
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and electronic structure analysis of the actinide allenylidenes, [{(NR2)3}An(CCCPh2)]− (An = U, Th; R = SiMe3)†
Greggory T.
Kent‡
 a,
Xiaojuan
Yu‡
b,
Guang
Wu
a,
Jochen
Autschbach
*b and
Trevor W.
Hayton
*a
a,
Xiaojuan
Yu‡
b,
Guang
Wu
a,
Jochen
Autschbach
*b and
Trevor W.
Hayton
*a
aDepartment of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106, USA. E-mail: hayton@chem.ucsb.edu
bDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260, USA. E-mail: jochena@buffalo.edu
First published on 4th October 2021
Abstract
The reaction of [AnCl(NR2)3] (An = U, Th, R = SiMe3) with in situ generated lithium-3,3-diphenylcyclopropene results in the formation of [{(NR2)3}An(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCPh2)] (An = U, 1; Th, 2) in good yields after work-up. Deprotonation of 1 or 2 with LDA/2.2.2-cryptand results in formation of the anionic allenylidenes, [Li(2.2.2-cryptand)][{(NR2)3}An(CCCPh2)] (An = U, 3; Th, 4). The calculated 13C NMR chemical shifts of the Cα, Cβ, and Cγ nuclei in 2 and 4 nicely reproduce the experimentally assigned order, and exhibit a characteristic spin–orbit induced downfield shift at Cα due to involvement of the 5f orbitals in the Th–C bonds. Additionally, the bonding analyses for 3 and 4 show a delocalized multi-center character of the ligand π orbitals involving the actinide. While a single–triple–single-bond resonance structure (e.g., An–C
CCPh2)] (An = U, 1; Th, 2) in good yields after work-up. Deprotonation of 1 or 2 with LDA/2.2.2-cryptand results in formation of the anionic allenylidenes, [Li(2.2.2-cryptand)][{(NR2)3}An(CCCPh2)] (An = U, 3; Th, 4). The calculated 13C NMR chemical shifts of the Cα, Cβ, and Cγ nuclei in 2 and 4 nicely reproduce the experimentally assigned order, and exhibit a characteristic spin–orbit induced downfield shift at Cα due to involvement of the 5f orbitals in the Th–C bonds. Additionally, the bonding analyses for 3 and 4 show a delocalized multi-center character of the ligand π orbitals involving the actinide. While a single–triple–single-bond resonance structure (e.g., An–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CPh2) predominates, the AnCCCPh2 resonance form contributes, as well, more so for 3 than for 4.
C–CPh2) predominates, the AnCCCPh2 resonance form contributes, as well, more so for 3 than for 4.
Introduction
Despite the significant advancements made in actinide–carbene chemistry in the past decade,1–6 every example reported thus far has relied on ancillary chelators or heteroatom-containing substituents to stabilize the An–C multiple bond.7–10 For example, the groups of Ephritikhine and Zi employed a pincer-type ligand to form [U{C(PPh2S)2}(BH4)2(THF)2] and [Th{C(PPh2S)2}2(DME), respectively,11,12 where two thiophosphinoyl pendant arms support the An–carbene interaction. In addition, Liddle et al. isolated the silyl-phosphino-carbene [Li(2.2.2-cryptand)][U{C(SiMe3)(PPh2)}(BIPMTMS)(Cl)] (BIPMTMS = C(PPh2NSiMe3)2), whose UC bond is stabilized by a P(III) substituent.13 Similarly, the uranium(IV) arsonium carbene complex [U(TrenTIPS)(CHAsPh3)] (TrenTIPS = N(CH2CH2NSiPri3)3) features stabilization by an As(V) substituent.14 These heteroatom substituents help dissipate the negative charge at the carbene carbon caused by the weak AnC π-bond, which itself results from the energetic mismatch between actinide and carbon valence orbitals combined with the relatively small rmax of the 5f orbitals.14 Without these substituents, the AnC bond would likely be too reactive to isolate.
Because of the requirement for heteroatom substituents, no isolable “Schrock-type” actinide alkylidenes, i.e., AnCR2 (R = H, alkyl, aryl), are known,7,9,15 although they have been observed in inert gas matrices.16–22 Even vinylidene and allenylidene complexes, which should be less reactive than alkylidenes, are unknown, in part due to the lack of viable synthetic routes. Allenylidenes are especially informative in this regard, as they are typically made by H2O elimination from a propargyl alcohol – a route that is problematic for actinide organometallics given their high sensitivity to water.23,24
Herein, we report the synthesis of the actinide allenyl complexes [{(NR2)3}An(CHCCPh2)] (An = U, 1; Th, 2), formed via salt metathesis with lithium-3,3-diphenylcyclopropene. Subsequent deprotonation of 1 and 2 results in the formation of the actinide allenylidene complexes, [Li(2.2.2-cryptand)][{(NR2)3}An(CCCPh2)] (An = U, 3; Th, 4). Significantly, 3 and 4 represent the first complexes with An–C multiple bonds that do not feature heteroatom stabilization.
Synthesis and characterization
Drawing inspiration from the groups of Hashmi and Binger,25,26 we sought to synthesize an An–cyclopropenyl complex, which we hypothesized could undergo thermal ring opening to form an An–allenyl complex. In fact, addition of in situ generated lithium-3,3-diphenylcyclopropene to an Et2O solution of [UCl(NR2)3] (R = SiMe3) does result in formation of the allenyl complex, 1, which can be isolated as dark-brown blocks in 72% yield after work up (Scheme 1).25 The thorium analogue 2 can be prepared in a similar fashion in 62% yield, via the reaction of [ThCl(NR2)3] with 1 equiv. of lithium-3,3-diphenylcyclopropene in Et2O. We hypothesize that the ring opening occurs after salt metathesis.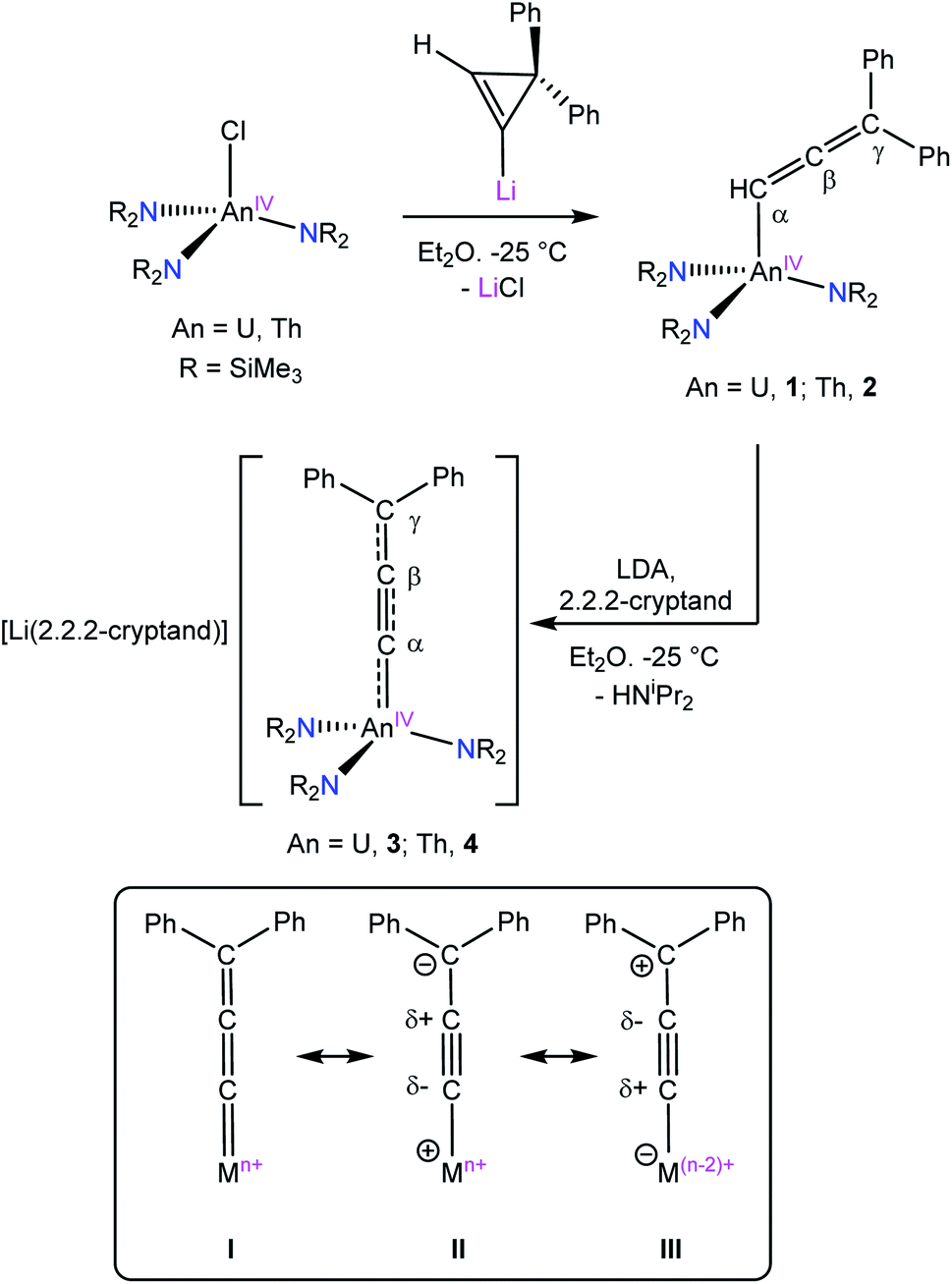 | ||
| Scheme 1 Synthesis of complexes 1–4. | ||
The 1H NMR spectrum of complex 1 in C6D6/THF-d8 features a resonance at −174.8 ppm assigned to the proton attached to the Cα carbon (Fig. S4†). The 1H NMR spectrum of 2 in C6D6/THF-d8 displays a resonance at 5.77 ppm assigned to the same ligand environment (Fig. S5†). Additionally, the 13C{1H} NMR spectrum of 2 features resonances at 139.4, 204.7, and 96.7 ppm assigned to the Cα, Cβ, and Cγ environments of the allenyl ligand, respectively (Fig. S6†). For comparison, the Cα and Cβ NMR shifts of 1,1-diphenylallene are 78.2 and 210.0 ppm, respectively,27 whereas the Cα, Cβ, and Cγ shifts of [OsCl2(NO)(CHCCPh2)(PiPr3)2] are 79.1, 199.1, and 101.0 ppm, respectively.28 We attribute the large Cα shift of 2 to spin–orbit coupling (SOC) effects (see below for more discussion).4,29–33 Finally, the IR spectra of 1 and 2 exhibit Cα–Cβ and Cβ–Cγ stretching modes at 1934/1871 and 1934/1869 cm−1, respectively (Table 1). For comparison, [OsCl2(NO)(CHCCPh2)(PiPr3)2] exhibits a single CC stretch at 1881 cm−1 in its IR spectrum.28
| Complex | ν(Cα–Cβ) (cm−1) | ν(Cβ–Cγ) (cm−1) |
|---|---|---|
| 1 | 1934 | 1871 |
| 2 | 1934 | 1869 |
| 3 | 2050 | 1911 |
| 4 | 2044 | 1921 |
Complexes 1 and 2 both crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one and two independent molecules in their asymmetric unit cells, respectively (Fig. 1). The An–C distances (1: 2.457(3); 2: 2.529(5), 2.536(5) Å) are consistent with those previously reported for An(IV)–C single bonds.34–37 Additionally, the longer distances observed for 2 reflect the increased ionic radius of Th(IV) vs. U(IV) (Table 2).38 The Cα–Cβ and Cβ–Cγ distances of the allenyl ligands, along with the Cα–Cβ–Cγ angles, are consistent with those previously reported for transition metal allenyl complexes.28,39–42 Furthermore, the An–Cα–Cβ angles (1: 133.2(2); 2: 132.0(4), 128.6(4)°) confirm that Cα is sp2 hybridized, consistent with our proposed formulation. Notably, 1 and 2 are the first reported f element allenyl complexes.
with one and two independent molecules in their asymmetric unit cells, respectively (Fig. 1). The An–C distances (1: 2.457(3); 2: 2.529(5), 2.536(5) Å) are consistent with those previously reported for An(IV)–C single bonds.34–37 Additionally, the longer distances observed for 2 reflect the increased ionic radius of Th(IV) vs. U(IV) (Table 2).38 The Cα–Cβ and Cβ–Cγ distances of the allenyl ligands, along with the Cα–Cβ–Cγ angles, are consistent with those previously reported for transition metal allenyl complexes.28,39–42 Furthermore, the An–Cα–Cβ angles (1: 133.2(2); 2: 132.0(4), 128.6(4)°) confirm that Cα is sp2 hybridized, consistent with our proposed formulation. Notably, 1 and 2 are the first reported f element allenyl complexes.
 | ||
| Fig. 1 Solid-state structure of 1 shown with 50% probability ellipsoids. Hydrogen atoms omitted for clarity. | ||
| Bond (Å, °) | 1 | 2 | 3·C5H12 | 4·C5H12 |
|---|---|---|---|---|
| An–Cα | 2.457(3) | 2.529(5), 2.536(5) | 2.305(8) | 2.368(16) |
| Cα–Cβ | 1.299(4) | 1.292(7), 1.288(7) | 1.221(11) | 1.23(2) |
| Cβ–Cγ | 1.329(4) | 1.327(7), 1.319(7) | 1.403(11) | 1.40(2) |
| Cγ–Cipso | 1.490(4), 1.474(4) | 1.492(7), 1.487(7), 1.497(7), 1.488(7) | 1.443(12), 1.468(12) | 1.47(2), 1.45(2) |
| An–Cα–Cβ | 133.2(2) | 132.0(4), 128.6(4) | 173.3(8) | 172.0(14) |
| Cα–Cβ–Cγ | 176.1(3) | 175.5(6), 176.5(6) | 176.7(9) | 174.6(16) |
| Σ(∠Cipso/β–Cγ–Cipso) | 359.0 | 360.0/359.9 | 359.9 | 359.9 |
Given the low proton affinity at Cα,43 we hypothesized that addition of base to 1 or 2 would yield an actinide–allenylidene. Gratifyingly, addition of 1 equiv. of LDA and 2.2.2-cryptand to 1 in Et2O results in the formation of 3, which can be isolated as dark purple blocks in 54% yield after work-up (Scheme 1). The thorium analogue 4 can be prepared in a similar fashion, via the reaction of 2 with 1 equiv. of LDA and 2.2.2-cryptand, in 46% yield as deep orange-red solid. Complexes 3 and 4 are the first reported f-element allenylidenes and are the first AnC complexes that do not employ heteroatoms or ancillary chelators to stabilize the AnC interaction.
The 1H NMR spectrum of 3 in C6D6/THF-d8 features a broad singlet at −1.60 ppm, assignable to the lone SiMe3 environment (Fig. S7†). The 1H NMR spectrum of 4 features a sharp singlet at 0.53 ppm, assignable to its SiMe3 environment (Fig. S7 and S8†), whereas its 13C{1H} NMR spectrum exhibits resonances at 205.4, 128.5, and 70.6 ppm. These resonances are assigned to the Cα, Cβ, and Cγ environments of the allenylidene ligand, respectively (Fig. S11†). Complexes 3 and 4 exhibit Cα–Cβ and Cβ–Cγ stretching modes at 2050/1911 and 2044/1921 cm−1, respectively, in their IR spectra (Table 1). These vibrational frequencies are higher than those observed for their respective precursors, suggesting an increase in both the Cα–Cβ and Cβ–Cγ bond orders upon deprotonation. For further comparison, the Os allenylidene complex, [Os(CCCPh2)(CH3CN)3(IPr)(PiPr3)][BF4]2, features a single CC band at 1929 cm−1 in its IR spectrum.44 Finally, the UV-vis spectrum of 4 in C6H6 features intense absorptions at 403 nm (ε = 8310 L mol−1 cm−1) and 537 nm (ε = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 030 L mol−1 cm−1) (Fig. S12†). The spectrum is qualitatively similar to that recorded for [CPh3]−,45 suggesting a similar electronic environment for Cγ (see below for more discussion).
030 L mol−1 cm−1) (Fig. S12†). The spectrum is qualitatively similar to that recorded for [CPh3]−,45 suggesting a similar electronic environment for Cγ (see below for more discussion).
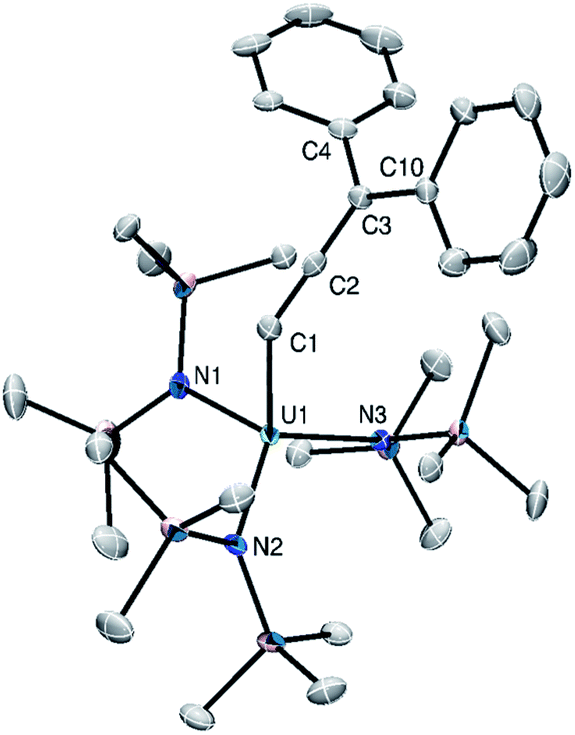
Complexes 3 and 4 crystallize in the monoclinic space group P21 as the pentane solvates, 3·C5H12 and 4·C5H12, respectively. They are isomorphous and crystallize as discrete cation–anion pairs (Fig. 2). The An–Cα distances in 3 and 4 are 2.305(8) and 2.368(16) Å, respectively (Table 2). These distances are among the shortest known An–C distances and suggest the presence of An–Cα multiple bond character. Additionally, these values are shortened by 0.15 Å from the An–Cα distances observed for their respective precursors. For comparison, the U–Cα distances in Cp3UCHPMe3 and [U{C(SiMe3)(PPh2)}(BIPMTMS)(DMAP)2] are 2.274(8) and 2.296(5) Å,13,46 respectively, whereas the Th–Cα distances in [(C5Me5)2ThCl(CHPPh3)] and [Th(CHPPh3)(NR2)3] are 2.3235(1) and 2.362(2) Å, respectively.4,5
 | ||
| Fig. 2 Solid-state structure of [Li(2.2.2-cryptand)][{(NR2)3}U(CCCPh2)] (3) shown with 50% probability ellipsoids. Hydrogen atoms, [Li(2.2.2-cryptand)], and pentane solvate omitted for clarity. | ||
Compared to 1 and 2, the Cα–Cβ distances in 3 and 4 are slightly shortened, whereas the Cβ–Cγ distances are slightly elongated. The Cα–Cβ distances are similar to those observed for the An–acetylide complexes [Th(CCH)(NR2)3] (1.173(12) Å) and [(NN′3)U(CCPh)] (1.212(5) Å, NN′3 = N(CH2CH2NSitBuMe2)3). However, the An–C distances in these examples are much longer, at 2.481(8) and 2.480(4) Å, respectively, reflecting their single bond character.47,48 The Cα–Cβ–Cγ angles in 3 and 4 remain unchanged, whereas the An–Cα–Cβ angles approach linear. In addition, the sum of angles around Cγ confirms that it is sp2 hybridized (Table 2). These metrical parameters are typical of the allenylidene ligand and can be rationalized by the contribution of resonance forms I and II to its electronic structure (Scheme 1, inset).49 For comparison, [Os(CCCPh2)(CH3CN)3(IPr)(PiPr3)][BF4]2 features Cα–Cβ and Cβ–Cγ distances of 1.246(8) and 1.362(9) Å, respectively.44
Electronic structure analysis
The An–allenylidene interaction in complexes 3 and 4 was analyzed via relativistic density functional theory (DFT).50–52,56 Complete computational details and results are provided in the ESI.† Based on Natural Localized Molecular Orbital (NLMO) analyses, complexes 3 and 4 exhibit strong π-delocalization (Fig. 3). Taking 4 as an example, the NLMO picture indicates triple bond character between Cα and Cβ, corresponding to resonance structure (RS) II in Scheme 1. However, three-center character involving thorium, Cα, and Cβ, denoted as π(Th–C) in Fig. 3 and Table S1,† also reveals an important contribution from RS I. The calculated natural charges for Th, Cα, Cβ, and Cγ are 1.52, −0.67, 0.05, and −0.22, respectively, whereas the averaged natural charge for three N atoms bound to Th is −1.65. The NLMO representing the lone pair (and negative charge) at Cγ is π-LP(Cγ), and is strongly delocalized over the phenyl groups, Cβ, and even Cα and Th (Fig. S3†). This delocalization further confirms that RS I contributes to the overall electronic structure. In addition, for complex 3, the Mulliken spin population of U is 2.3 (excess alpha over beta spin), beyond the two unpaired spins expected for the f2 configuration, indicating preferential alpha spin electron donation from ligand to metal. The spin populations for Cα, Cβ, and Cγ are −0.08, 0.02, −0.10, respectively; the remaining spin density in the ligand is further delocalized.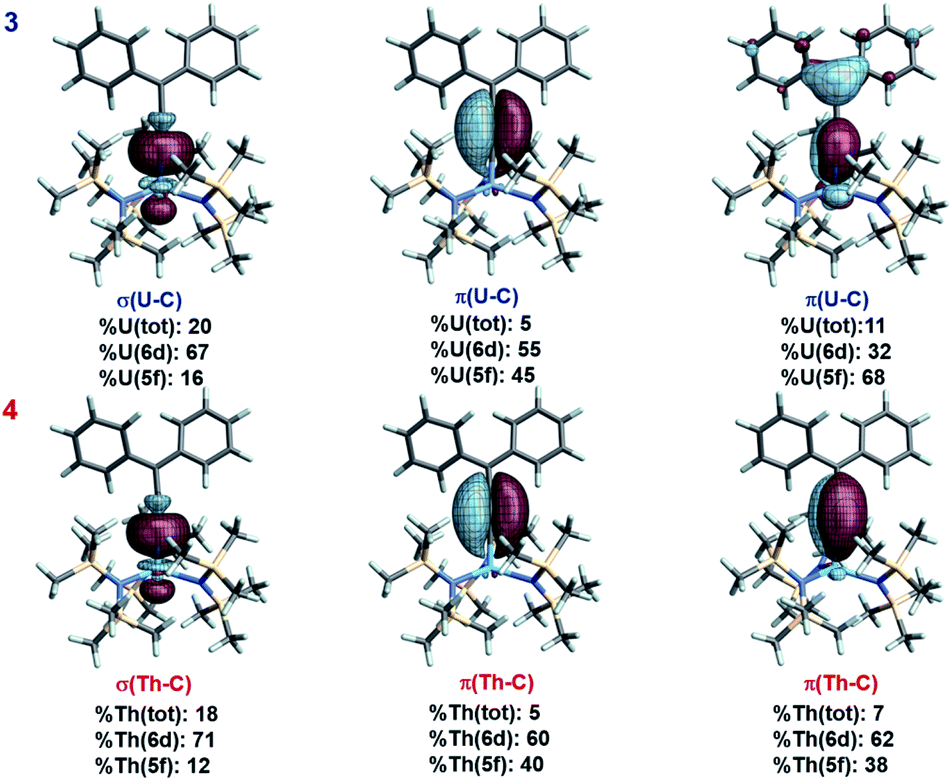 | ||
| Fig. 3 Isosurfaces (±0.03 a.u.) of selected NLMOs for 3 and 4. The α spin NLMOs of 3 are shown, weight-% metal character and 6d vs. 5f contributions at U are averaged over spins. | ||
An NLMO representing the π-component of a AnC double bond is clearly seen for both 3 and 4 (Fig. 3). This NLMO features multi-center character (3: 31% Cα, 28% Cβ, and 11% U; 4: 53% Cα, 37% Cβ, and 7% Th), and indicates that RS I is an important contributor to the overall electronic structure of both complexes, more so for 3 than for 4; although the metal weight in 4 is still significant. The other π-bonding NLMO in either complex has only 5% metal weight. Finally, the σ(An–C) bonds of 3 and 4 are represented by two-center two electron NLMOs with 20% and 18% total An weight for 3 and for 4, respectively (Fig. 3). These results are similar to the weights found in the uranium methanediide complex, [{C(PPh2S)2}U(BH4)2(THF)2].12
As seen in Table S2,† the Wiberg Bond Order (WBO) analyses are consistent with the conclusions drawn from the NLMO picture. For example, the An–Cα, Cα–Cβ, and Cβ–Cγ WBOs are 0.91, 2.36, and 1.31, respectively, for 4, and 0.98, 2.40, and 1.28, respectively, for 3, further suggesting RS I is more important for 3 than for 4. Interestingly, the An–Cα WBOs in 3 and 4 are notably larger than those of 1 (0.60) and 2 (0.57). The An–Cα WBOs in 3 and 4 are also larger than those of the An(IV) parent acetylides, [An(CCH)(NR2)3] (An = Th, WBO = 0.67; U, WBO = 0.71).48 Thus, the larger An–Cα WBOs evident in 3 and 4vs. [{(NR2)3}An(CHCCPh2)] vs. [An(CCH)(NR2)3] also supports the importance of resonance form I for these species, and confirms that they can be properly described as actinide carbenes.
An alternative way to examine the bonding in complexes 3 and 4 is offered by the quantum theory of atoms in molecules (QTAIM). This theory utilizes a variety of descriptors based on the topology of the electron density at a bond critical point (BCP).53 QTAIM data (Table S4†) suggest that the An–C bonds in 3 and 4 are polarised toward the ligand but possess covalent character, and, for 3, the results are nearly identical to the data reported for [U(BIPMMes)(Cl)2(THF)2] (BIPMMes = {C(PPh2NMes)2}).3 Furthermore, the QTAIM data suggest that Th–C bond in 4 is somewhat less covalent than the U–C bond in 3, consistent with the NLMO analysis.
13C chemical shift analysis
An NLMO analysis of the 13C NMR shielding for complexes 2 and 4 was performed using the computational methods reported in ref. 50–52 and 54,55,63. Data reported here are from scalar relativistic and spin-orbit (SO) DFT calculations with the PBE functional. Additional data provided in the ESI† show that the calculated shifts do not vary strongly with the functional used in the calculations (Table S3†). We also performed a shielding analysis of allene (H2CCCH2),57 for comparison with complex 2. For all compounds, the diamagnetic and paramagnetic contributions to the shieldings were combined. We confirmed that the observed variations in the carbon shielding and chemical shift come from the (usually negative) paramagnetic shielding mechanism, involving magnetic coupling between occupied and unoccupied orbitals,58 along with SO effects. The diamagnetic shielding per carbon is essentially constant, as usual. More details are provided in the ESI.†
Our calculated shielding constants for allene agree well with those reported by Wiberg et al. who analyzed the system in great detail (Table S3†).57 Effects from SO coupling (SOC) are very minor, as expected for an organic molecule without a heavy element. Cβ is strongly deshielded, by almost 150 ppm, relative to the methylene carbons. The primary reason for this difference is a strong paramagnetic deshielding from both π(C–C) NLMOs for Cβ (Table S5†). Although, the σ-bond NLMOs also contribute somewhat to the large shielding difference between the central and terminal carbons.
The calculated chemical shifts for complex 2 agree reasonably well with the experimental data (Tables S3 and S6†). For example, the calculated Cα shift for 2 is 144 ppm (expt. = 139.4 ppm). The replacement of CγH2 in allene by CγPh2 in 2 and the bonding of Cα to Th has a noticeable effect on most of the NLMO shielding contributions, leading to an overall decrease of the Cα and Cγ shielding, relative to allene, and a modest increase (13–17 ppm) of the Cβ shielding. The shielding patterns and contributions remain allene-like, however. This conclusion is further buttressed by the WBOs for Cα–Cβ (2.0) for Cβ–Cγ (1.6). The former value corresponds exactly to the expected bond order, whereas the latter reflects the aforementioned delocalization of π(Cβ–Cγ) onto Cipso. The main difference to allene is the inequivalency of Cα and Cγ. The shielding difference is −13 ppm in the calculations without SOC, and is primarily caused by more negative contributions from σ(Cα–Th) and σ(Cα–H) to the Cα shielding vs. the σ(Cγ–Cipso) contributions to the Cγ shielding, and a more negative contribution of σ(Cα–Cβ) to the Cα shielding vs. σ(Cβ–Cγ) contributing to the Cγ shielding. These differences are partially counteracted by a more positive allene-like Cα shielding from π(Cα–Cβ) compared to the Cγ shielding from π(Cβ–Cγ) (Table S6†). The delocalization onto Cipso (Fig. S3†) evidently enhances the Cγ paramagnetic deshielding relative to allene. With SOC effects included, the difference between the Cα and Cγ shielding becomes −39 ppm, as a result of the Th 5f (and 6d) AO contributions in σ(Cα–Th) and an associated SOC deshielding in the Cα core. The situation is reminiscent of the SOC effects on the shielding of nitrogen atoms bound to Th that we identified recently.59,60
The calculated 13C chemical shifts for complex 4 (Tables S3 and S7†) also agree reasonably well with the experimental data. For example, the calculated Cα shift for complex 4 is 211 ppm (expt. = 205.4 ppm). This value includes a 36 ppm deshielding contribution due to SOC, which is about 10 ppm larger in magnitude than that calculated for Cα in complex 2 as a result of the stronger σ(An–C) covalency in 4. The calculations also reproduce the experimentally assigned chemical shift ordering Cα > Cβ > Cγ, in 4, which is different from complex 2, for which the shifts are Cβ > Cγ > Cα. The increased Cβ shielding (smaller chemical shift) in 4 compared to 2 partially reflects the formal Cα–Cβ triple bond, according to RS II. However, Cβ in 4 is still substantially deshielded relative to Cβ in an authentic alkyne, such as PhCCH (77.2 ppm chemical shift),61 consistent with delocalization according to RS I. The different ordering is the result of two effects: (1) Cβ in 4 has triple bond character with a concomitant increase in magnetic shielding; and (2) the stronger Cα–Th covalency lowers the shielding of Cα in 4, compared to 2, via the combined effects of greater paramagnetic deshielding due to stronger Th–C bonding, and a stronger SOC deshielding (Tables S6 and S7†). Finally, the SOC induced deshielding for Cβ in 4 (−9 ppm) is much larger than that calculated for complex 2 (−4 ppm), which shows independently from the NLMO analysis that the delocalization involves Th, where most of the SOC originates.
Conclusions
In summary, reaction of [AnCl(NR2)3] with in situ generated lithium-3,3-diphenylcyclopropene affords the first actinide–allenyl complexes, [{(NR2)3}An(CHCCPh2)] (An = U, Th). Subsequent treatment with LDA and 2.2.2-cryptand results in the formation of the actinide–allenylidene complexes, [Li(2.2.2-cryptand)][{(NR2)3}An(CCCPh2)] (An = U, Th), which represent the first non-heteroatom supported carbene complexes of the actinides. Importantly, their isolation suggests that other actinide cumulenylidene complexes could be isolable, provided a viable synthetic route is available. Quantum chemical calculations give a detailed picture of the actinide–allenylidene interaction, which features partial AnC double bond character. Additionally, the Cα chemical shift in the two Th complexes exhibit SOC-induced deshielding due to 5f orbital participation in the Th–C bonds. The larger deshielding in the allenylidene complex vs. the allenyl is consistent with its greater Th–C covalency.
Going forward, we plan to explore the reactivity of our actinide allenylidene for comparison with the late transition metal allenylidenes, which will provide further insight into their electronic structure and potentially uncover new modes of allenylidene reactivity. The latter point is significant because the polarity of the carbon atoms within the actinide allenylidene unit is reversed relative to that observed in the late transition metals (e.g., resonance form III, Scheme 1).23,24,62
Data availability
Raw experimental and computational data will be made available free of charge upon request.Author contributions
G. T. K., G. W., and T. W. H. performed the experimental synthesis, characterization, and analysis of the data. X. Y. and J. A. performed the theoretical calculations and analyzed the data. All authors jointly wrote the manuscript. J. A. and T. W. H. secured the funding for the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the US Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Biosciences, and Geosciences Division under Contract DE-SC-0001861. J. A. acknowledges support for the theoretical component of this study by the U.S. Department of Energy, Office of Science, Heavy Element Chemistry program, grant DE-SC0001136. We thank the Center for Computational Research (CCR) at the University of Buffalo for providing computational resources.References
- S. Fortier, J. R. Walensky, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 6894–6897 CrossRef CAS PubMed.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Moro, E. S. Davies, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 2383–2386 CrossRef CAS PubMed.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Chem.–Eur. J., 2013, 19, 7071–7083 CrossRef CAS PubMed.
- D. E. Smiles, G. Wu, P. Hrobárik and T. W. Hayton, Organometallics, 2017, 36, 4519–4524 CrossRef CAS.
- P. Rungthanaphatsophon, P. Huang and J. R. Walensky, Organometallics, 2018, 37, 1884–1891 CrossRef CAS.
- P. Rungthanaphatsophon, A. Bathelier, L. Castro, A. C. Behrle, C. L. Barnes, L. Maron and J. R. Walensky, Angew. Chem., Int. Ed., 2017, 56, 12925–12929 CrossRef CAS PubMed.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- M. Gregson, A. J. Wooles, O. J. Cooper and S. T. Liddle, Comments Inorg. Chem., 2015, 35, 262–294 CrossRef CAS.
- T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC.
- J. T. Boronski, J. A. Seed, A. J. Wooles and S. T. Liddle, Chem. Commun., 2021, 57, 5090–5093 RSC.
- W. Ren, X. Deng, G. Zi and D.-C. Fang, Dalton Trans., 2011, 40, 9662–9664 RSC.
- T. Cantat, T. Arliguie, A. Noël, P. Thuéry, M. Ephritikhine, P. L. Floch and N. Mézailles, J. Am. Chem. Soc., 2009, 131, 963–972 CrossRef CAS PubMed.
- E. Lu, J. T. Boronski, M. Gregson, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2018, 57, 5506–5511 CrossRef CAS PubMed.
- J. A. Seed, H. R. Sharpe, H. J. Futcher, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2020, 59, 15870–15874 CrossRef CAS PubMed.
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed.
- J. T. Lyon and L. Andrews, Inorg. Chem., 2005, 44, 8610–8616 CrossRef CAS PubMed.
- J. T. Lyon and L. Andrews, Inorg. Chem., 2006, 45, 1847–1852 CrossRef CAS PubMed.
- J. Li, H.-S. Hu, J. T. Lyon and L. Andrews, Angew. Chem., Int. Ed., 2007, 46, 9045–9049 CrossRef CAS PubMed.
- B. O. Roos, R. Lindh, H.-G. Cho and L. Andrews, J. Phys. Chem. A, 2007, 111, 6420–6424 CrossRef CAS PubMed.
- J. T. Lyon, L. Andrews, P.-Å. Malmqvist, B. O. Roos, T. Yang and B. E. Bursten, Inorg. Chem., 2007, 46, 4917–4925 CrossRef CAS PubMed.
- H.-G. Cho, J. T. Lyon and L. Andrews, J. Phys. Chem. A, 2008, 112, 6902–6907 CrossRef CAS PubMed.
- J. T. Lyon, L. Andrews, H.-S. Hu and J. Li, Inorg. Chem., 2008, 47, 1435–1442 CrossRef CAS PubMed.
- M. I. Bruce, Chem. Rev., 1998, 98, 2797–2858 CrossRef CAS PubMed.
- V. Cadierno and J. Gimeno, Chem. Rev., 2009, 109, 3512–3560 CrossRef CAS PubMed.
- F. F. Mulks, P. W. Antoni, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 1810–1821 CrossRef CAS.
- P. Binger, P. Müller, R. Wenz and R. Mynott, Angew. Chem., Int. Ed., 1990, 29, 1037–1038 CrossRef.
- J.-S. Wang, L. Yao, J. Ying, X. Luo and X.-F. Wu, Org. Chem. Front., 2021, 8, 792–798 RSC.
- H. Werner, R. Fluegel, B. Windmueller, A. Michenfelder and J. Wolf, Organometallics, 1995, 14, 612–618 CrossRef CAS.
- E. A. Pedrick, P. Hrobárik, L. A. Seaman, G. Wu and T. W. Hayton, Chem. Commun., 2016, 52, 689–692 RSC.
- A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 13185–13192 CrossRef CAS PubMed.
- K. C. Mullane, P. Hrobárik, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2019, 58, 4152–4163 CrossRef CAS PubMed.
- G. B. Panetti, D.-C. Sergentu, M. R. Gau, P. J. Carroll, J. Autschbach, P. J. Walsh and E. J. Schelter, Nat. Commun., 2021, 12, 1713 CrossRef CAS PubMed.
- L. A. Seaman, P. Hrobárik, M. F. Schettini, S. Fortier, M. Kaupp and T. W. Hayton, Angew. Chem., Int. Ed., 2013, 52, 3259–3263 CrossRef CAS PubMed.
- N. S. Settineri, M. E. Garner and J. Arnold, J. Am. Chem. Soc., 2017, 139, 6261–6269 CrossRef CAS PubMed.
- S. Fortier, B. C. Melot, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2009, 131, 15512–15521 CrossRef CAS PubMed.
- L. A. Seaman, J. R. Walensky, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 3556–3564 CrossRef CAS PubMed.
- A. C. Behrle, A. J. Myers, P. Rungthanaphatsophon, W. W. Lukens, C. L. Barnes and J. R. Walensky, Chem. Commun., 2016, 52, 14373–14375 RSC.
- R. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- C. J. Elsevier, H. Kleijn, J. Boersma and P. Vermeer, Organometallics, 1986, 5, 716–720 CrossRef CAS.
- R. S. Keng and Y. C. Lin, Organometallics, 1990, 9, 289–291 CrossRef CAS.
- A. Wojcicki and C. E. Shuchart, Coord. Chem. Rev., 1990, 105, 35–60 CrossRef CAS.
- C. E. Shuchart, R. R. Willis and A. Wojcicki, J. Organomet. Chem., 1992, 424, 185–198 CrossRef CAS.
- I. V. Alabugin, in Stereoelectronic Effects, 2016, pp. 183–213, DOI:10.1002/9781118906378.ch7.
- R. Castarlenas, M. A. Esteruelas, R. Lalrempuia, M. Oliván and E. Oñate, Organometallics, 2008, 27, 795–798 CrossRef CAS.
- E. Buncel and B. Menon, J. Org. Chem., 1979, 44, 317–320 CrossRef CAS.
- R. E. Cramer, M. A. Bruck, F. Edelmann, D. Afzal, J. W. Gilje and H. Schmidbaur, Chem. Ber., 1988, 121, 417–420 CrossRef CAS.
- B. S. Newell, A. K. Rappé and M. P. Shores, Inorg. Chem., 2010, 49, 1595–1606 CrossRef CAS PubMed.
- G. T. Kent, X. Yu, C. Pauly, G. Wu, J. Autschbach and T. W. Hayton, Inorg. Chem., 2021 DOI:10.1021/acs.inorgchem.1c02064.
- C. Coletti, A. Marrone and N. Re, Acc. Chem. Res., 2012, 45, 139–149 CrossRef CAS PubMed.
- J. Autschbach and E. Zurek, J. Phys. Chem. A, 2003, 107, 4967–4972 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CAS.
- S. K. Wolff, T. Ziegler, E. v. Lenthe and E. J. Baerends, J. Chem. Phys., 1999, 110, 7689–7698 CrossRef CAS.
- R. F. W. Bader, T. S. Slee, D. Cremer and E. Kraka, J. Am. Chem. Soc., 1983, 105, 5061–5068 CrossRef CAS.
- J. Autschbach, J. Chem. Phys., 2008, 128, 164112 CrossRef PubMed.
- J. Autschbach and S. Zheng, Magn. Reson. Chem., 2008, 46, S45–S55 CrossRef PubMed.
- E. J. Baerends, T. Ziegler, J. Autschbach, D. Bashford, A. Bérces, F. Bickelhaupt, C. Bo, P. Boerrigter, L. Cavallo and D. Chong, 2014, http://www.scm.com.
- K. B. Wiberg, J. D. Hammer, K. W. Zilm and J. R. Cheeseman, J. Org. Chem., 1999, 64, 6394–6400 CrossRef CAS.
- R. V. Viesser, L. C. Ducati, C. F. Tormena and J. Autschbach, Chem. Sci., 2017, 8, 6570–6576 RSC.
- S. L. Staun, D.-C. Sergentu, G. Wu, J. Autschbach and T. W. Hayton, Chem. Sci., 2019, 10, 6431–6436 RSC.
- D.-C. Sergentu, G. T. Kent, S. L. Staun, X. Yu, H. Cho, J. Autschbach and T. W. Hayton, Inorg. Chem., 2020, 59, 10138–10145 CrossRef CAS PubMed.
- B. Wrackmeyer and K. Horchler, Prog. Nucl. Magn. Reson. Spectrosc., 1990, 22, 209–253 CrossRef CAS.
- S. W. Roh, K. Choi and C. Lee, Chem. Rev., 2019, 119, 4293–4356 CrossRef CAS PubMed.
- J. Autschbach, Mol. Phys., 2013, 111, 2544–2554 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, computational results, and spectral data for complexes 1–4. CCDC 2098903–2098906. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04666g |
| ‡ G. T. K. and X. Y. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |