
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDepolymerization of supramolecular polymers by a covalent reaction; transforming an intercalator into a sequestrator†
Kasper M.
Vonk
,
E. W.
Meijer
 and
Ghislaine
Vantomme
*
and
Ghislaine
Vantomme
*
Institute for Complex Molecular Systems, Laboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: g.vantomme@tue.nl
First published on 29th September 2021
Abstract
Controlling the reciprocity between chemical reactivity and supramolecular structure is a topic of great interest in the emergence of molecular complexity. In this work, we investigate the effect of a covalent reaction as a trigger to depolymerize a supramolecular assembly. We focus on the impact of an in situ thiol–ene reaction on the (co)polymerization of three derivatives of benzene-1,3,5-tricarboxamide (BTA) monomers functionalized with cysteine, hexylcysteine, and alkyl side chains: Cys-BTA, HexCys-BTA, and a-BTA. Long supramolecular polymers of Cys-BTA can be depolymerized into short dimeric aggregates of HexCys-BTAvia the in situ thiol–ene reaction. Analysis of the system by time-resolved spectroscopy and light scattering unravels the fast dynamicity of the structures and the mechanism of depolymerization. Moreover, by intercalating the reactive Cys-BTA monomer into an unreactive inert polymer, the in situ thiol–ene reaction transforms the intercalator into a sequestrator and induces the depolymerization of the unreactive polymer. This work shows that the implementation of reactivity into supramolecular assemblies enables temporal control of depolymerization processes, which can bring us one step closer to understanding the interplay between non-covalent and covalent chemistry.
Introduction
In the field of supramolecular polymerisation, much progress has been achieved in understanding the assembly properties and pathway selection of the products obtained.1,2 Often the construction of these structures is divided into two unconnected steps: first the covalent synthesis of the building blocks followed by the study of their self-assembly properties in solution. In contrast, concomitant covalent modifications of structures held together by non-covalent interactions are widespread in living systems.3 This connection between biological materials with chemical reaction networks results in the spatio-temporal control needed to sustain the complex functions of life. A fascinating example on how nature controls dynamic supramolecular systems by incorporating covalent reactions is the dynamic instability of microtubules that governs their cyclic growth and shrinkage.4,5 Microtubules consist of α/β-tubulin molecules that can polymerize into long fibres by incorporation of guanosine di- or triphosphate (GDP or GTP) on the growing end of the tubules. Hydrolysis of GTP into GDP destabilizes the fibre and triggers depolymerization. Addition of new GTP to the stack stabilizes the fibre and initiates the growth cycle again.4,5 Integration of this fine level of control and precision over aggregation mechanisms by combining covalent and non-covalent synthesis would be of great interest to tackle the challenge of recyclability of supramolecular materials. Inspired by these systems, the interplay between structure and reactivity can serve as a stepping stone towards the design of functional and life-like 1D supramolecular materials.6–8Recently, chemists have combined covalent and non-covalent synthesis for the construction of complex molecular systems.9 Methods to construct these complex systems require implementing chemical reactivity into synthetic supramolecular assemblies. Inclusion of chemical functionality in the assembled building blocks enables in situ covalent modification, which induces changes in the structural unit and consequently impacts the assembly properties. In supramolecular polymerization, the lability of the non-covalent interactions makes the structures intrinsically dynamic and sensitive to changes in molecular functionalities and external conditions. Therefore, aside from increase in temperature,10–13 addition of cosolvent,14,15 and use of supramolecular additives,16–19 the modification of structural units by covalent reactions would provide an alternative method to control supramolecular polymerization. Elegant examples have been presented in which adaptation, regulation and replication can be demonstrated by integrating chemical reactions with polymerization processes.20–24 Complementarily, deepening our understanding on the molecular mechanisms at stake and structure–property relationships is essential as the complexity of molecular systems is ever growing, in particular in the cooperative supramolecular polymerization of reactive multi-component systems.
The assembly of benzene-1,3,5-tricarboxamides derivatised with amino-ester side chains (ester-BTAs) has been reported in great details.25,26 Studies have demonstrated that the nature of the side chains dramatically influences the structure, length, and mechanism of formation of the aggregates. Ester-BTAs are known for their ability to self-assemble into helical stacks in apolar solvent by formation of an intermolecular three-fold hydrogen bonding between the amides or into dimeric structures by six-fold hydrogen bonding of the ester carbonyls depending on the nature of the side chains, the concentration and the temperature of the solutions (Scheme 1).25 Recently, these chiral ester-BTA have been shown to intercalate into achiral assemblies and generate homochiral helices, indicating the effectiveness of ester-BTAs in steering the co-assembly process.27–30
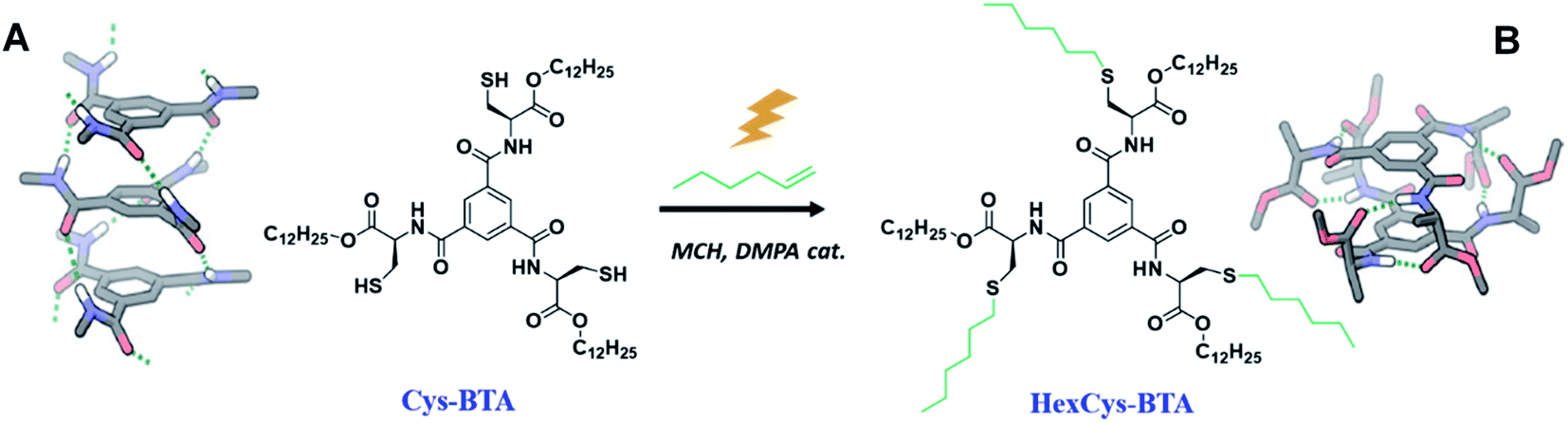 | ||
| Scheme 1 An overview of the structures of benzene-1,3,5-tricarboxamides Cys-BTA and HexCys-BTA and the transformation induced by the thiol–ene reaction. (A) The molecular structure of the helical stack formed by Cys-BTA derived from X-ray data31 and (B) the dimeric hydrogen-bonded structure formed by HexCys-BTA derived from DFT calculations.25 | ||
Here, we report on the controlled depolymerization of supramolecular 1D polymers into well-defined dimers triggered by a covalent reaction on the side chains of the monomer. For the purpose of this study, we selected Cys-BTA, a monomer of BTA derived from cysteine with a thiol containing residue. In the assembly of this monomer, a morphological change from stacks to dimeric species is induced by performing a covalent reaction on these reactive thiol groups. The changes in both the hydrogen bond pattern and the size of the aggregates were analysed by spectroscopic and light scattering techniques to follow the kinetics of the reaction. A detailed analysis was conducted to understand how the differences in molecular structure of the monomers (reagents and products) and the kinetics of the thiol–ene reaction affect the depolymerization and the overall mechanism of the process. Moreover, an unreactive achiral alkyl-BTA monomer copolymerized with a Cys-BTA additive was also depolymerized upon performing the thiol–ene reaction on Cys-BTA. These results demonstrate how an unreactive monomer in a multi-component system can be depolymerized by a covalent reaction through competitive interactions with a reactive sequestrator.
Results and discussion
Chiral Cys-BTA and HexCys-BTA were synthesized in four and five steps with 57% and 31% yield respectively, starting from the commercial protected N-Fmoc-S-trityl-cysteine amino acid and 1,3,5-benzenetricarbonyltrichloride (see ESI† for details). The synthesis starts by the esterification of the commercial N-Fmoc-S-trityl-cysteine amino acid with 1-dodecanol. The optically pure N-Fmoc-S-trityl-cysteine dodecyl ester is subsequently deprotected by removal of the Fmoc group and reacted with 1,3,5-benzenetricarbonyltrichloride. The removal of the trityl protective group yields the Cys-BTA, as a gum-like solid. Finally, HexCys-BTA was synthesized by a photo-initiated thiol–ene reaction on Cys-BTA. All compounds were purified by column chromatography and fully characterized by 1H and 13C NMR spectroscopy and MALDI (Fig. S1–S6†).Homoaggregation of Cys-BTA and dimerization of HexCys-BTA
We first investigated the homopolymerization properties of Cys-BTA and HexCys-BTA in both bulk and apolar solution. Measurements in bulk show that both Cys-BTA and HexCys-BTA assemble into helical stacks (Table 1, Fig. S7 and S8†). In solution, Cys-BTA forms both long supramolecular polymers and dimers depending on the concentration, whereas HexCys-BTA only assembles into dimers (Table 1). The details of the combined NMR, IR, UV and CD spectroscopy analysis of the assembly in solution are presented in the paragraphs below.| Assembly in bulk | Assembly in solution | ||||
|---|---|---|---|---|---|
| Structure | T c | Structure | c* | Viscosity | |
| a Crystallization transition temperature (Tc) determined with DSC using a cooling rate of 5 K min−1. b The critical concentration (c*) from which stacks predominate over dimers in MCH solution investigated at 20 °C. c No transition between dimers and stacks have been observed in the range of concentrations 0.05–10 mM at 20 °C. | |||||
| Cys-BTA | Stacks | 170 °C | Dimers + stacks | 0.150 mM | Viscous |
| HexCys-BTA | Stacks | 130 °C | Dimers | —c | Fluid |
Fig. 1 shows that the IR spectrum of Cys-BTA in 1 mM methylcyclohexane (MCH) solution displays an N–H stretch at about ≈3229 cm−1, the unbonded ester at ≈1748 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O amide I at ≈1644 cm−1, and the amide II at ≈1558 cm−1. The unbonded ester and bonded carbonyl of the amide suggests the presence of one-dimensional polymers stabilized by helically ordered, intermolecular hydrogen-bonds. In contrast, the IR spectrum of HexCys-BTA presents vibrations for the NH stretch at ≈3388 cm−1, the bonded ester at ≈1737 cm−1, the free amide I at ≈1675 cm−1, and the free amide II at ≈1527 cm−1. In this case, the bands imply the presence of dimers with intermolecular hydrogen bond formation between the N–H protons and the ester carbonyls. The formation of helically-ordered polymers and dimeric structures for Cys-BTA and HexCys-BTA, respectively, are in line with previous studies on the aggregation of these ester-BTAs.25 From the IR data, the critical concentration (c*) for polymerization of Cys-BTA was determined to be approximately 0.150 mM in MCH at 20 °C (Fig. S9†), below which the dominant species shifted from the helical assemblies to the dimers. HexCys-BTA remained in its dimeric form irrespective of concentration, indicating that the steric hindrance posed by the hexyl group prevents further aggregation even at high concentrations.
O amide I at ≈1644 cm−1, and the amide II at ≈1558 cm−1. The unbonded ester and bonded carbonyl of the amide suggests the presence of one-dimensional polymers stabilized by helically ordered, intermolecular hydrogen-bonds. In contrast, the IR spectrum of HexCys-BTA presents vibrations for the NH stretch at ≈3388 cm−1, the bonded ester at ≈1737 cm−1, the free amide I at ≈1675 cm−1, and the free amide II at ≈1527 cm−1. In this case, the bands imply the presence of dimers with intermolecular hydrogen bond formation between the N–H protons and the ester carbonyls. The formation of helically-ordered polymers and dimeric structures for Cys-BTA and HexCys-BTA, respectively, are in line with previous studies on the aggregation of these ester-BTAs.25 From the IR data, the critical concentration (c*) for polymerization of Cys-BTA was determined to be approximately 0.150 mM in MCH at 20 °C (Fig. S9†), below which the dominant species shifted from the helical assemblies to the dimers. HexCys-BTA remained in its dimeric form irrespective of concentration, indicating that the steric hindrance posed by the hexyl group prevents further aggregation even at high concentrations.
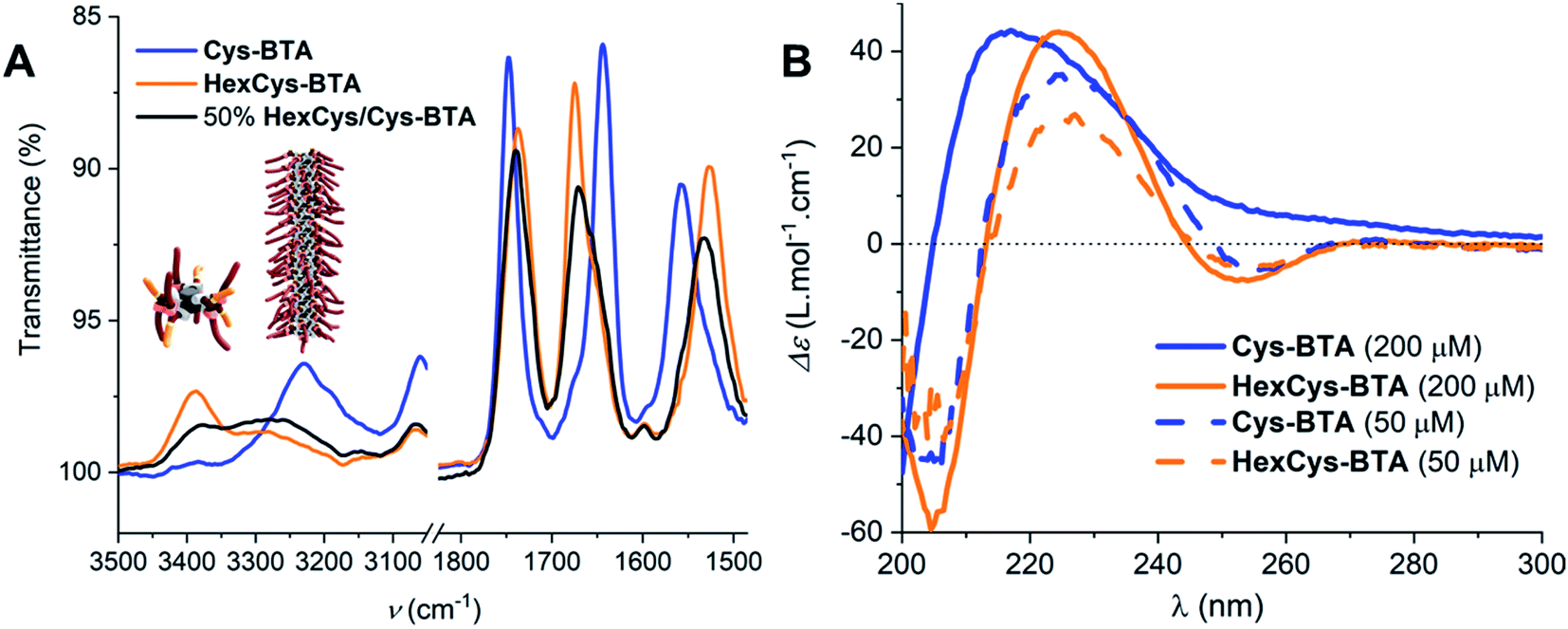 | ||
| Fig. 1 (A) IR spectra of Cys-BTA (blue), HexCys-BTA (orange), and 50 mol% Cys-BTA/HexCys-BTA (black) at 1 mM in MCH at 20 °C. (B) CD spectra of Cys-BTA (blue) and HexCys-BTA (orange) at 50 μM and 200 μM in MCH at 20 °C. | ||
CD spectroscopy provides further evidence for the existence of the assemblies determined by IR spectroscopy. Fig. 1 shows that at 50 μM in MCH, both Cys-BTA (dashed blue trace) and HexCys-BTA (dashed orange trace) display the chiroptical signatures of dimers (maxima at λ− = 205 nm, λ+ = 225 nm, and λ− = 255 nm). While no change in the shape of the CD spectrum of HexCys-BTA (orange trace) occurred upon increasing the concentration to 0.2 mM, the spectrum of Cys-BTA (blue trace) shows both a shift for the maximum λ+ = 225 nm to 220 nm and the disappearance of the minimum at λ− = 255 nm. This transition indicates the formation of helical stacks at a concentration of 0.2 mM. Moreover, NMR spectroscopy of 5 mM MCH solution of Cys-BTA (Fig. S10A†) confirms the aggregation into large assemblies as only broad signals are observed and no signals related to protons of the BTA core are visible. In contrast, 5 mM MCH solution of HexCys-BTA shows well-resolved signals, confirming the formation of well-defined dimers because of the diastereotopicity of the H1 and H2 protons in the ester-bonded dimer (Fig. S10B†).25,32 Additionally, in the more polar solvent CHCl3, a single signal is observed at 4.2 ppm indicating the presence of only monomers in solution (Fig. S10C†). In MCH, splitting of this same signal at 4.2 ppm occurs which is characteristic of six-folded hydrogen bonding. Additionally, fitting of the scattering curves to a cylindrical model with a fixed radius of 6 nm also provided us with an approximate length of 180 nm for the helical Cys-BTA assembly (Fig. S11†).33 Variable temperature measurement in CD also revealed that at a concentration of 200 μM in MCH, the homodimer of HexCys-BTA remains stable between 10 °C and 90 °C, whereas the CD spectrum of Cys-BTA at the same concentration shifts from a helical to a dimeric shape around 40 °C (Fig. S12†). The lack of isodichroic point for Cys-BTA further supports the presence of multiple species (monomers, short aggregates and stacks) co-existing in solution.24 The relatively low temperature required for this change in morphology is indicative of the instability of the helical assembly at this specific concentration (Fig. S12†).
To understand the mechanism of dimerization in more detail, we explored the possibility of heteroaggregates formation between HexCys-BTA and Cys-BTA (Fig. 2). This study is similar to previously reported results on the mixing of dimeric ester-BTA and polymeric alkyl-BTA.27,33 IR and CD data point to the exclusive formation of heterodimers between HexCys-BTA and Cys-BTA above 50 mol% of HexCys-BTA (Fig. 2). Decrease of the ratio of HexCys-BTA below 50 mol% leads to the co-existence of heterodimers, homodimers of HexCys-BTA and Cys-BTA dominated fibres with intercalated HexCys-BTA. In details, the comparison of the IR spectra of 1 mM MCH solutions of Cys-BTA/HexCys-BTA mixtures (blue to orange traces in Fig. 2B) shows a transition from helical stacks to dimeric species above 50 mol% of HexCys-BTA indicating the interaction between the two monomers. Furthermore, it shows that addition of 50 mol% of HexCys-BTA is sufficient to depolymerize the helical assembly by formation of heterodimers. Even at 10 mol% of HexCys-BTA, a small band at 1675 cm−1 can be observed, which corresponds to the formation of dimers and thus indicates the ability of HexCys-BTA to act as a sequestrator by stabilizing the free Cys-BTA monomers.24 The CD spectra of the Cys-BTA/HexCys-BTA mixtures show a similar trend where a small negative minimum at λmin = 255 nm can be observed, which indicates the presence of dimers (Fig. 2A).
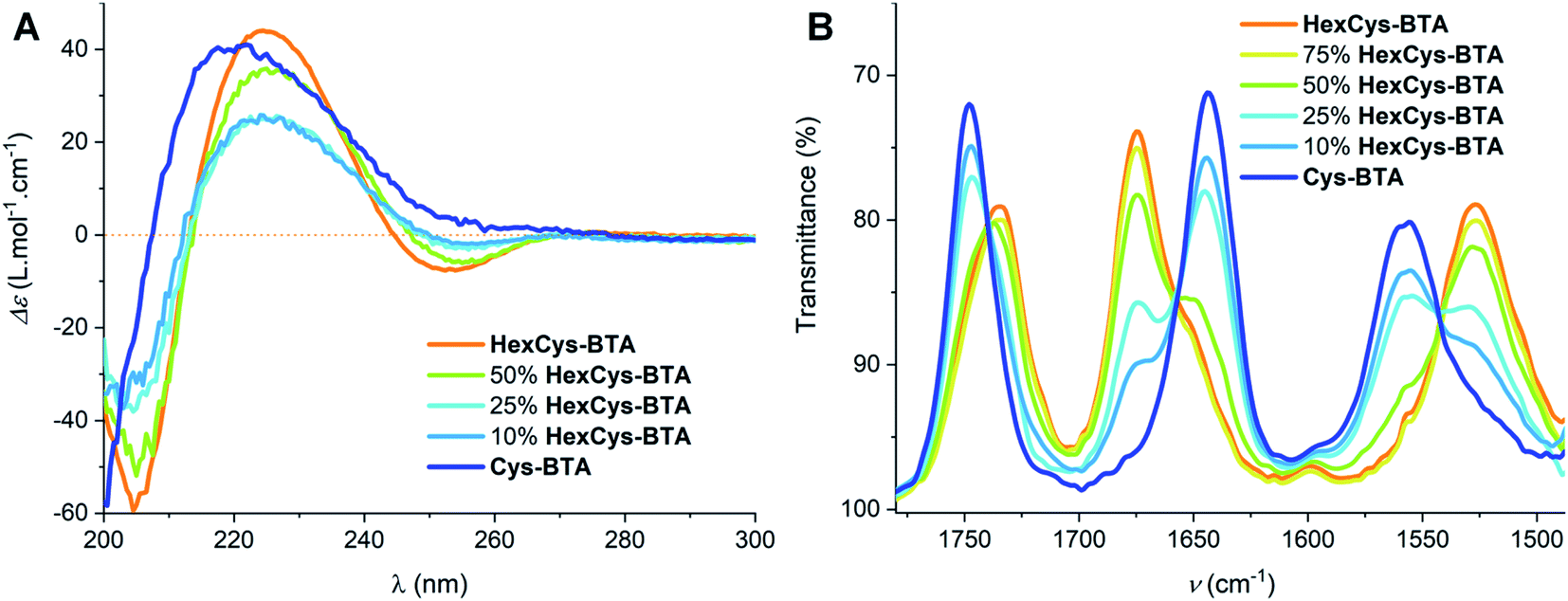 | ||
| Fig. 2 (A) CD spectra of different ratios of HexCys-BTA and Cys-BTA (percentages are given in moles of HexCys-BTA mixed with Cys-BTA in solution) at a total concentration of 0.2 mM in MCH at 20 °C. (B) The IR spectra of different ratios of Cys-BTA/HexCys-BTA mixtures at 1 mM in MCH at 20 °C. | ||
Supramolecular depolymerization of Cys-BTA 1D fibres into HexCys-BTA dimers upon covalent reaction
The difference in the aggregation properties of the Cys-BTA helical polymers and HexCys-BTA dimers prompted us to investigate the in situ depolymerization of Cys-BTA aggregates over the course of a thiol–ene reaction yielding dimers of HexCys-BTA (Scheme 2). The thiol–ene reaction was selected due to its compatibility with both the apolar solvent and temperature range for the existence of the helical BTA stacks.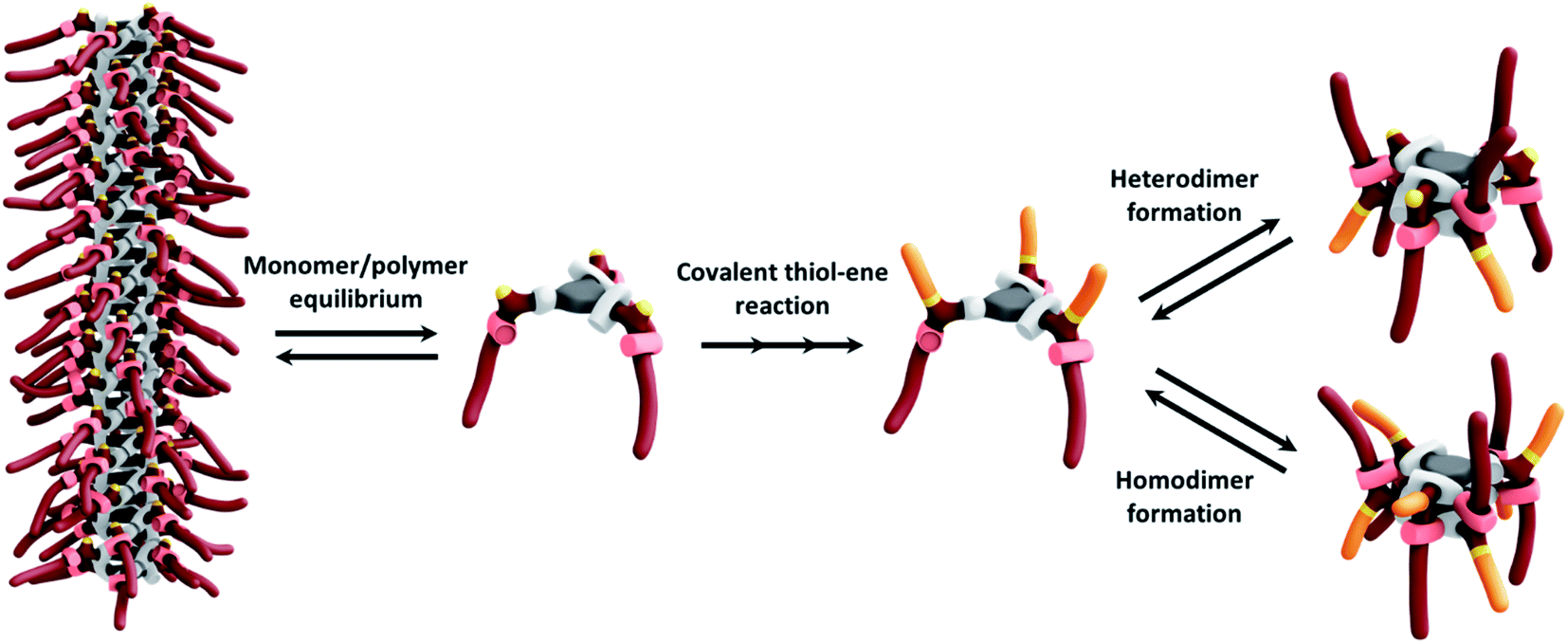 | ||
| Scheme 2 Schematic overview of the supramolecular depolymerization of Cys-BTA polymers by thiol–ene reaction on the side chains of Cys-BTA. In solution, the monomer and polymer are in equilibrium and occurrence of the covalent thiol–ene reaction decreases the Cys-BTA monomer concentration and forms HexCys-BTA, which favours the formation of homo- and heterodimers. | ||
To elucidate in detail the efficiency of the in situ depolymerization, we conducted a series of experiments analysed by NMR, IR, CD, and static light scattering (SLS) (Fig. 3–5). Addition of 0.3 eq. of catalyst 2,2-dimethoxy-2-phenyl acetophenone (DMPA) and 3 eq. of 1-hexene (stoichiometric amount) to a solution of Cys-BTA (MCH, 1 mM) and subsequent irradiation with UV light for 10 minutes, results in a shift in the CD signals from the helical assembly to the chiroptical signatures of dimers (Fig. 3A). This observation suggests the in situ conversion of Cys-BTA to HexCys-BTA. 1H-NMR spectroscopy confirms this in situ conversion, as new and sharper signals corresponding to the formation of HexCys-BTA emerge in the spectrum (Fig. S13C†) attributed to short defined aggregates of HexCys-BTA (Fig. S13D†).
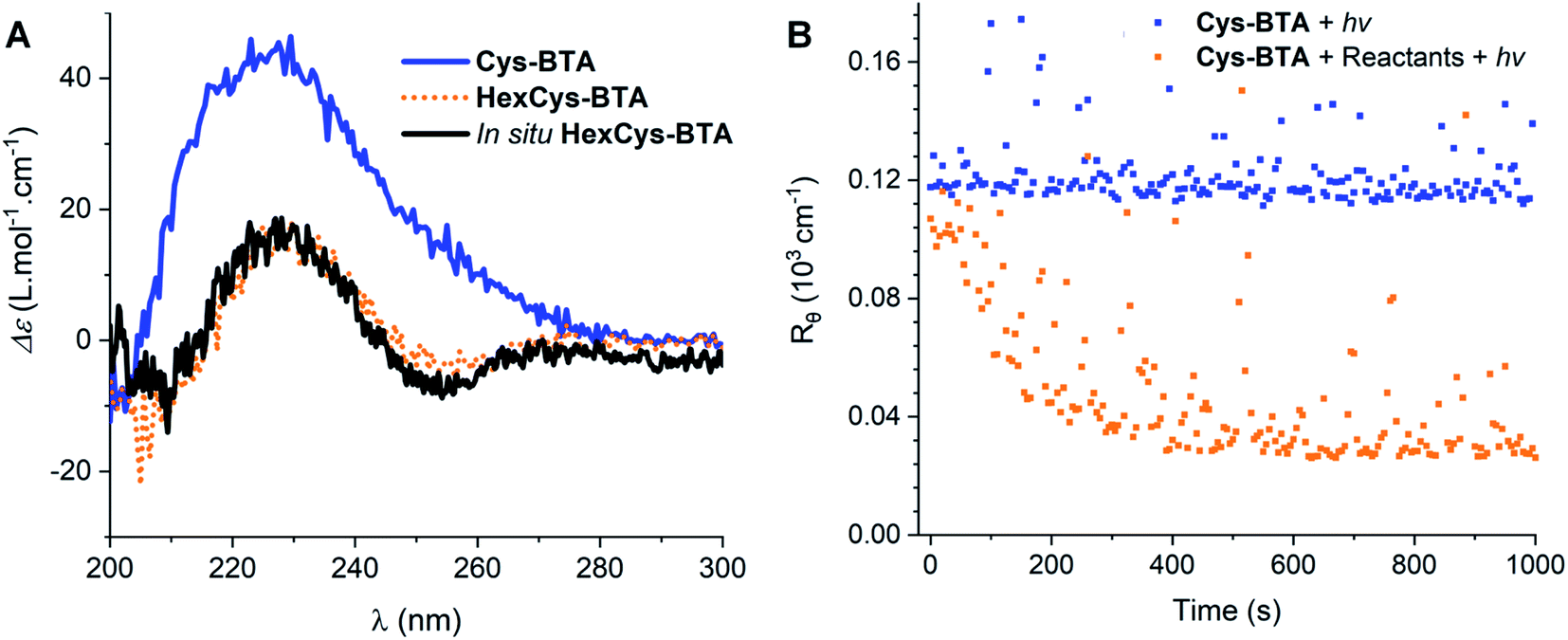 | ||
| Fig. 3 (A) CD spectra of Cys-BTA (blue), HexCys-BTA (orange) and the in situ formed HexCys-BTA (black) after 20 minutes exposure to UV light (λ = 365 nm, 70 mW cm−2) measured at 1 mM in MCH at 20 °C. (B) SLS data showing the change in particle size over UV irradiation time (λ = 365 nm, 70 mW cm−2) of the mixture of Cys-BTA with reactants (orange) and without reactants (blue) measured at 0.5 mM in MCH at 20 °C. | ||
Kinetics experiments give valuable information on the mechanism of the depolymerization. Interestingly, by combining different characterization techniques, both changes in structural morphologies and chemical conversion can be followed over time at various concentrations. Time-resolved IR spectroscopy was used to provide insights into the changes from polymers to dimers and chemical conversion over time (Fig. 4), while kinetics of the reaction followed by SLS (Fig. 3B) and CD (Fig. S14†) gave the change in the aggregate size and morphology, respectively. Over 20 minutes irradiation of a 1 mM MCH solution containing Cys-BTA, 1-hexene (3 eq.), and DMPA (0.3 eq.), a shift in the distinctive bands of the NH stretch (from ≈3240 cm−1 to ≈3389 cm−1) and the three carbonyl bands (between 1500 and 1800 cm−1) was observed in IR spectroscopy and attributed to the change in hydrogen bonding pattern from helical assemblies to dimers (Fig. 4). Moreover, the band at ν = 2559 cm−1, distinctive of the unreacted thiol moiety, allows to follow the quantitative conversion of free thiol over 20 minutes (Fig. 4). Kinetics of the conversion of the thiol and the loss of polymers show a similar shape (Fig. S15†). However, the disappearance of these polymers exhibits a lag time (about 2 minutes) in comparison to the thiol conversion, indicating that the thiol–ene reaction does not immediately result in a change of the assembly. The presence of such a lag time was already noted on the kinetics of “sergeant and soldiers” experiments with BTA, which is attributed to the exchange dynamics between the two components.34 The depolymerization of Cys-BTA elongated structures over the course of the thiol–ene reaction was also confirmed by SLS (Fig. 3B and S11†). Without the catalyst and 1-hexene, no change in size of the aggregates was observed upon UV irradiation, whereas when these reactants are present a decrease in fibre length was observed after 5 minutes, until the fibres disappear. Combined with the fact that the thiol–ene reaction, unlike most radical reactions, is not affected by oxygen in the air,35 it indicates that the observed changes over time are the result of the thiol–ene reaction between the terminal thiol unit on Cys-BTA side chain and the double bond of hexene to form HexCys-BTA. Conversion of Cys-BTA into HexCys-BTA decreases the free monomer concentration of Cys-BTA and induces the formation of heterodimers Cys-BTA/HexCys-BTA, which therefore pushes the thermodynamic equilibrium of formation of Cys-BTA polymers towards depolymerization.
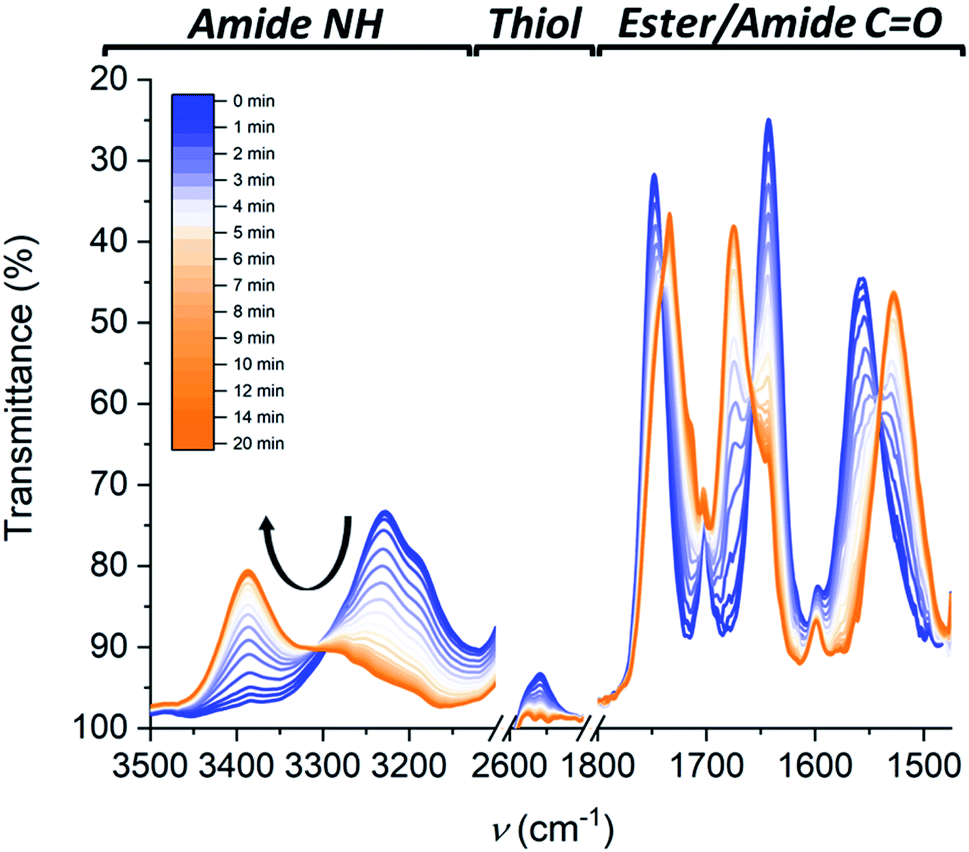 | ||
| Fig. 4 The change in FT-IR spectra over time following the thiol–ene reaction in the Cys-BTA solution from the characteristic bands of polymers (blue) to the bands of dimers (orange). The spectra are taken at a concentration of 10 mM in MCH at 20 °C under UV light irradiation (λ = 365 nm, 70 mW cm−2). Each spectrum was taken at an interval of 30 seconds from the blue to the orange traces over the course of 20 minutes. | ||
The normalized kinetic profiles followed at different concentrations between the different techniques were compared (ranging from 0.2 to 10 mM MCH solutions from CD, IR, and SLS kinetic data, Fig. 5A) and no major difference in kinetics was observed. The data were fitted to first order kinetics, which is in correspondence with the usual dependence on the thiol concentration for the thiol–ene reaction between a large variety of thiol/alkene pairs.36,37 Due to the relatively high light intensities used in this study (70 W cm−2), we assume that the concentration of free radicals reaches its steady state quickly and ultimately be of negligent impact. Furthermore, we assume that thus the chain transfer step (movement of the radical from the formed sulfide to a new thiol group) can be considered as the rate-limiting step.38,39 When the reaction is performed with low light intensity (365 nm, ∼7 mW cm−2), and followed by IR, the kinetics of thiol conversion and polymers disappearance overlap with approximately 50% conversion of the thiol after 23 minutes of irradiation corresponding to 50 mol% of the polymer disappearance (Fig. S17†). This result is surprising because the presence of 50 mol% HexCys-BTA with Cys-BTA normally results in the formation of heterodimers. We attribute this difference to the formation of asymmetric mono- and di-substituted Cys-BTA, which are less bulky than HexCys-BTA and could therefore more easily intercalate into the Cys-BTA stacks.
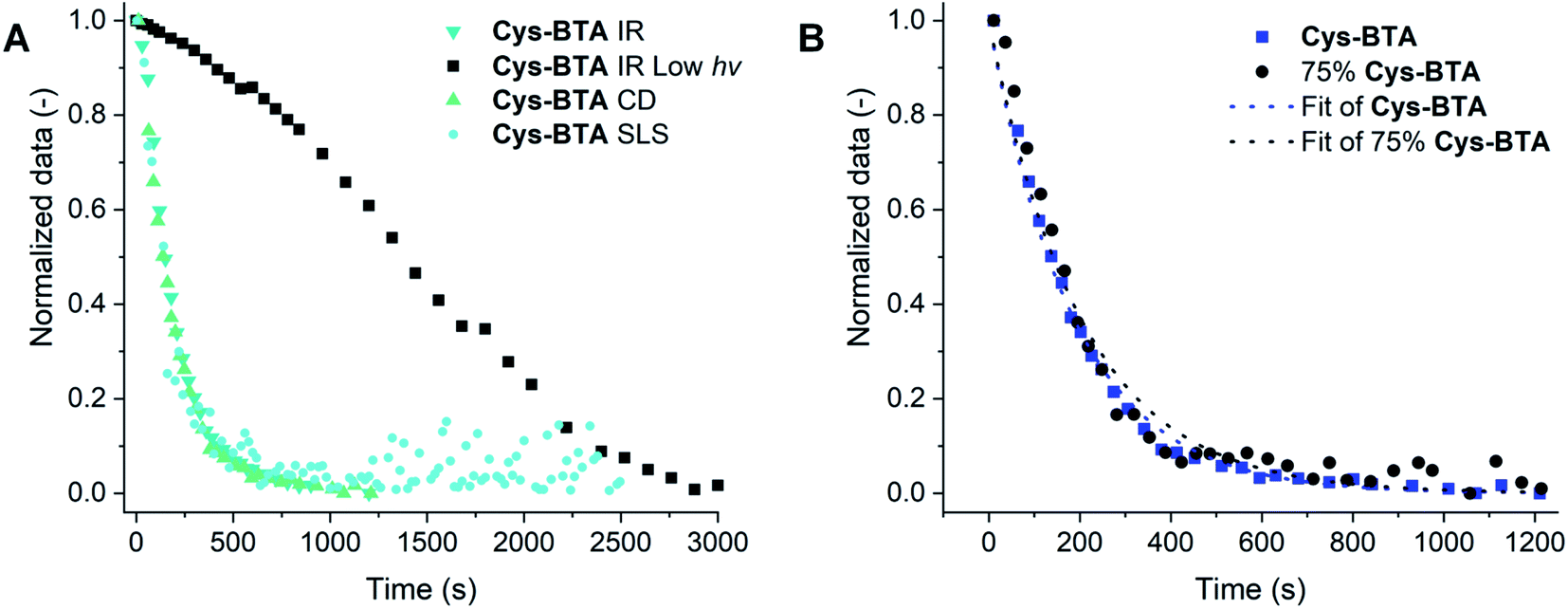 | ||
| Fig. 5 (A) The normalized kinetics of the IR (ν = 2559 cm−1, 10 mM in MCH, 70 mW cm−2 or 2 mM in MCH, 7 mW cm−2 for IR low light intensity), CD (at λ = 214 nm, 0.2 mM in MCH), and SLS data (0.5 mM in MCH) showing the change in signal intensities over time during UV irradiation at 20 °C. Fitting of the data is given in Fig. S16.† (B) The normalized data of the change in CD intensity at λ = 214 nm during the thiol–ene reaction performed on pure Cys-BTA (blue) and on the 3/1 Cys-BTA/a-BTA mixture (75 mol% Cys-BTA, black) at 0.2 mM in MCH at 20 °C. The dotted lines are the fitted data to first order kinetics (see ESI†). | ||
The depolymerization of the supramolecular aggregates can also be probed on a macroscopic scale. The ability of BTAs with alkyl side chains to form gels in apolar solvent is a well-known feature.40 The addition of catalyst and 3 eq. of hexene to a gel of Cys-BTA (40 mM in MCH) and irradiation with UV light resulted in an immediate loss in viscosity and the self-supporting properties of the gel (Fig. S18†). This loss of gel property is attributed to the in situ thiol–ene reaction, which initiates the covalent modification of Cys-BTA to form HexCys-BTA and results in the disassembly of the helical structures into dimers.
Depolymerization of inactive polymers by competitive interactions
Going further, after establishing the depolymerization of Cys-BTA polymers into dimers via the thiol–ene reaction, we investigated how this covalent modification can influence the polymerization of an inert monomer copolymerized with Cys-BTA. Indeed, the polymerization of achiral alkyl a-BTA can be controlled by copolymerization with a second component, such as a chain capper or a sequestrator.18,33,41 Previous work from our group19 has shown that copolymerizing the inert a-BTA polymers with a small amount of responsive chain capper can lead to a large decrease in polymer length. Because of this sensitivity of supramolecular systems to additives and changes in molecular structures, we hypothesized that the covalent transformation of Cys-BTA into HexCys-BTA should affect the overall cooperative assembly of the inactive monomer a-BTA. In “sergeant-and-soldiers” experiments34 between the soldiers a-BTA and either the sergeants Cys-BTA or HexCys-BTA, two different trends are observed. The presence of 4 mol% of either Cys-BTA or HexCys-BTA showed a helical bias in stacking of a-BTA, which points to the intercalation of these monomers into the a-BTA stacks (Fig. S19†). However, in a second trend at higher ratio of sergeants (>50 mol%), while Cys-BTA still intercalates into a-BTA polymers, in the case of HexCys-BTA a sequestration by formation of heterodimers of HexCys-BTA/a-BTA and homodimers of HexCys-BTA is obtained (Fig. S19–S21†). As a result, the in situ thiol–ene reaction of a 3/1 Cys-BTA/a-BTA mixture shows conversion from mainly polymers to mainly HexCys-BTA/a-BTA heterodimers, as followed by observing the decrease of the helical aggregates CD signal over irradiation time (Fig. 5B). Comparison of the decay in CD intensity of the helical aggregates for the pure Cys-BTA and the 3/1 Cys-BTA/a-BTA mixture display similarly shaped curves, with reaction rate constants of approximately k ≈ 5 × 10−3 s−1 underlying the high dynamicity of this system. This demonstrates how an unreactive monomer in a multi-component system can be depolymerized by a covalent reaction through competitive interactions with a reactive sequestrator.Conclusions
In conclusion, we have been able to induce depolymerization of supramolecular fibres by introducing covalent synthesis into the system. By performing the covalent reaction on the side chain of the monomer, the hydrogen bond pattern for assembly changes from formation of cooperative polymers to short, ordered aggregates, which was followed by spectroscopy and light scattering. The depolymerization of the supramolecular fibres into ordered dimers was achieved by the use of a photo-initiated thiol–ene reaction, which allowed for additional temporal control over the reaction. The results indicate that supramolecular systems are sensitive to small alterations in molecular structure, as small changes in steric hindrance and polarity have been shown to have a large effect on morphologies. Detailed analysis has shown that usage of 10% of the modified BTA can induce partial depolymerization of the helical assembly, but optimally 50% is used to depolymerize the assembly into heterodimers. These ester-BTAs can also be used to induce depolymerization of inactive polymer, which shows that stabilization of the inactive monomer by competitive interactions with a sequestrator plays an important role in the depolymerization process. Application of these types of covalent steps on non-covalent assemblies creates a new set of tools for exploring the world of supramolecular total synthesis and come closer to the complexity of natural supramolecular systems.Data availability
All experimental procedures characterization data and Fig. S1–S21 can be found in the ESI.†Author contributions
K. M. V. performed the experiments. K. M. V., E. W. M. and G. V. interpreted the results. G. V. supervised the overall research. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by The Netherlands Organization for Scientific Research (NWO-VENI Grant 722.017.003), the Dutch Ministry of Education, Culture and Science (Gravity Program 024.001.035) and the European Research Council (H2020-EU.1.1., SYNMAT project, ID 788618). The ICMS Animation Studio is acknowledged for providing the artwork.References
- T. Aida, E. W. Meijer and S. I. Stupp, Sci. 80, 2012, 335, 813–817 CrossRef CAS PubMed.
- M. Hartlieb, E. D. H. Mansfield and S. Perrier, Polym. Chem., 2020, 11, 1083–1110 RSC.
- C. T. Walsh, Posttranslational Modification of Proteins, Roberts & Company Publishers, Greenwood Village, 2006 Search PubMed.
- M. Igaev and H. Grubmüller, PLoS Comput. Biol., 2020, 16, e1008132 CrossRef CAS PubMed.
- T. Mitchison and M. Kirschner, Nature, 1984, 312, 237–242 CrossRef CAS PubMed.
- A. R. Studart, Angew. Chem., Int. Ed., 2015, 54, 3400–3416 CrossRef CAS PubMed.
- N. Huebsch and D. J. Mooney, Nature, 2009, 462, 426–432 CrossRef CAS PubMed.
- P. Fratzl and R. Weinkamer, Prog. Mater. Sci., 2007, 52, 1263–1334 CrossRef CAS.
- T. Schnitzer and G. Vantomme, ACS Cent. Sci., 2020, 6, 2060–2070 CrossRef CAS PubMed.
- P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS PubMed.
- D. D. Prabhu, K. Aratsu, Y. Kitamoto, H. Ouchi, T. Ohba, M. J. Hollamby, N. Shimizu, H. Takagi, R. Haruki, S. Adachi and S. Yagai, Sci. Adv., 2018, 4, 8466 CrossRef PubMed.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS PubMed.
- H. Gao, L. Gao, J. Lin, Y. Lu, L. Wang, C. Cai and X. Tian, Macromolecules, 2020, 53, 3571–3579 CrossRef CAS.
- R. P. M. Lafleur, X. Lou, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Chem. Sci., 2018, 9, 6199–6209 RSC.
- P. A. Korevaar, C. Schaefer, T. F. A. De Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- F. Lortie, S. Boileau, L. Bouteiller, C. Chassenieux and F. Lauprêtre, Macromolecules, 2005, 38, 5283–5287 CrossRef CAS.
- S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570–10577 CrossRef CAS PubMed.
- G. M. ter Huurne, P. Chidchob, A. Long, A. Martinez, A. R. A. Palmans and G. Vantomme, Chem. – Eur. J., 2020, 26, 9964–9970 CrossRef CAS PubMed.
- E. Weyandt, G. M. Ter Huurne, G. Vantomme, A. J. Markvoort, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 6295–6303 CrossRef CAS PubMed.
- G. Monreal Santiago, K. Liu, W. R. Browne and S. Otto, Nat. Chem., 2020, 12, 603–607 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. Van Esch, Sci. 80, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- S. Dhiman, A. Jain and S. J. George, Angew. Chem., Int. Ed., 2017, 56, 1329–1333 CrossRef CAS PubMed.
- D. Núñez-Villanueva and C. A. Hunter, Acc. Chem. Res., 2021, 54, 1298–1306 CrossRef PubMed.
- N. Singh, B. Lainer, G. J. M. Formon, S. De Piccoli and T. M. Hermans, J. Am. Chem. Soc., 2020, 142, 4083–4087 CrossRef CAS PubMed.
- A. Desmarchelier, B. G. Alvarenga, X. Caumes, L. Dubreucq, C. Troufflard, M. Tessier, N. Vanthuyne, J. Idé, T. Maistriaux, D. Beljonne, P. Brocorens, R. Lazzaroni, M. Raynal and L. Bouteiller, Soft Matter, 2016, 12, 7824–7838 RSC.
- M. Raynal, Y. Li, C. Troufflard, C. Przybylski, G. Gontard, T. Maistriaux, J. Idé, R. Lazzaroni, L. Bouteiller and P. Brocorens, Phys. Chem. Chem. Phys., 2021, 23, 5207–5221 RSC.
- M. A. Martínez-Aguirre, Y. Li, N. Vanthuyne, L. Bouteiller and M. Raynal, Angew. Chem., Int. Ed., 2021, 60, 4183–4191 CrossRef PubMed.
- A. Desmarchelier, X. Caumes, M. Raynal, A. Vidal-Ferran, P. W. N. M. Van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2016, 138, 4908–4916 CrossRef CAS PubMed.
- J. M. Zimbron, X. Caumes, Y. Li, C. M. Thomas, M. Raynal and L. Bouteiller, Angew. Chem., Int. Ed., 2017, 56, 14016–14019 CrossRef CAS PubMed.
- Y. Li, X. Caumes, M. Raynal and L. Bouteiller, Chem. Commun., 2019, 55, 2162–2165 RSC.
- S. Cantekin, T. F. A. De Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- X. Caumes, A. Baldi, G. Gontard, P. Brocorens, R. Lazzaroni, N. Vanthuyne, C. Troufflard, M. Raynal and L. Bouteiller, Chem. Commun., 2016, 52, 13369–13372 RSC.
- G. Vantomme, G. M. Ter Huurne, C. Kulkarni, H. M. M. Ten Eikelder, A. J. Markvoort, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 18278–18285 CrossRef CAS PubMed.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Macromolecules, 2003, 36, 7964–7969 CrossRef CAS.
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS PubMed.
- K. W. E. Sy Piecco, A. M. Aboelenen, J. R. Pyle, J. R. Vicente, D. Gautam and J. Chen, ACS Omega, 2018, 3, 14327–14332 CrossRef CAS PubMed.
- M. Claudino, M. Jonsson and M. Johansson, RSC Adv., 2013, 3, 11021–11034 RSC.
- P. J. M. Stals, J. F. Haveman, A. R. A. Palmans and A. P. H. J. Schenning, J. Chem. Educ., 2009, 86, 230–233 CrossRef CAS.
- E. Weyandt, M. F. J. Mabesoone, L. N. J. de Windt, E. W. Meijer, A. R. A. Palmans and G. Vantomme, Org. Mater., 2020, 02, 129–142 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04545h |
| This journal is © The Royal Society of Chemistry 2021 |