enriched unreacted aryl-pyridyl-sulfoximine precursors and C(aryl)–H alkenylation/arylation products in good yields with high enantioselectivity (up to >99% ee), and selectivity factor up to >200. The coordination preference of the directing group, ligand effect, geometry constraints, and the transient six-membered concerted-metalation–deprotonation species dictate the stereoselectivity; DFT studies validate this hypothesis.
Introduction
The directing group (DG) assisted desymmetrization of prochiral C–H bonds provides a suitable way to construct carbon, phosphorus, silicon, and sulfur centered functionalized chiral molecules.1–3 However, this approach requires achiral precursors with two identical enantiotopic groups, which prevents its application for broad synthetic benefits. On the other hand, kinetic resolution (KR) of C–H bonds offers booming advantages for making functionalized enantioenriched molecules. In this regard, Yu's pioneering work on DG assisted chiral amino acid (MPAA) enabled Pd-catalyzed carbon centered KR of arene C–H bonds through alkenylation, arylation, and/or iodination is undoubtedly a breakthrough (Fig. 1A).4 In spite of this success, the related strategy of Pd-catalyzed heteroatom centered KR of arenes remains to be explored, although exceedingly appealing. | ||
| Fig. 1 Kinetic resolution via C–H activation (free energies at 343.15 K in DCE). | ||
Sulfoximines, which are configurationally stable motifs with S-stereogenecity, are found in molecules of medicinal importance and agrochemicals.5 Notably, sulfoximines have emerged as chiral auxiliaries and DG for C–H functionalizations.6 The syntheses of enantioenriched sulfoximines have invariably relied on resolution techniques, stereoselective imination, and oxidation processes.7,8
Elegant enantioselective and KR routes to sulfoximines have been independently developed by Cramer, Li, Shi, and others, but all these approaches rely on Rh/Ru-catalyzed [4 + 2] annulation of diazoesters/sulfoxonium ylides and aryl-sulfoximines in the presence of specially designed ligands.9 On our side, we have devised an expedient Pd-catalyzed C–H functionalization method for KR of 2-pyridylaryl sulfoximines, using Pd(II) catalyst and MPAA ligand, via C(aryl)–H arylation and olefination (Fig. 1B). The concept relies on kinetically regulated concerted-metalation–deprotonation (CMD) step of C(aryl)–H activation (k1 ≫ k2, Fig. 1B) through preferred coordination of pyridine over imine to Pd–MPAA (Fig. 1C-I)10 and ligand geometry CMDPyr-DS over CMDPyr-DR (Fig. 1C-II). The transformation is general, constructing a wide array of enantiomerically enriched C-olefinated/arylated aryl-pyridyl-S-sulfoximines.
Results and discussion
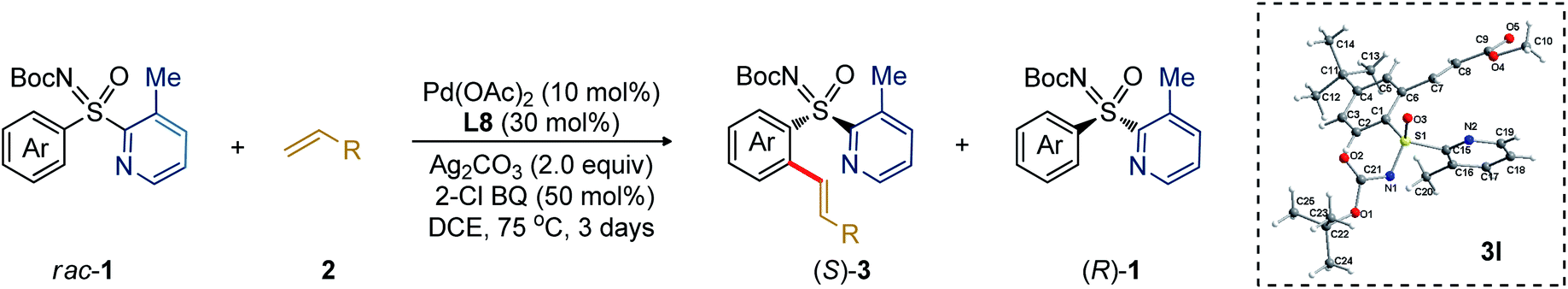
The study was initiated with the non-substituted N-Boc-phenyl-2-pyridyl sulfoximine rac-1a-1 and ethyl acrylate (2a; 0.6 equiv.) in presence of Pd(OAc)2 (10 mol%), Boc-L-Phe-OH (L1; 30 mol%), Ag2CO3 (2.0 equiv.) in ClCH2CH2Cl (1,2-DCE) at 75 °C (Table 1a). The desired C2-alkenylation product (S)-3a-1 (18%, conversion after 3 days) along with precursor (R)-1a-1 were obtained in 75% ee and 17% ee, respectively, exhibiting a low selectivity factor (s) of 8. This encouraging result unfolded our curiosity about examining the effect of other ligands. None of the N-Boc, N-acetyl-, and N-imide-protected commercially available α-amino acid ligands (L2–L6) with distinct side chains were effective. Assuming the additional coordination ability of the easily modifiable OH group in threonine, various N,O-protected threonine ligands were tested. The reaction s factor was improved a little for (S)-3a-1 to 12 and 11 when Boc-L-Thr(t-Bu)-OH (L7) and Boc-L-Thr(Bn)-OH (L8) were used, respectively. Electronic perturbation in the O-benzyl moiety did not have any impact on the enantioselectivity (L9 and L10). The use of 2a (2.0 equiv.) in presence of ligand L8 improved the conversion (50%) with (S)-3a-1 (70% ee) (entry 13).|
|
|||||
|---|---|---|---|---|---|
| (b) Additive screening | |||||
| Entry | Additives | C | eec (%) | s | |
| (R)-1a-1 | (S)-3a-1 | ||||
| a Reaction conditions:rac-1a-1 (0.1 mmol), ethyl acrylate 2a (0.6 equiv.), Pd(OAc)2 (10 mol%), ligand (30 mol%), Ag2CO3 (2.0 equiv.), 2-Cl-BQ (0.3 equiv.), 1,2-DCE (1.0 mL), N2, 75 °C, 3 days. b Calculated conversion, C = eeSM/(eeSM + eePR). c Determined by chiral HPLC analysis. d Selectivity (s) = ln[(1 − C)(1 − eeSM)]/ln[(1 − C)(1 + eeSM)]. e 2a (2.0 equiv.) used. | |||||
| 1 | BQ | 18 | 17 | 78 | 10 |
| 2 | 2-Chloro BQ | 39 | 50 | 77 | 13 |
| 3 | 2,5-dichloro BQ | 35 | 40 | 75 | 10 |
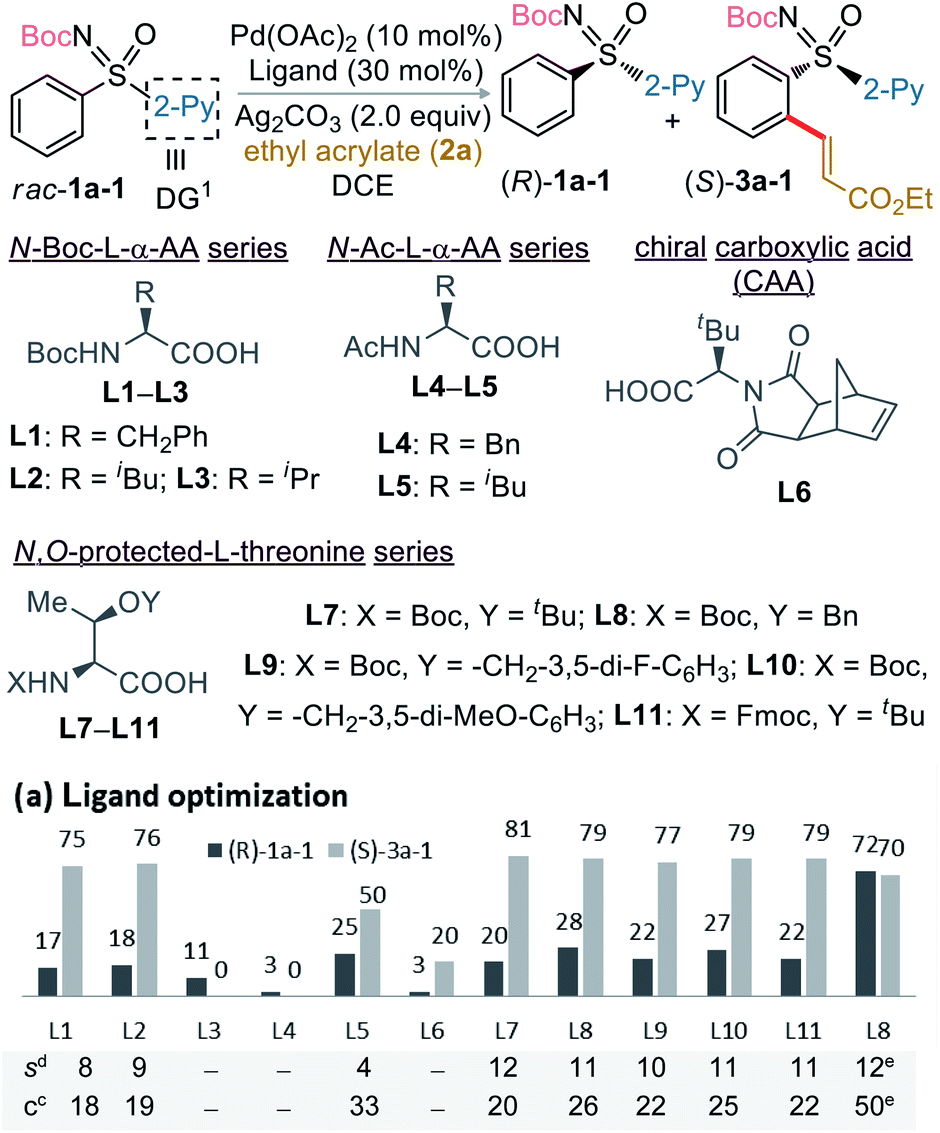
To enhance sulfoximine resolution efficiency while maintaining conversion (∼50%; Table 1a), we scrutinized the co-oxidant effect (Table 1b).11,12 2-Chlorobenzoquinone (2-Cl-BQ) was found to be the best, providing (S)-3a-1 in 77% ee with 39% conversion (s factor of 13; entry-2, Table 1b). Next, sulfoximines having various N-protecting groups (PG) were screened; the results are shown in Table 2a. None of the N–Me/Piv/Cbz-protected sulfoximines were found effective.
| (a) Protecting group screening | |||||
|---|---|---|---|---|---|
|
|
|||||
| Entry | PG | C | eec (%) | s | |
| (R)-1a-1 | (S)-3a-1 | ||||
| a Reaction conditions: rac-1a-1 (0.1 mmol), ethyl acrylate 2a (0.6 equiv.), Pd(OAc)2 (10 mol%), ligand (30 mol%), Ag2CO3 (2.0 equiv.), 2-Cl-BQ (0.3 equiv.), 1,2-DCE (1.0 mL), N2, 75 °C, 3 days. b Calculated conversion, C = eeSM/(eeSM + eePR). c Determined by chiral HPLC analysis. d Selectivity (s) = ln[(1 − C)(1 − eeSM)]/ln[(1 − C)(1 + eeSM)]. e Olefin (2.0 equiv.) used. f Instead of 2a, methyl acrylate was used. g 50 mol% 2-Cl-BQ was used. | |||||
| 1 | Me | 0 | — | — | — |
| 2 | Piv | 45 | 54 | 66 | 8 |
| 3 | Cbz | 20 | 21 | 85 | 14 |
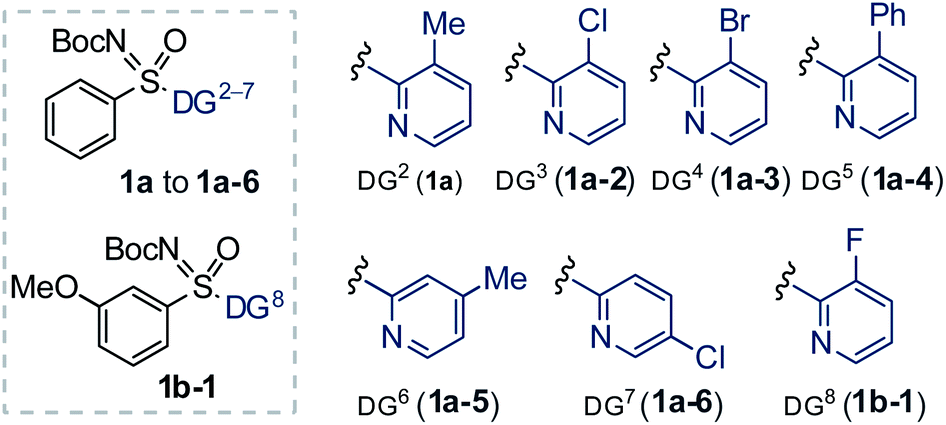
Next, we studied the DG effect (Table 2b). Thus, various substituted 2-pyridyl containing sulfoximines were independently subjected to 2a. After several trials, the 3-methyl pyridyl DG was found superior, affording the alkenylation resolution species (S)-3a in 96% ee with s factor of 58, although conversion was limited to 17% (entry 1). On the other hand, 3-Cl/Br substituted pyridyl DG were unsuccessful (entries 2 and 3). While trace of desired olefination product (S)-3s with 96% ee was noticed from the reaction of 3-phenyl-pyridyl (DG5) bearing phenyl sulfoximine (1a-4) with methyl acrylate (2b, 0.6 equiv.; entry 4). The 4-Me/5-Cl substituted pyridyl DG were ineffective, providing a lower selectivity factor (s) of 12 and 11, respectively (entries 5 and 6). No desired olefination product was obtained when 3-F substituted (DG8) pyridyl group was used (entry 7). The reaction conversion was improved to 22% when 2a (2.0 equiv.) was employed under the reaction shown in entry 1 (entry 8). The identical transformation with 2b (2.0 equiv.) could enhance the conversion to 27% (entry 9). Finally, a 50 mol% loading of 2-Cl-BQ led to (S)-3b (96% ee, s factor of 85 with 34% conversion; entry 10), which was found optimum.
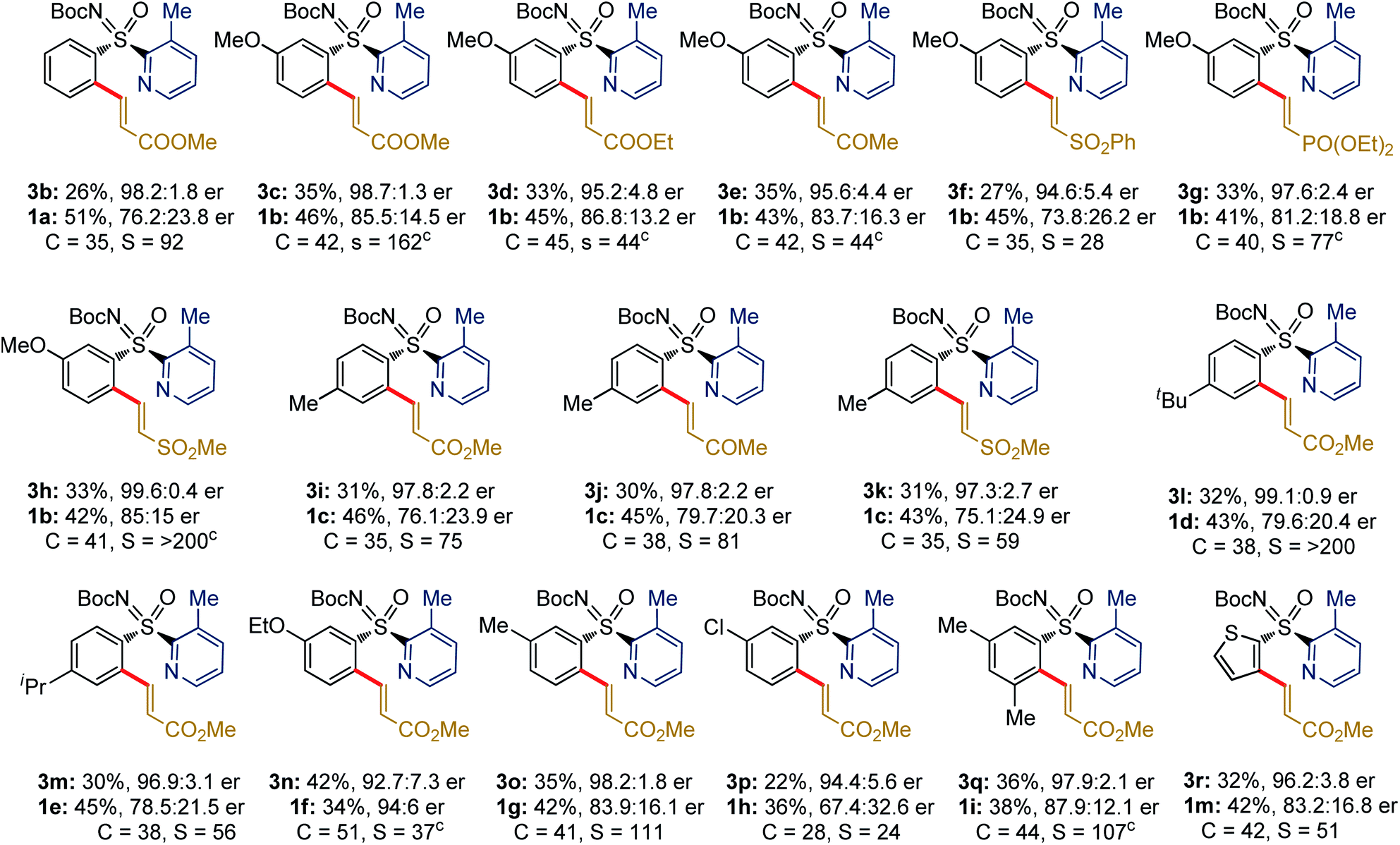
The generality of the Pd-catalyzed C–H alkenylative KR of sulfoximines was then surveyed (Table 3). Compound 3b (98.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8 er) was isolated in 26% yield. The alkenylation occurred at the less-hindered arene C–H bond and the chiral sulfoximines 3c and 3d were obtained with s factors of 162 and 44, respectively. The catalytic system was compatible with common functional groups, such as ketone, sulfone, and phosphate in the alkene, providing access to 3e (95.6:4.4 er), 3f (94.6:5.4 er), and 3g (97.6:2.4 er). Notably, the reaction of methyl vinyl sulfone with 1b displayed an exceptional s factor of >200 for compound 3h. The reaction of p-(Me/tBu/iPr)-substituted aryl sulfoximines with 2b/vinyl-ketone (2c)/vinyl-sulfone (2f) smoothly delivered 3i–m in excellent enantioselectivity and s factor of 56 to >200. The m-substituted electron donating (OEt, Me) and chloro-bearing aryl-sulfoximines underwent olefination with 2b to give the desired products 3n–p with s factor of 24 to 111. Even the sterically hindered m,m′-dimethyl substituted aryl sulfoximine 1i reacted well, yielding 3q (36%, 97.9:2.1 er, s factor of 107). The reaction of heteroaryl bearing 2-thiophenyl-2-pyridylsulfoximines (1m) with 2b afforded the olefination product 3r (32%, 96.2:3.8 er, s factor of 51).13
:1.8 er) was isolated in 26% yield. The alkenylation occurred at the less-hindered arene C–H bond and the chiral sulfoximines 3c and 3d were obtained with s factors of 162 and 44, respectively. The catalytic system was compatible with common functional groups, such as ketone, sulfone, and phosphate in the alkene, providing access to 3e (95.6:4.4 er), 3f (94.6:5.4 er), and 3g (97.6:2.4 er). Notably, the reaction of methyl vinyl sulfone with 1b displayed an exceptional s factor of >200 for compound 3h. The reaction of p-(Me/tBu/iPr)-substituted aryl sulfoximines with 2b/vinyl-ketone (2c)/vinyl-sulfone (2f) smoothly delivered 3i–m in excellent enantioselectivity and s factor of 56 to >200. The m-substituted electron donating (OEt, Me) and chloro-bearing aryl-sulfoximines underwent olefination with 2b to give the desired products 3n–p with s factor of 24 to 111. Even the sterically hindered m,m′-dimethyl substituted aryl sulfoximine 1i reacted well, yielding 3q (36%, 97.9:2.1 er, s factor of 107). The reaction of heteroaryl bearing 2-thiophenyl-2-pyridylsulfoximines (1m) with 2b afforded the olefination product 3r (32%, 96.2:3.8 er, s factor of 51).13
|
|
|---|
| a Reaction conditions: rac-1 (0.25 mmol), olefin (2.0 equiv.), Pd(OAc)2 (10 mol%), L8 (30 mol%), Ag2CO3 (2.0 equiv.), 2-Cl-BQ (0.5 equiv.), 1,2-DCE (2.5 mL), 75 °C, 3 days. b Yield of the isolated olefinated product. c Olefin (1.8 equiv.) was used and reaction was performed for 1.5 days. Calculated conversion, C = eeSM/(eeSM + eePR). Selectivity (s) = ln[(1 − C)(1 − eeSM)]/ln[(1 − C)(1 + eeSM)]. |
|
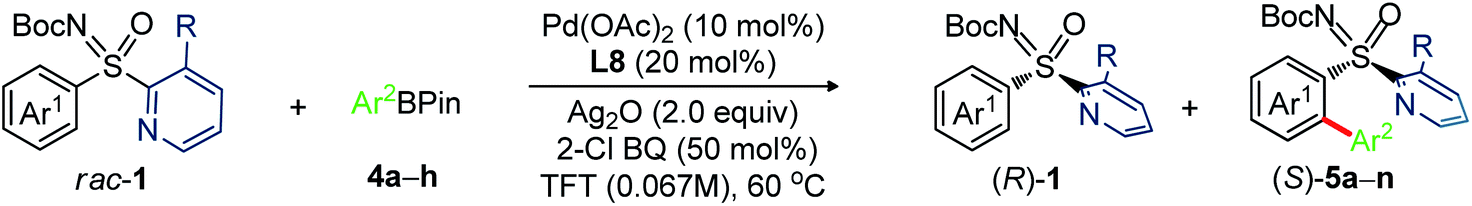
Next, we investigated the feasibility of Pd-catalyzed C–H arylative KR of sulfoximines (Table 4).14 The reaction of N-Boc-3-methoxyphenyl-2-(3-methylpyridyl) sulfoximine (1b) with (4-CF3)Ph-Bpin (4a; 2.0 equiv.) was performed under the catalytic conditions of entry 10, Table 2. Pleasingly, the desired product (S)-5a was obtained in 94% ee with s factor of 39 along with the recovery of (R)-1b in 20% ee and 18% conversion (entry 1). The oxidant Ag2O played a vital role; the conversion was increased to 51% (entry 2). Carrying out the reaction at 60 °C enhanced the s factor to 50 (entry 3). The s factor was raised to 64 with reaction conversion 41% and 94% ee of (S)-5a, when trifluorotoluene (TFT) was used (entry 4). Performing the reaction with 20 mol% L8 improved the outcome (entry 5). Importantly, reaction concentration from 0.1 M to 0.067 M led to (S)-5a (94% ee) and (R)-1b (88% ee) with 48% conversion and s factor of 95 (entry 6); this catalytic system was thus able to provide a balanced outcome.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Deviation | Conversion (c) | ee (%) | S | |
| (R)-1b | (S)-5a | ||||
| a Reaction conditions: 1b (0.1 mmol), 4a (2.0 equiv.), Pd(OAc)2 (10 mol%), L8 (30 mol%), Ag2CO3 (2.0 equiv.), 2-Cl-BQ (0.5 equiv.), DCE (1.0 mL), N2, 75 °C, 3 days. b Ag2O oxidant. c Reaction was performed at 60 °C. d TFT was used instead of 1,2-DCE. e L8 (20 mol%) was used. Calculated conversion, C = eeSM/(eeSM + eePR). Selectivity (s) = ln[(1 − C)(1 − eeSM)]/ln[(1 − C)(1 + eeSM)]. | |||||
| 1 | None | 18 | 20 | 94 | 39 |
| 2 | Ag2O instead Ag2CO3 | 51 | 88 | 86 | 38 |
| 3b | 60 °C instead 75 °C | 43 | 70 | 92 | 50 |
| 4b,c | TFT instead 1,2-DCE | 41 | 66 | 94 | 64 |
| 5b,c,d | 20 mol% ligand | 46 | 80 | 94 | 79 |
| 6b,c,d,e | 0.067 M TFT | 48 | 88 | 94 | 95 |
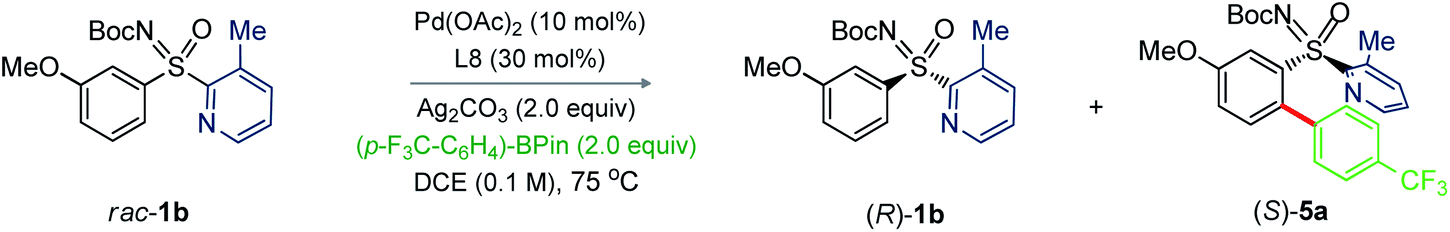
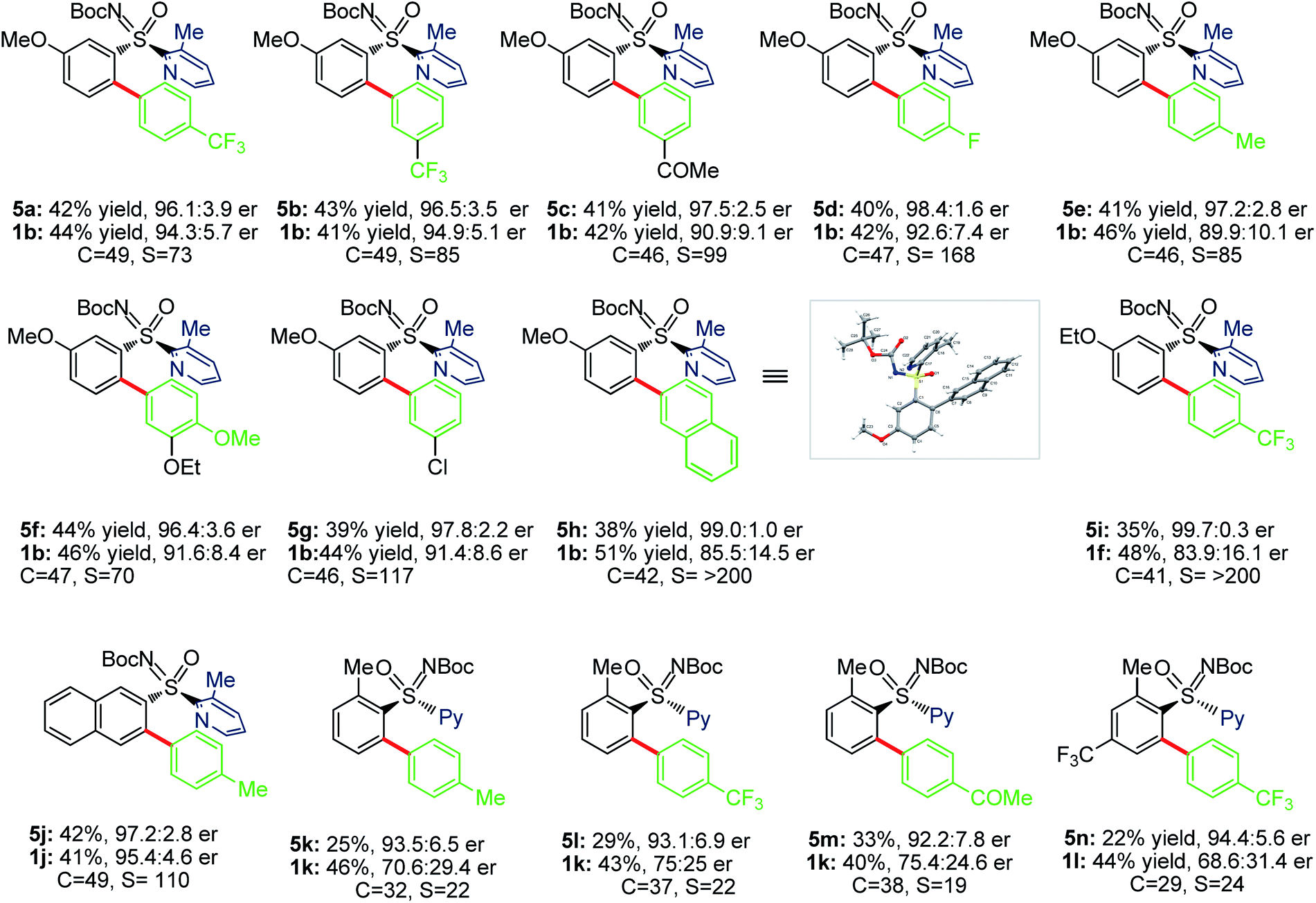
We next probed sulfoximines KR via enantioselective C–H arylation with arylpinacol boronate esters (Table 5). The reaction of 1b with various arylpinacol boronate esters having electron withdrawing groups [p-CF3 (4a), m-CF3 (4b), m-COMe (4c), and p-F (4d)], electron donating groups [p-Me (4e) and p-OMe-m-OEt (4f))] at the aryl motif independently led to the arylative resolution products 5a (96.1:3.9 er, 42%), 5b (96.5:3.5 er, 43%), 5c (97.5:2.5 er, 41%), 5d (98.4:1.6 er, 40%), 5e (97.2:2.8 er, 41%), and 5f (96.4:3.6 er, 44%), respectively, with s factor of 70–168 and conversion 46–49%. Moreover, the precursor (R)-1b was isolated in 41–46% yield with good enantioselectivity. The labile –Cl group was tolerated under the Pd-catalytic system, making 5g (97.8:2.2 er, 39%) with an s factor of 117. Notably, π-conjugated naphthyl-enabled sulfoximine resolution product 5h (99.0:1.0 er, s factor of >200) was reliably accessed. Next, the arylation of m-OEt-phenyl bearing sulfoximine 1f with 4a provided 5i (>99% ee) with s factor of >200. Likewise, 5j (97.2:2.8 er, s factor of 110) was made from the arylation of 2-naphthyl containing sulfoximine 1j with 4e. The sterically bulky o-tolyl enabled sulfoximines 1k and 1l were successful in undergoing arylation with 4a/4c/4e to afford 5k–n in good enantioselectivity; the moderate s factors of 19–24 and conversions (c = 29–38%) are considered suitable.
|
|
|---|
| a Reaction conditions: rac- 1 (0.2 mmol), 4 (2.0 equiv.), Pd(OAc)2 (10 mol%), L8 (20 mol%), Ag2O (2.0 equiv.), 2-Cl-BQ (0.5 equiv.), TFT (3.0 mL), 60 °C, 3 days. b Yield of the isolated arylation product. Calculated conversion, C = eeSM/(eeSM + eePR). Selectivity (s) = ln[(1 − C)(1 − eeSM)]/ln[(1 − C)(1 + eeSM)]. |
|
We performed a theoretical study to unveil the reaction mechanism (Fig. 2 and 3).14–16 The MPAA ligand coordination to the metal center lowers the energy barrier of the CMD step, forming a semiplanar five membered ring.15 We believe the CMD step could be the main responsible for the kinetic resolution. This hypothesis has been previously validated by Wu et al., who also focused their study on the CMD as determining step.15 Based on their findings, and considering the plane defined by the coordination of MPAA to the Pd, the bulky α-side chain of the ligand (above the plane) pushes the N-Boc moiety down to avoid steric hindrance (Fig. 2 and 1-C-II). Thus, sulfoximine phenyl group coordination complex with Pd–MPAA can point upward (U) or downward (D) on the plane, with R or S configurations. This translates to four possible CMDs: CMDPyr-UR, CMDPyr-US, CMDPyr-DR, and CMDPyr-DS. The CMDs adopt a 6-membered palladacycle with twisted boat conformation.
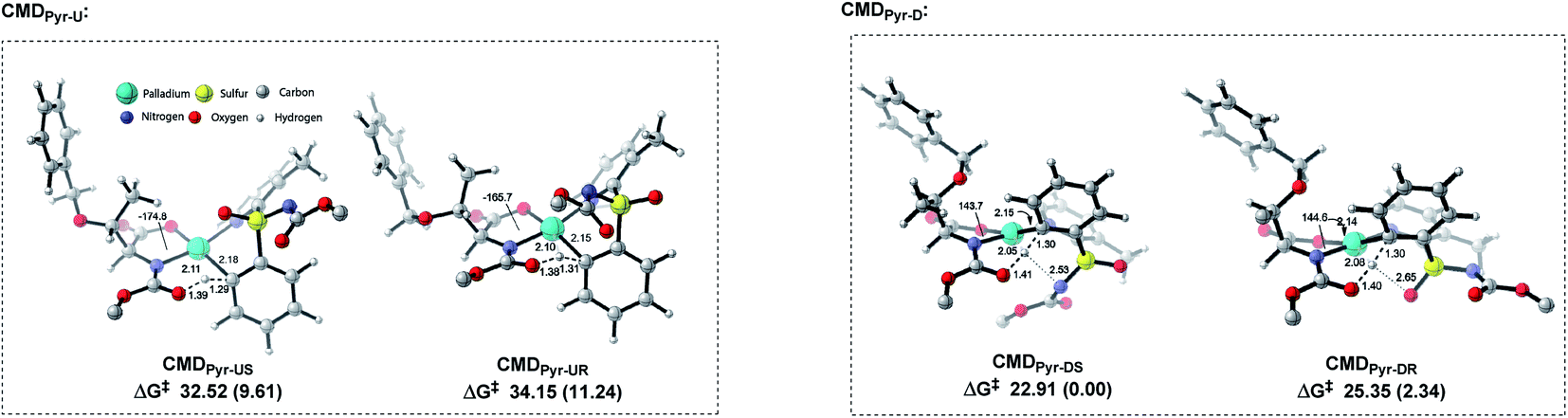 | ||
| Fig. 2 Transition structures for each CMDPyr approach. tert-Butyl group from NBoc removed from all models to simplify visualization. Relative free energies in parentheses (ΔΔG‡), distances in Å. | ||
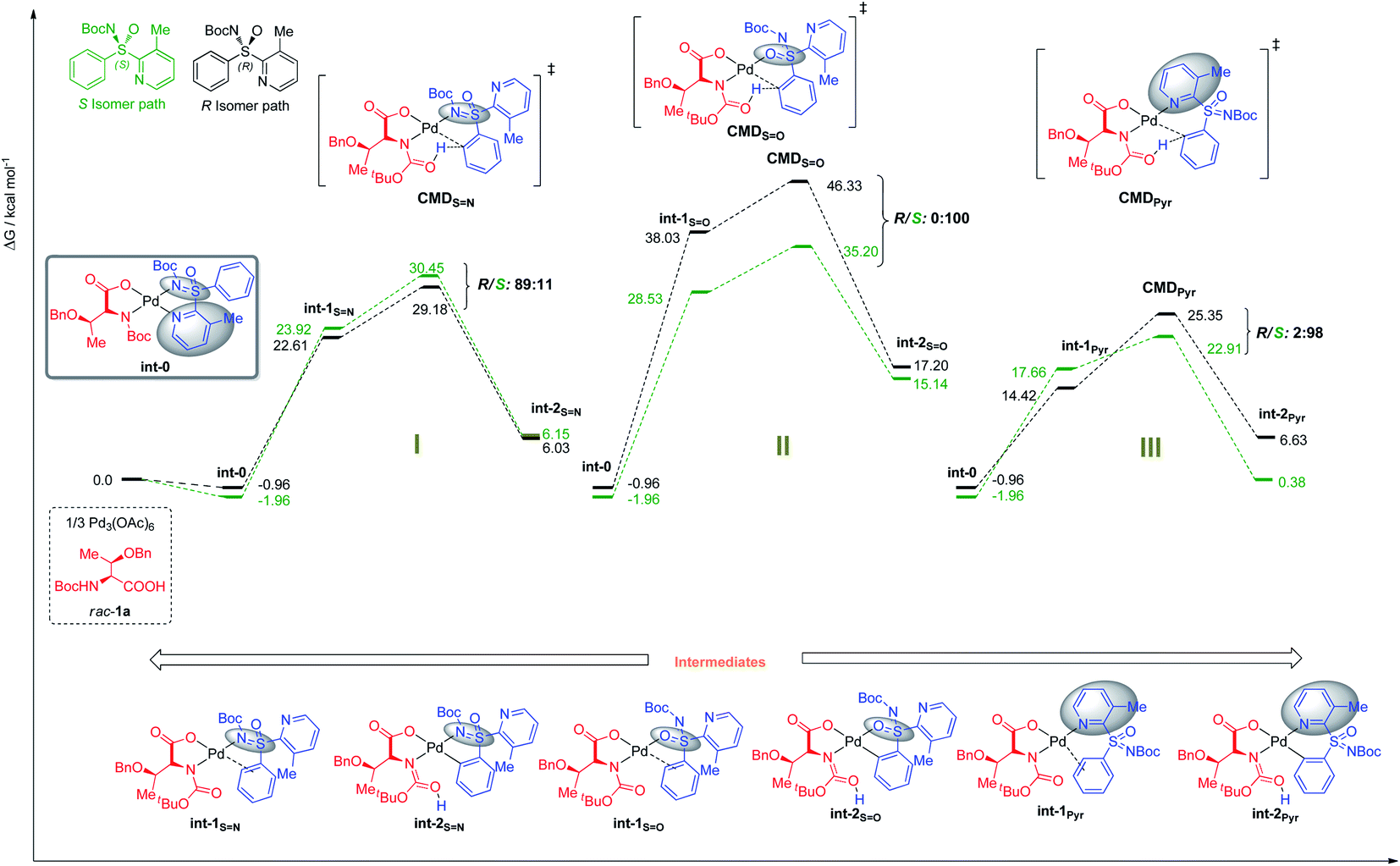 | ||
| Fig. 3 Free energies at 343.15 K in DCE in kcal mol−1. | ||
In case of upward phenyl group linkage (CMDPyr-UR and CMDPyr-US), the sulfur atom and its substituents are located above the plane; while these substituents are below the plane for CMDPyr-DR and CMDPyr-DS. In agreement with Wu's observations,15 the C1–N2–Pd–O3 dihedral angle for CMDPyr-UR and CMDPyr-US is ca. 170°, which generates a high steric interaction when compared with the ca. 140° for CMDPyr-DR and CMDPyr-DS. These latter are favored by hydrogen bond interactions, making the combination of steric and electronic effects accounting for a difference of nearly 10 kcal mol−1 in each enantiomer (Fig. 2).
The preference for the S configuration by ∼2.5 kcal mol−1 over the R isomer, lies in a steric clash of the NBoc group with the methyl group from the pyridine moiety and in consequence with the phenyl group causing an energetically demanding arrangement. The coordination of both ‘N’ atoms in sulfoximine 1a forms int-0 with the displacement of acetic acid, where the S-configuration at sulfur is 1.0 kcal mol−1 more stable than the R one (Fig. 3). Prior to deprotonation, a cis coordination of aryl group to the N-protected moiety of the MPAA-ligated intermediate occurs. This assists the CMD process by establishing the absolute configuration of the sulfur motif. This calculation fully complies with the experimental observations of the resolution selectivity (calc. 98:2, exp. 98:2; Fig. 3-III). Notably, the experimentally observed S-int-2Pyr is thermodynamically favored over R-int-2Pyr isomer by 6 kcal mol−1. In retrospect, the CMD transition states of int-1S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (Fig. 3-I) and int-1S=O (Fig. 3-II) lie much higher than int-1Pyr (Fig. 3-III), and their respective ΔG≠ do not coincide with the experimental findings. Of note, the CMD process through int-1pyr is endergonic (Fig. 3-III); thus, the calculated R/S ratio is relevant if the next steps display lower free energies of activation than the CMDPyr transition states. However, the system becomes too large to study the insertion step; simplification is therefore needed. Since we aim to distinguish the absolute configuration at the sulfur atom, a monodentate ligand for example, acetyl-L-alanine instead of bulky mono-protected threonine moiety was used for modelling purposes.11 The olefin insertion with metalated sulfoximine (made by the coordination of SN and Pyr) is next considered (Fig. 4). The corresponding SN coordination with R configuration Int-3SN is found most stable (Fig. 4). The detailed analysis of transition states (INS) occurred in the CMD revealed that the pyridine directed insertion (INSPyr) involves lowest energy barriers (17.41 kcal mol−1 for the S isomer and 21.80 for the R isomer); see Fig. 4. This results a final selectivity >99:1 (Fig. 4). This exergonic step, thus, funnels the reaction without affecting the ratio earlier dictated by the CMD. Interestingly, both INSPyr and INSsN structures are same (ignoring configuration); since both DGs (SN and Pyr) are coordinated to the metal center in their corresponding products (int-4; Fig. 4).
N (Fig. 3-I) and int-1S=O (Fig. 3-II) lie much higher than int-1Pyr (Fig. 3-III), and their respective ΔG≠ do not coincide with the experimental findings. Of note, the CMD process through int-1pyr is endergonic (Fig. 3-III); thus, the calculated R/S ratio is relevant if the next steps display lower free energies of activation than the CMDPyr transition states. However, the system becomes too large to study the insertion step; simplification is therefore needed. Since we aim to distinguish the absolute configuration at the sulfur atom, a monodentate ligand for example, acetyl-L-alanine instead of bulky mono-protected threonine moiety was used for modelling purposes.11 The olefin insertion with metalated sulfoximine (made by the coordination of SN and Pyr) is next considered (Fig. 4). The corresponding SN coordination with R configuration Int-3SN is found most stable (Fig. 4). The detailed analysis of transition states (INS) occurred in the CMD revealed that the pyridine directed insertion (INSPyr) involves lowest energy barriers (17.41 kcal mol−1 for the S isomer and 21.80 for the R isomer); see Fig. 4. This results a final selectivity >99:1 (Fig. 4). This exergonic step, thus, funnels the reaction without affecting the ratio earlier dictated by the CMD. Interestingly, both INSPyr and INSsN structures are same (ignoring configuration); since both DGs (SN and Pyr) are coordinated to the metal center in their corresponding products (int-4; Fig. 4).
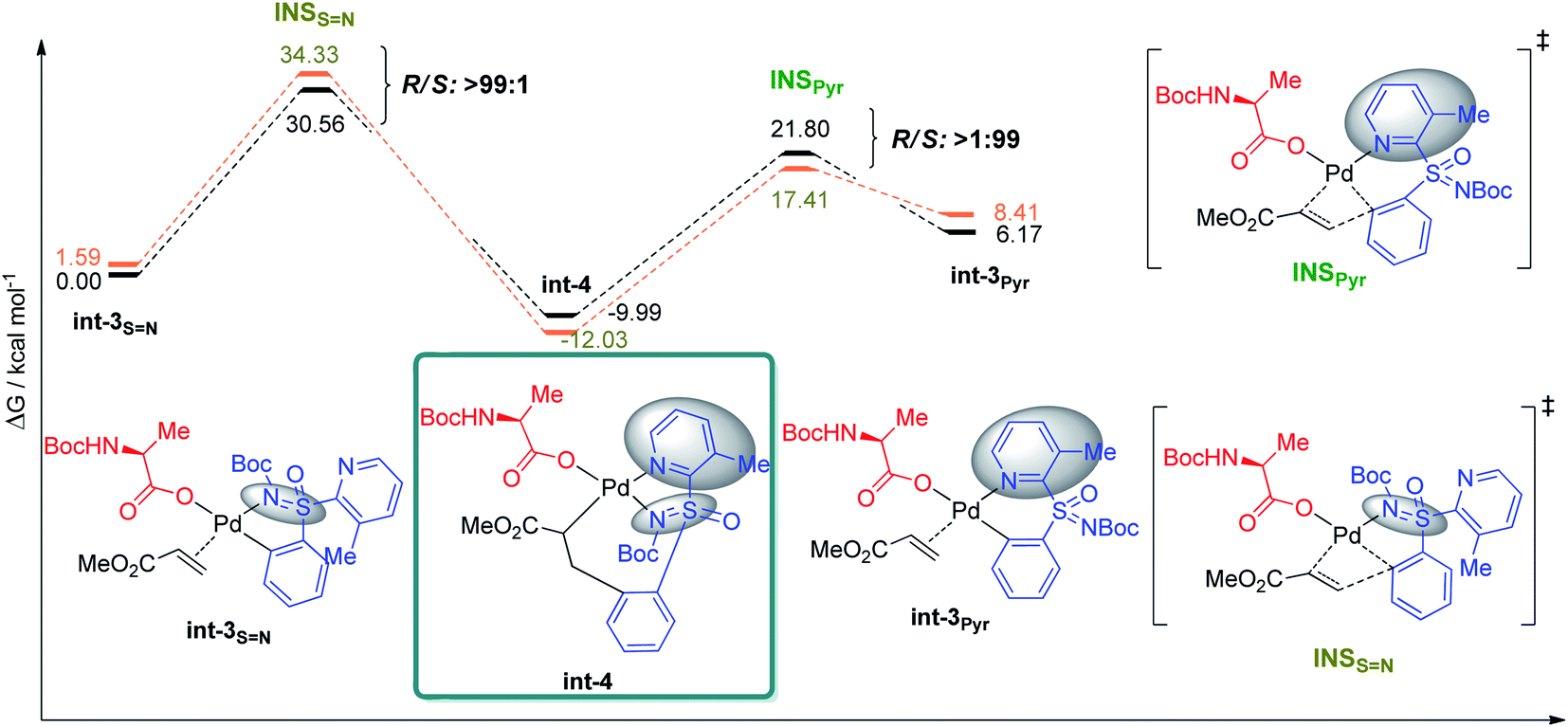 | ||
| Fig. 4 Comparison of the different energy profiles for the insertion step (INS). Energies relative to the most stable int-3 system. Free energies at 343.15 K in DCE in kcal mol−1. R/S selectivity highlighted at the transition state of each diagram (orange = S; black = R). | ||
The synthetic potential of chiral sulfoximine was next probed (Scheme 1). The trifluoroacetic acid (TFA) mediated N-Boc deprotection of (R)-1b provided chiral sulfoximine (R)-6 (>88% ee). Next, reduction of (R)-6 led to chiral sulfoxide (R)-7 (90% ee) when exposed to t-BuNO2 at rt for 2 h. The N-Boc deprotection and intramolecular Michael cyclization to the activated olefin-moiety of (S)-3c smoothly delivered 8 (as a single diastereomer) in 95% ee. A TFA assisted N-Boc deprotection and oxidative intramolecular C–N bond formation of (S)-5d furnished (S)-9 (93% ee, 62% yield).
 | ||
| Scheme 1 Derivatization of chiral products. | ||
Conclusions
In summary, a Pd(II)-catalyzed pyridyl substituted KR of sulfoximines through C(aryl)–H alkenylation and arylation has been revealed. The transformation addresses the inherent challenges in the KR of coordinatively active pyridyl-enabled sulfoximines (highly susceptible to TM-catalyst quenching) with no prochiral center in the presence of chiral amino acid MPAA ligands and Pd(II)-catalyst. The common functional groups were tolerated under Pd-catalysis exhibiting good substrate scope for C–H alkenylative and arylative sulfoximines KR products in high enantioselectivity with s factor up to >200. In-depth DFT studies uncover the salient features of coordination selectivity of pyridyl-group over sulfoximine imine.Data availability
Data for all compounds (experimental details, characterization and copies of 1H and 13C NMR, HRMS spectra and HPLC data) in this manuscript are available in the ESI.† The computational details, additional computations, energies and coordinates of the computed intermediates and transition states are also provided in the ESI.† Crystallographic data for compound 3l and 5h have been deposited at the CCDC 2088890 and 2109210, respectively.Author contributions
K. M., S. S., and A. K. S. designed and investigated. K. M. and S. S. performed the experiments. N. G. and V. G. performed the DFT studies. K. G. and M. S. reviewed the experimental data. A. K. S. and V. G. wrote the paper. Review, editing & supervision done by A. K. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank University of Hyderabad UoH-IoE for financial support (UOH/IOE/RC1-20-006). K. M, S. S, M. S, and K. G thank CSIR, India, for fellowship. VG thanks UPSaclay, Ecole Polytechnique and CNRS for financial support. NG thanks ANR-18-CE07-0012 for grant. This work was granted access to the HPC resources of CINES under the allocation 2020-A0070810977 made by GENCI.Notes and references
- For recent reviews on enantioselective C–H functionalization, see: (a) J. Wencel-Delord and F. Colobert, Chem.–Eur. J., 2013, 19, 14010–14017 CrossRef CAS PubMed; (b) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC; (c) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, 759 CrossRef CAS PubMed; (d) J. Diesel and N. Cramer, ACS Catal., 2019, 9, 9164 CrossRef CAS.
- (a) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS PubMed; (b) M. R. Albicker and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 9139 CrossRef CAS PubMed; (c) B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 460 CrossRef CAS PubMed; (d) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 7865 CrossRef CAS PubMed; (e) X. F. Cheng, Y. Li, Y. M. Su, F. Yin, J. Y. Wang, J. Sheng, H. U. Vora, X. S. Wang and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed; (f) L. Chu, X. C. Wang, C. E. Moore, A. L. Rheingold and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 16344 CrossRef CAS PubMed; (g) T. Lee, T. W. Wilson, R. Berg, P. Ryberg and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 6742 CrossRef CAS PubMed; (h) B. N. Laforteza, K. S. L. Chan and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 11143 CrossRef CAS PubMed; (i) D. Grosheva and N. Cramer, ACS Catal., 2017, 7, 7417 CrossRef CAS; (j) J. Wang, D. Gao, J. Huang, S. Tang, Z. Xiong, H. Hu, S.-L. You and Q. Zhu, ACS Catal., 2017, 7, 3832 CrossRef CAS; (k) H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Nature, 2018, 558, 581 CrossRef CAS PubMed; (l) L. Yang, M. Neuburger and O. Baudoin, Angew. Chem., Int. Ed., 2018, 57, 1394 CrossRef CAS PubMed; (m) L. Lin, S. Fukagawa, D. Sekine, E. Tomita, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2018, 57, 12048 CrossRef CAS PubMed; (n) D. Grosheva and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 13644 CrossRef CAS PubMed.
- (a) D. Gwon, S. Park and S. Chang, Tetrahedron, 2015, 71, 4504 CrossRef CAS; (b) Z.-Q. Lin, W.-Z. Wang, S.-B. Yan and W.-L. Duan, Angew. Chem., Int. Ed., 2015, 54, 6265 CrossRef CAS PubMed; (c) L. Liu, A.-A. Zhang, Y. Wang, F. Zhang, Z. Zuo, W.-X. Zhao, C.-L. Feng and W. Ma, Org. Lett., 2015, 17, 2046 CrossRef CAS PubMed; (d) G. Xu, M. Li, S. Wang and W. Tang, Org. Chem. Front., 2015, 2, 1342 RSC; (e) Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS PubMed; (f) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364 CrossRef CAS PubMed; (g) S. X. Li, Y. N. Ma and S. D. Yang, Org. Lett., 2017, 19, 1842 CrossRef CAS PubMed; (h) Y.-S. Jang, M. Dieckmann and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 15088 CrossRef CAS PubMed; (i) Z. Wang and T. Hayashi, Angew. Chem., Int. Ed., 2018, 57, 1702 CrossRef CAS PubMed; (j) Y.-S. Jang, Ł. Wozniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS PubMed; (k) Y.-C. Zhu, Y. Li, B.-C. Zhang, F.-X. Zhang, Y.-N. Yang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5129 CrossRef CAS PubMed; (l) W. Liu, W. Yang, J. Zhu, Y. Guo, N. Wang, J. Ke, P. Yu and C. He, ACS Catal., 2020, 10, 7207 CrossRef CAS.
- (a) K.-J. Xiao, L. Chu, G. Chen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 7796 CrossRef CAS PubMed; (b) K.-J. Xiao, L. Chu and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 2856 CrossRef CAS PubMed; (c) L. Chu, K.-J. Xiao and J.-Q. Yu, Science, 2014, 346, 451 CrossRef CAS PubMed.
- (a) J. A. Sirvent and U. Lücking, ChemMedChem, 2017, 12, 476 CrossRef; (b) M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225 CrossRef CAS PubMed.
- (a) S. G. Pyne and Z. Dong, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 95, 425 CrossRef; (b) K. Mukherjee, M. Shankar, K. Ghosh and A. K. Sahoo, Org. Lett., 2018, 20, 1914 CrossRef CAS PubMed; (c) M. Shankar, R. K. Rit, S. Sau, K. Mukherjee, V. Gandon and A. K. Sahoo, Chem. Sci., 2020, 11, 10770 RSC.
- (a) J. Wang, M. Frings and C. Bolm, Angew. Chem., Int. Ed., 2013, 52, 8661 CrossRef CAS PubMed; (b) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2003, 42, 5487 CrossRef CAS PubMed.
- (a) D. J. Cram, J. Day, D. R. Rayner, D. M. von Schriltz, D. J. Duchamp and D. C. Garwood, J. Am. Chem. Soc., 1970, 92, 7369 CrossRef CAS; (b) J. Wang, M. Frings and C. Bolm, Chem.–Eur. J., 2014, 20, 966 CrossRef CAS PubMed; (c) S. Dong, M. Frings, H. Cheng, J. Wen, D. Zhang, G. Raabe and C. Bolm, J. Am. Chem. Soc., 2016, 138, 2166 CrossRef CAS PubMed.
- (a) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 15539 CrossRef CAS PubMed; (b) B. Shen, B. Wan and X. Li, Angew. Chem., Int. Ed., 2018, 57, 15534 CrossRef CAS PubMed; (c) M. Brauns and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 8902 CrossRef CAS PubMed; (d) T. Zhou, P.-F. Qian, J.-Y. Li, Y.-B. Zhou, H.-C. Li, H.-Y. Chen and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 6810 CrossRef CAS PubMed; (e) Y. Tang and S. J. Miller, J. Am. Chem. Soc., 2021, 143, 9230 CrossRef CAS PubMed.
- To examine the DG capability of imine group in sulfoximine, a number of N-protected diphenyl sulfoximines have been tested. However, no reaction was detected when a N-Boc protected diphenyl sulfoximine was exposed to the reaction conditions. On the other hand, a N-Me protected sulfoximine provided the desired arylation product in 51% ee [see the ESI†].
- D. G. Musaev, A. Kaledin, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 1690 CrossRef CAS PubMed.
- Since we have focused on the stereoselectivity of the process, BQ was not incorporated in our computations, nor the additives that lead to the (chiral mono-N-protected amino acid)-Pd(II) complex. This point has been clarified in the ESI†.
- Effort to prepare furanyl-2-pyridyl sulfoximine was failed.
- See the ESI†.
- G.-J. Cheng, P. Chen, T.-Y. Sun, X. Zhang, J.-Q. Yu and Y.-D. Wu, Chem.–Eur. J., 2015, 21, 11180 CrossRef CAS PubMed.
- G.-J. Cheng, Y.-F. Yang, P. Liu, P. Chen, T.-Y. Sun, G. Li, X. Zhang, K. N. Houk, J.-Q. Yu and Y.-D. Wu, J. Am. Chem. Soc., 2014, 136, 894 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of the 1H NMR, 13C NMR and HRMS data for all products. CCDC 2088890 and 2109210. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04299h |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |