DOI:
10.1039/D1SC04910K
(Edge Article)
Chem. Sci., 2021,
12, 14281-14287
Chromium carbides and cyclopropenylidenes†
Received
6th September 2021
, Accepted 8th October 2021
First published on 8th October 2021
Abstract
Carbon tetrabromide can be reduced with CrBr2 in THF to form a dinuclear carbido complex, [CrBr2(thf)2)][CrBr2(thf)3](μ-C), along with formation of [CrBr3(thf)3]. An X-ray diffraction (XRD) study of the pyridine adduct displayed a dinuclear structure bridged by a carbido ligand between 5- and 6-coordinate chromium centers. The carbido complex reacted with two equivalents of aldehydes to form α,β-unsaturated ketones. Treatment of the carbido complex with alkenes resulted in a formal double-cyclopropanation of alkenes by the carbido moiety to afford spiropentanes. Isotope labeling studies using a 13C-enriched carbido complex, [CrBr2(thf)2)][CrBr2(thf)3](μ-13C), identified that the quaternary carbon in the spiropentane framework was delivered by carbide transfer from the carbido complex. Terminal and internal alkynes also reacted with the carbido complex to form cyclopropenylidene complexes. A solid-state structure of the diethylcyclopropenylidene complex, prepared from 3-hexyne, showed a mononuclear cyclopropenylidene chromium(III) structure.
Introduction
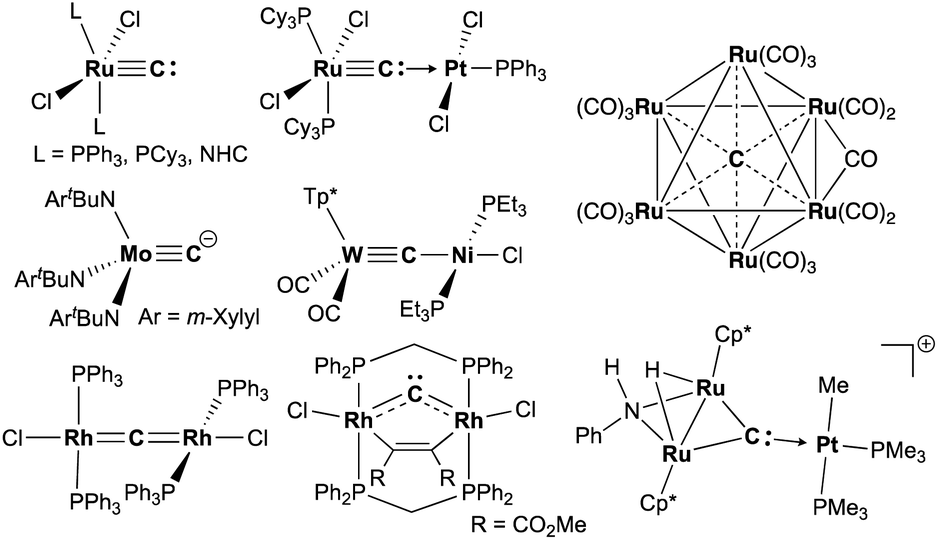
Carbide is a special ligand in organometallic,1–3 materials,4–6 and bioinorganic chemistry,7–9i.e. the core structure in nitrogenases.7 Despite numerous studies on stable interstitial carbido clusters2,3,10–13 and carbido materials4–6 as well as gas-phase generation of metal carbides,14–16 examples of more exposed and reactive molecular metal carbides, such as mononuclear, dinuclear, or trinuclear complexes, are still rare.1 As shown in Fig. 1, several types of molecular metal carbido species have been reported. Terminally bound carbide species can be categorized into two types based on their electronic structures. The neutral carbido complexes [(L)2Cl2M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C:] (M = Ru,17–19 Os;20 L = PPh3, PCy3, NHC), originally synthesized by Heppert in 2002,17 can form dative bonding Lewis pairs with various transition metals.21–25 The anionic terminal carbide [(ArtBuN)3MoC]− (Ar = m-Xylyl) has been prepared by Cummins and co-workers via deprotonation of a terminal methylidyne complex.26 Akin to the neutral terminal carbides,17–21 the anionic carbides also bind various metals to form dinuclear structures, but those dinuclear carbido complexes have a metallocarbyne character.27–31 In addition to those two types of dinuclear carbido complex, dimetallacumulene structures of dinuclear μ-carbides have been reported generally with late-transition metals.32–38 Recently, Hill and co-workers have reported a bent dimetallacumulene, which has a rather dimetallocarbene character, called dirhoda-heterocyclic carbene.39 The dimetallocarbene species39,40 can bind various Lewis acids to form trinuclear μ3-carbides, which have been investigated by Takemoto and Matsuzaka.41,42
C:] (M = Ru,17–19 Os;20 L = PPh3, PCy3, NHC), originally synthesized by Heppert in 2002,17 can form dative bonding Lewis pairs with various transition metals.21–25 The anionic terminal carbide [(ArtBuN)3MoC]− (Ar = m-Xylyl) has been prepared by Cummins and co-workers via deprotonation of a terminal methylidyne complex.26 Akin to the neutral terminal carbides,17–21 the anionic carbides also bind various metals to form dinuclear structures, but those dinuclear carbido complexes have a metallocarbyne character.27–31 In addition to those two types of dinuclear carbido complex, dimetallacumulene structures of dinuclear μ-carbides have been reported generally with late-transition metals.32–38 Recently, Hill and co-workers have reported a bent dimetallacumulene, which has a rather dimetallocarbene character, called dirhoda-heterocyclic carbene.39 The dimetallocarbene species39,40 can bind various Lewis acids to form trinuclear μ3-carbides, which have been investigated by Takemoto and Matsuzaka.41,42
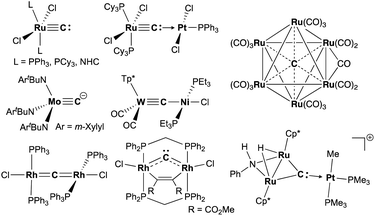 |
| | Fig. 1 Examples of metal carbido complexes: neutral terminal carbide, anionic terminal carbide, dative bonding μ-carbide, metallocarbyne, dimetallacumulene, dimetallocarbene, trinuclear μ3-carbide, and interstitial carbide. | |
Carbido complexes have been generally prepared by deprotonation of methylidynes,26,40,42 metallation of halocarbynes,28,43 or multiple C–X bond cleavages of various carbide sources, such as CO,10,44–46 CI4,32 CHCl3,35 and CS2.36,37 Although functionalization of lithiocarbynes [(OC)2(Tp*)MCLi(thf)n] (M = Mo, W; Tp* = tris(3,5-dimethyl-1-pyrazolyl)borate), prepared by lithiation of bromocarbynes [(OC)2(Tp*)MCBr],43 has been widely explored by Hill and co-workers,43,47–50 reactivity studies on carbide transfer of carbido ligands as a C1 source have not been developed. Herein, we report reduction of CBr4 with CrBr2 and structural characterization of a chromium carbide as well as a reactivity study for carbide transfer.
Results and discussion
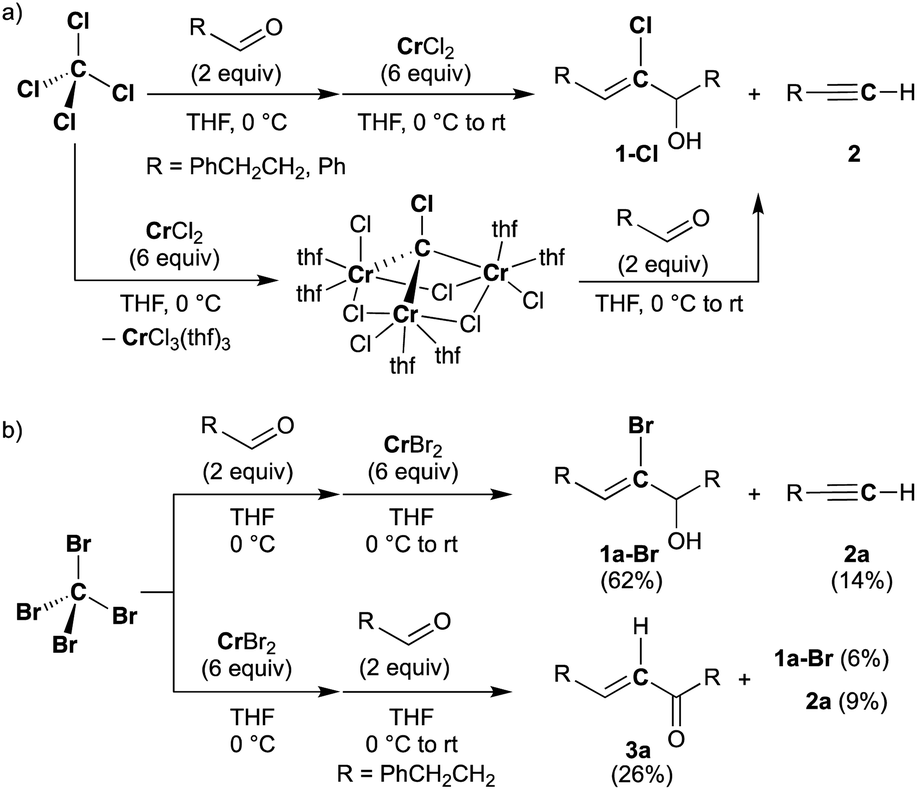
Halocarbyne and carbide transfer to aldehydes by the CX4–CrX2 reagent (X = Cl, Br)
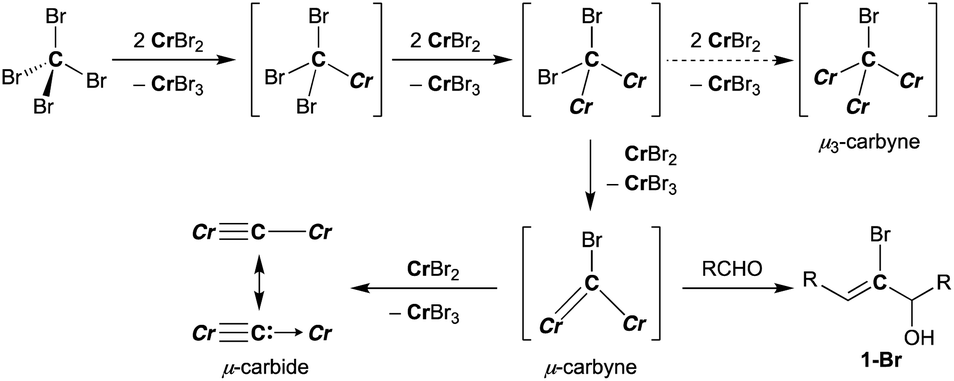
Falck and Mioskowski have reported a chlorocarbyne transfer reaction to aldehydes by treatment with a mixture of CCl4 and CrCl2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio.51 Recently, we have achieved characterization of a trinuclear chromium chlorocarbyne complex, [CrCl(thf)2]3(μ3-CCl)(μ-Cl)3, obtained from the CCl4–CrCl2 reagent,52 and the chlorocarbyne complex indeed underwent chlorocarbyne transfer to aldehydes to afford chloroallylic alcohols 1-Cl (Scheme 1a). In addition to formation of 1-Cl, Corey–Fuchs-type homologation products 2,53 terminal alkynes, were also formed via Cl-abstraction as a formal carbide transfer from the chlorocarbyne complex. A bromocarbyne transfer reaction has also been reported for the bromide analogues CBr4 and CrBr2,51 which was prepared in situ by reduction of CrBr3 with LiAlH4. We have revisited the classical protocol of preparation of CrBr2 by treatment of chromium(0) with hydrobromic acid.54 Although the Cr2+ ion is still fairly reducing (Cr3+/Cr2+: −0.424 V vs. SHE), a blue solid of the chromium(II) bromide hydrate [CrBr2(H2O)6] was readily precipitated out from an aqueous mixture of chromium(0) powder and hydrobromic acid at 0 °C.55 Having a pure solid of anhydrous CrBr2 in hand, we demonstrated the bromocarbyne transfer to aldehydes by use of the isolated CrBr2. To our surprise, in addition to formation of the bromoallylic alcohol 1a-Br and terminal alkyne 2a, an α,β-unsaturated ketone 3a was also formed (Scheme 1b). Interestingly, pre-mixing CBr4 and CrBr2 in THF prior to treatment with aldehydes resulted in the formation of the α,β-unsaturated ketone 3a as a major product. Unfortunately, formation of unidentifiable products by further reactions of 3a with THF56,57 promoted by some low-valent chromium species lowered the yields of 3a. Combinations of different halogens CCl4–CrBr2 and CBr4–CrCl2 were also attempted, but a mixture of all four products 1a-Cl, 1a-Br, 2a, and 3a was formed in both cases due to halogen-scrambling.
:6 ratio.51 Recently, we have achieved characterization of a trinuclear chromium chlorocarbyne complex, [CrCl(thf)2]3(μ3-CCl)(μ-Cl)3, obtained from the CCl4–CrCl2 reagent,52 and the chlorocarbyne complex indeed underwent chlorocarbyne transfer to aldehydes to afford chloroallylic alcohols 1-Cl (Scheme 1a). In addition to formation of 1-Cl, Corey–Fuchs-type homologation products 2,53 terminal alkynes, were also formed via Cl-abstraction as a formal carbide transfer from the chlorocarbyne complex. A bromocarbyne transfer reaction has also been reported for the bromide analogues CBr4 and CrBr2,51 which was prepared in situ by reduction of CrBr3 with LiAlH4. We have revisited the classical protocol of preparation of CrBr2 by treatment of chromium(0) with hydrobromic acid.54 Although the Cr2+ ion is still fairly reducing (Cr3+/Cr2+: −0.424 V vs. SHE), a blue solid of the chromium(II) bromide hydrate [CrBr2(H2O)6] was readily precipitated out from an aqueous mixture of chromium(0) powder and hydrobromic acid at 0 °C.55 Having a pure solid of anhydrous CrBr2 in hand, we demonstrated the bromocarbyne transfer to aldehydes by use of the isolated CrBr2. To our surprise, in addition to formation of the bromoallylic alcohol 1a-Br and terminal alkyne 2a, an α,β-unsaturated ketone 3a was also formed (Scheme 1b). Interestingly, pre-mixing CBr4 and CrBr2 in THF prior to treatment with aldehydes resulted in the formation of the α,β-unsaturated ketone 3a as a major product. Unfortunately, formation of unidentifiable products by further reactions of 3a with THF56,57 promoted by some low-valent chromium species lowered the yields of 3a. Combinations of different halogens CCl4–CrBr2 and CBr4–CrCl2 were also attempted, but a mixture of all four products 1a-Cl, 1a-Br, 2a, and 3a was formed in both cases due to halogen-scrambling.
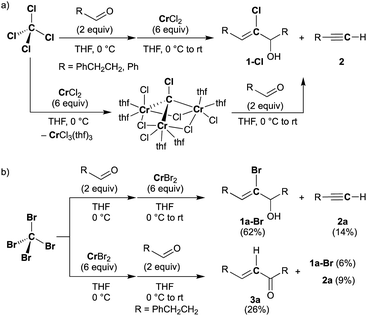 |
| | Scheme 1 Reactions of aldehydes with the CX4–CrX2 reagent ((a) X = Cl, (b) X = Br). | |
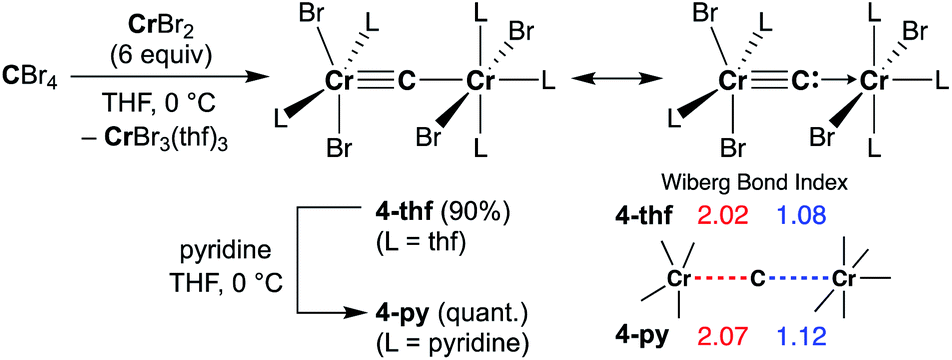
Reduction of CBr4 with CrBr2
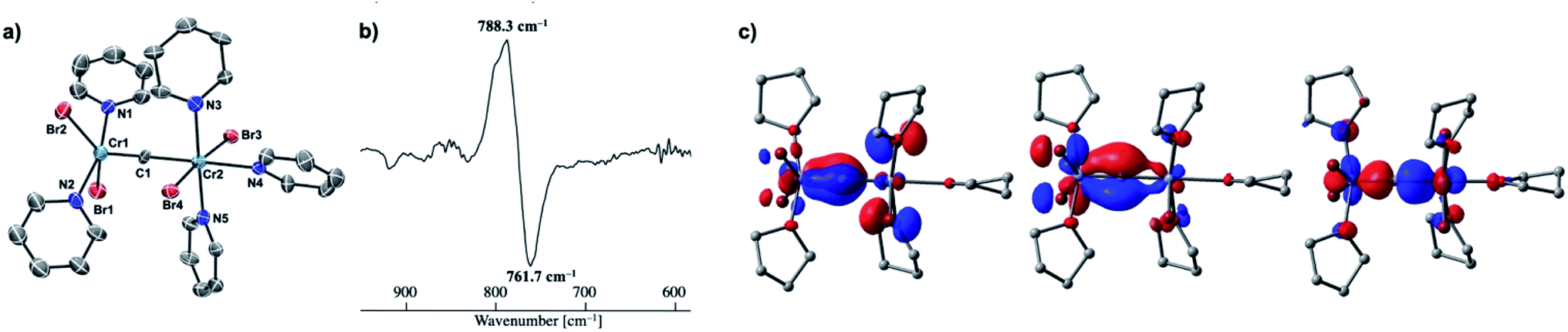
Given the idea of formation of the α,β-unsaturated ketone 3a, we inquired if the more reactive tetrahalomethane CBr4 could be further reduced by chromium(II) to form a chromium carbide species rather than a bromocarbyne species. Akin to the reduction of CCl4 with CrCl2 recently reported by our group,52 treatment of CBr4 with 6 equivalents of CrBr2 in THF at 0 °C produced [CrBr3(thf)3]58 as a brown solid along with a green supernatant. After removal of [CrBr3(thf)3], a green solid was obtained from the green supernatant and identified as paramagnetic 4-thf in 90% yield (Scheme 2). The green product 4-thf is stable in the solid state at room temperature, but slightly unstable in solution even at low temperature (−78 °C) to form [CrBr3(thf)3] and some unidentified chromium species. An XRD study of 4-thf revealed a dinuclear structure, namely [CrBr2(thf)2)][CrBr2(thf)3](μ-C), where a carbido ligand is bridging between 5- and 6-coordinate chromium centers (Fig. S35†). Upon crystallization of 4-thf to obtain a better crystal for the XRD study, complex 4-thf gradually decomposed in solution to form a precipitate of [CrBr3(thf)3]. The conclusive structural characterization was performed with a pyridine adduct, 4-py, which was obtained quantitatively by addition of pyridine to 4-thf in THF. The molecular structure of 4-py (Fig. 2a) still maintains a dinuclear chromium moiety bridged by a carbido ligand in a linear fashion (Cr1–C1–Cr2 = 174.5(6)°). The 5-coordinate chromium center is best described as distorted square pyramidal (τ5 = 0.29)59 with the carbido ligand on the apical position along with a short Cr1–C1 bond (1.634(10) Å), while the other chromium center represents a distorted octahedral geometry with a long Cr2–C1 bond (2.035(10) Å). Akin to the reported mononuclear neutral carbides and dinuclear dative bonding carbides, two X-type ligands (halides) and two L-type ligands (THF, pyridine, PR3 or NHC) are transoid to each other on the basal positions in the 5-coordinate environment, but the τ5 values of 4-thf (τ5 = 0.41) and 4-py (τ5 = 0.29) are larger than those of the mononuclear neutral carbides (τ5 = 0.07–0.16)17–20 and dinuclear dative bonding carbides (τ5 = 0.01–0.23)21–25 of ruthenium and osmium. Given the S = 2 nature of 4-thf in THF by Evans' method (μeff = 4.84 μB)60 as well as in the solid state (μeff = 5.03),55 the Wiberg bond indices of Cr1–C1 and Cr2–C1 bonds were calculated as 2.02 and 1.08, respectively, for 4-thf and 2.07 and 1.12 for 4-py. Bonding analyses of the neutral terminal carbides [(Me3P)2Cl2MC:] (M = Fe, Ru) have been previously reported by Krapp, Pandey, and Frenking.61 Although the Wiberg bond indices were ca. 2 for the M–C bond in the neutral terminal carbides, an MC triple bond character has been shown with two π- and one σ-type bonding orbitals. Molecular orbitals of 4-thf as well as 4-py also depicted two π-bonding interactions of the carbido ligand more delocalized around the 5-coordinate chromium center and a three-center two-electron [Cr–C–Cr] σ-interaction (Fig. 2c). Therefore, the canonical structure of the carbido complex 4 could be better described as a dative bonding μ-carbide21–25 than the metallocarbyne character.27–31
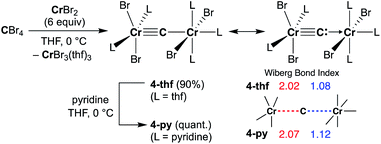 |
| | Scheme 2 Synthesis of 4-thf by reduction of CBr4 with CrBr2 and ligand-exchange with pyridine to form 4-py. | |
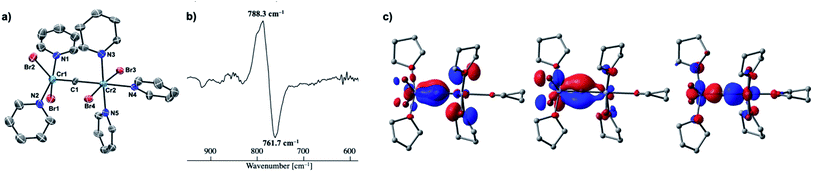 |
| | Fig. 2 (a) Solid-state structure of 4-py with thermal ellipsoids at the 50% probability level. Hydrogen atoms and solvated THF molecules have been omitted for clarity; (b) difference IR spectrum of 4-thf and 4-13C (4-thf: positive, 4-13C: negative); (c) molecular orbitals (isovalue = 0.05) of 4-thf representing Cr–C bonding interactions (left and middle: π-interactions, right: σ-interaction). | |
A plausible pathway of formation of the carbido complex 4-thf from CBr4 and CrBr2 is shown in Scheme 3. Akin to other Cr–C bond formations by reduction of haloalkanes with chromium(II) halides,62–66 single-electron reductions of C–Br bonds and subsequent radical coupling should proceed to form Cr–C bonds in the CBr4–CrBr2 system. As shown in Schemes 1b and 3, the transient bromocarbyne species could be trapped by aldehydes to afford the bromoallylic alcohols 1-Br.51 However, as far as we have attempted to isolate the bromocarbyne intermediate, no chromium species other than the carbido complex 4-thf and [CrBr3(thf)3] could be obtained by limiting the stoichiometry of CrBr2 to CBr4 or controlling the reaction temperature. Although the trinuclear μ3-chlorocarbyne complex [CrCl(thf)2]3(μ3-CCl) (μ-Cl)3 was obtained from the CCl4–CrCl2 system,52 a similar trinuclear framework bridged by bromides may be difficult for the bromocarbyne ligand to bridge in a μ3-fashion due to a larger ionic radius of bromine. Therefore, the transient dinuclear bromocarbyne could be further reduced by CrBr2 to cleave the last C–Br bond rather than forming a μ3-carbyne scaffold bridged by bromides, resulting in the formation of the dinuclear carbido complex 4-thf.
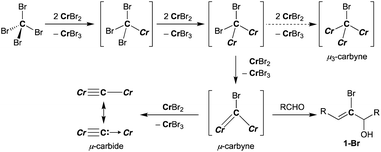 |
| | Scheme 3 Reduction of C–Br bonds with CrBr2 and formation of 4-thf (Cr = CrBr2(thf)n). | |
Isotope labeling studies
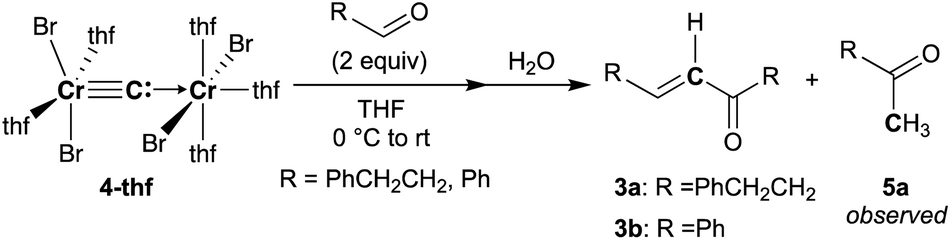
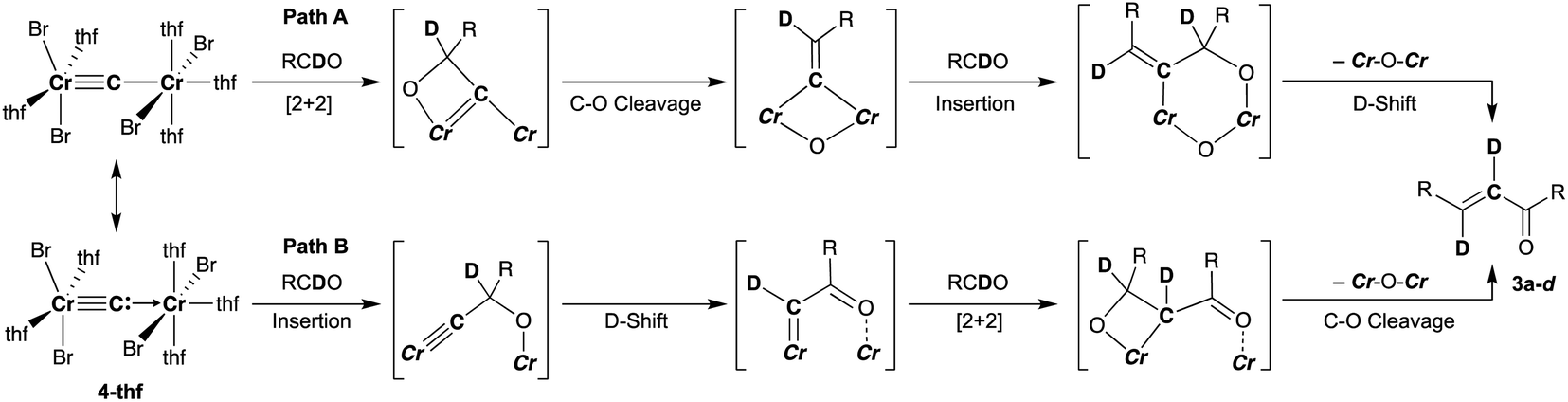
To spectroscopically confirm the carbido ligand delivered from CBr4, the isotopologue [CrBr2(thf)2)][CrBr2(thf)3](μ-13C) (4-13C) was prepared from 13CBr4. An IR spectrum of 4-13C revealed an absorption of the [Cr–C–Cr] 3-centered vibration at 762 cm−1, which was red-shifted from 788 cm−1 observed in the unlabeled carbide 4-thf (Fig. 2b). Measurement of a 13C NMR spectrum for 4-13C was also attempted, but 13C NMR signals other than solvents could not be located probably due to the paramagnetic feature of the chromium carbide. Akin to the in situ preparation of the CBr4–CrBr2 reagent, the isolated carbido complex 4-thf readily reacted with 2 equivalents of aldehydes (Scheme 4) to form the α,β-unsaturated ketones 3a (22%) and 3b (20%).55 Accordingly, a 13C-labeled α,β-unsaturated ketone 3a-13C was also prepared by use of 4-13C. The NMR spectrum of 3a-13C displayed an enriched 13C NMR signal selectively on the α-position at 130.85 ppm, which was coupled with α-H at 6.10 ppm (1JCH = 157 Hz). The deuterium labeling study was also demonstrated using a deuterated aldehyde, 3-phenylpropanal-d. The 2H NMR spectrum of 3a–d displayed deuterium signals on both α- and β-positions at 6.15 ppm and 6.88 ppm, respectively, implying that some H-shift event took place from the aldehyde to the α-carbon of the α,β-unsaturated ketone, which was delivered from the carbido ligand. As illustrated in Scheme 5, two plausible pathways to give the α,β-unsaturated ketones 3 could be considered from two canonical structures of 4-thf. Path A shows [2 + 2]-cycloaddition of the first aldehyde to the CrC bond, while Path B represents insertion of the aldehyde into the Cr–C or dative bond. Analyses of the quenched reaction mixture of aldehydes with 4-thf as well as the pre-mixed CBr4–CrBr2 revealed the formation of ketone 5a. The ketone 5a could be formed by hydrolysis of one of the intermediates represented in Path B, implying that the insertion pathway B is the more likely pathway.67
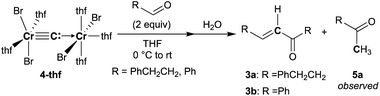 |
| | Scheme 4 Reactions of 4-thf with aldehydes. | |
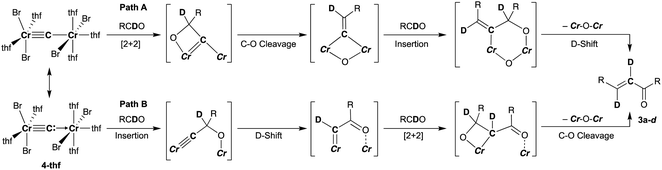 |
| | Scheme 5 Two plausible pathways to afford the α,β-unsaturated ketone 3 by reaction of 4-thf with aldehyde based on the isotope labeling studies. | |
Double-cyclopropanation by complex 4-thf to alkenes
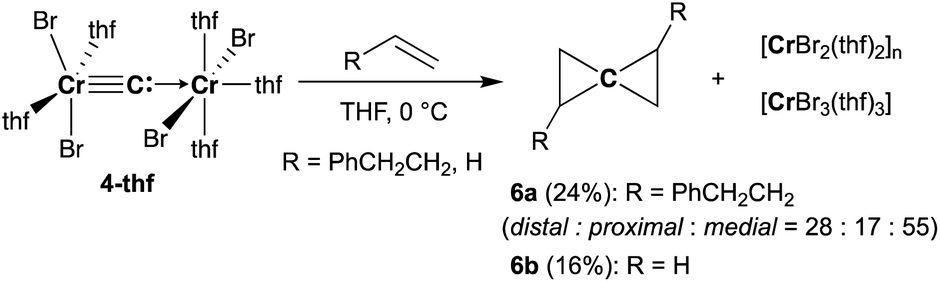
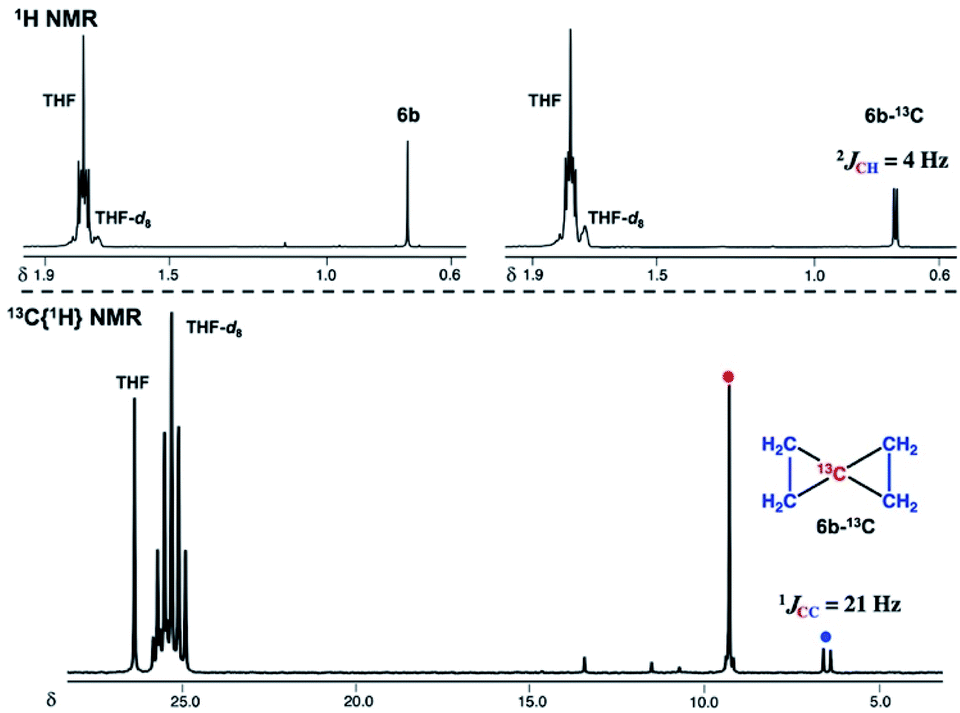
To gain further insight into the electronic structure of the dinuclear carbide 4, we hypothesized that a reactivity study of 4-thf with unsaturated substrates, such as alkenes or alkynes, would give some idea about which canonical structure of 4-thf is more dominant. A terminal alkene, 4-phenyl-1-butene, gradually reacted with 4-thf, resulting in precipitation of a mixture of [CrBr2(thf)2]n68 and [CrBr3(thf)3] (Scheme 6). After work-up, analyses by NMR and GC-MS spectroscopy revealed formation of spiropentane 6a (24% isolated yield) as a mixture of diastereomers69 (distal:proximal:medial = 28:17:55),55 and the spiropentane 6a was also prepared in 22% yield (distal:proximal:medial = 32:20:48) by the CBr4–CrBr2 reagent prepared in situ (Fig S15†). Addition of ethylene (1 atm) to a THF solution of 4-thf resulted in the formation of the parent spiropentane (6b). To conclusively confirm the formation of spiropentanes 6a and 6b by carbide transfer from 4-thf, the 13C-enriched carbide 4-13C was treated with 4-phenyl-1-butene and ethylene, respectively. As a result, the quaternary carbons in the spiropentane skeleton were indeed 13C-enriched in the 13C NMR spectra (Fig. 3) of 6a-13C (19.51 ppm, 21.19 ppm, and 21.22 ppm) and 6b-13C (9.30 ppm).
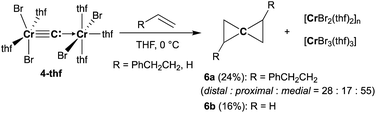 |
| | Scheme 6 Double-cyclopropanation by complex 4-thf to alkenes. | |
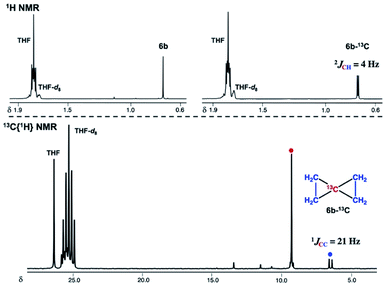 |
| | Fig. 3 Top: 1H NMR spectra of 6b (left) and 6b-13C (right). Bottom: 13C{1H} NMR spectrum of 6b-13C. | |
Spiropentane70,71 is still a challenging framework to assemble by organic synthetic methods such as reduction of tetrakis(1-haloalkyl)methanes72,73 due to multiple side-reactions and isomerization. Formal “double-cyclopropanation” by a carbido moiety to alkenes has been reported in the gas phase by use of carbon vapor,74,75 which was generated by arc discharge, but isomerization of the resulting spiropentanes also proceeded under such harsh conditions. Although the yields of the obtained spiropentanes 6a and 6b are still low, the carbido complex 4-thf underwent “double-cyclopropanation” to alkenes similar to the carbon vapor but without isomerization.
Synthesis of cyclopropenylidene complexes
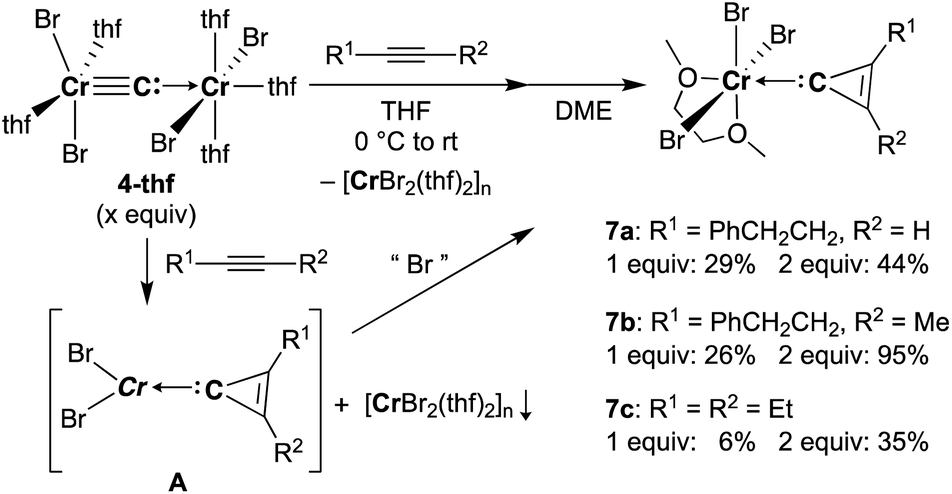
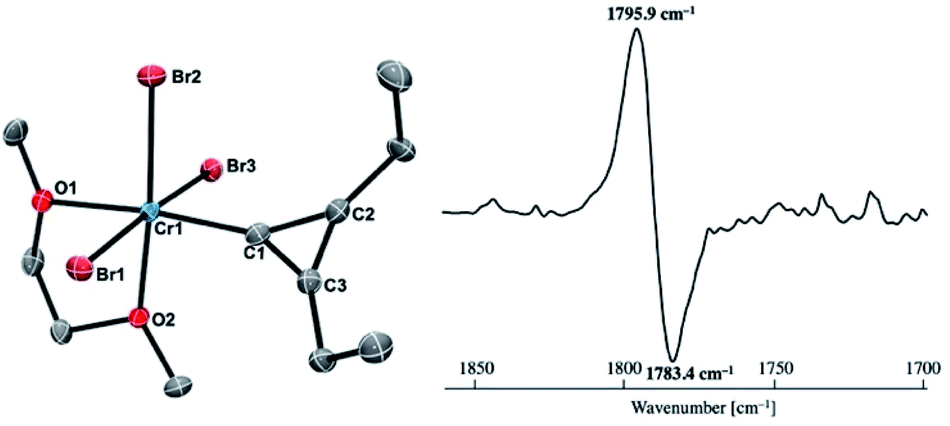
Reaction of 4-thf with alkynes smoothly proceeded to form a precipitate of [CrBr2(thf)2]n. After removal of [CrBr2(thf)2]n, paramagnetic green products 7a and 7b were obtained from a DME solution (Scheme 7). A single crystal suitable for an XRD study was obtained by crystallization of the 3-hexyne analogue 7c (μeff = 3.69 μB) from a DME solution layered with hexane. As shown in Fig. 4, a solid-state structure of 7c displayed a cyclopropenylidene ligand on a chromium(III) bromide [mer-CrBr3(dme)] along with a typical Cr(III)–carbene dative bond (2.039(3) Å).76–78 Akin to the reported chromium cyclopropenylidene complexes79–81 as well as the free cyclopropenylidenes,82,83 the cyclopropenylidene complex 7c also has a slightly shorter C–C bond (C2–C3: 1.351(4) Å) on the backbone than the other C–C bonds (C1–C2: 1.383(4) Å, C1–C3: 1.395(4) Å) in the cyclopropenylidene unit, implying delocalization of p-electrons over the 3-membered ring. IR spectra revealed a characteristic absorption of the cyclopropenylidene unit at 1796 cm−1, which was red-shifted to 1783 cm−1 by the 13C-enriched cyclopropenylidene 7c-13C. In contrast to the similar IR absorption observed in 7b/7b-13C at 1809/1796 cm−1, the IR spectra of the mono-substituted cyclopropenylidene complexes 7a/7a-13C delivered from the terminal alkyne showed two absorptions of the cyclopropenylidene ligand at 1351/1325 cm−1 and 1740/1720 cm−1 (Fig. S27†).
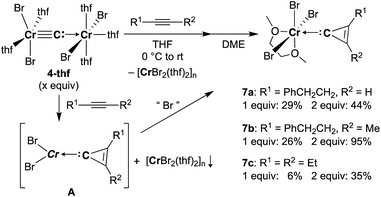 |
| | Scheme 7 Synthesis of cyclopropenylidene complexes. | |
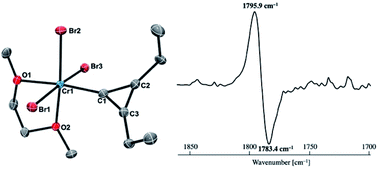 |
| | Fig. 4 (left) Solid-state structure of complex 7c with thermal ellipsoids at the 50% probability level. Hydrogen atoms have been omitted for clarity. (right) Difference IR spectrum (7c: positive, 7c-13C: negative). | |
Despite the tetrabromide complex 4-thf reacting with alkynes, a mixture of tribromide 7 and dibromide [CrBr2(thf)2]n was obtained. Thus, the third bromide in 7 could be provided by disproportionation of the resulting chromium(II) cyclopropenylidene A or another equivalent of carbide 4. Although the source of bromide in the formation of 7 is still unclear, use of two equivalents of carbide 4 to alkynes dramatically improved the yields of cyclopropenylidene complexes 7a (1 equiv.: 29% yield; 2 equiv.: 44% yield), 7b (1 equiv.: 26% yield; 2 equiv.: 95% yield), and 7c (1 equiv.: 6% yield; 2 equiv.: 35% yield). Note that addition of [CrBr3(thf)3] into the reaction was also attempted, but the yields of the cyclopropenylidene complexes were not improved.
Formation of a cyclopropenylidene unit by reaction of the ruthenium carbido complex [(Ph3P)2Cl2RuC:] with alkyne has been reported by Johnson and co-workers.84 The ruthenium carbide reacted only with an electron-deficient alkyne, dimethylacetylene dicarboxylate (DMAD), to form a cyclopropenylidene complex. In contrast to the ruthenium carbide [(Ph3P)2Cl2RuC:], our chromium carbide 4-thf reacted with electron-rich alkynes to form cyclopropenylidene complexes 7a, 7b, and 7c, while no reaction of 4-thf with DMAD in THF was observed along with a gradual decomposition of unreacted 4-thf in solution. Addition of alkenes as well as DMAD to cyclopropenylidene complexes 7a, 7b, and 7c has also been attempted, but identifiable products could not be obtained.
Conclusions
This work has shown that the CBr4–CrBr2 reagent possesses bromocarbyne and carbide transfer abilities to aldehydes. In the CBr4–CrBr2 reagent, the first example of a chromium carbido complex was isolated and structurally characterized. DFT calculations and NBO analyses of the carbido complex proposed a dative bonding μ-carbide character. Reactivity studies on carbide transfer of the carbido ligand with aldehydes and alkenes as well as isotope labeling studies have been demonstrated. It is noteworthy that a formal double-cyclopropanation of the carbido complex to alkenes might support the dative bond character of the carbido moiety. In addition, rare examples of cyclopropenylidene complexes have been prepared by treatment of the carbido complex with terminal and internal alkynes. Further investigation of carbide transfer is in progress.
Data availability
All experimental data, NMR spectra, FT-IR spectra, UV-Vis spectra, GC-MS data, crystallographic data, and computational analyses are provided in the ESI.†
Author contributions
K. Irifune prepared compounds and carried out reactions. T. Kurogi performed spectroscopy, crystallography, and computational studies and analyzed the data. T. Kurogi and K. Takai supervised this study and wrote the manuscript. All authors discussed the results and contributed to the peparation of the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by a Grant-in-Aid for Scientific Research (No. 18H03911) from MEXT, Japan. The DFT calculations were performed at the Research Center for Computational Science, Okazaki, Japan. We thank Prof. Hiroyuki Kawaguchi and Dr Yutaka Ishida for FT-IR and UV-vis measurements and elemental analyses of chromium compounds at Tokyo Institute of Technology. We also thank Ms. Chihiro Isonaka and Mr Takahiro Enoki at Okayama University for work on the preliminary study.
Notes and references
- S. Takemoto and H. Matsuzaka, Coord. Chem. Rev., 2012, 256, 574–588 CrossRef CAS.
- B. F. G. Johnson, J. Lewis, W. J. H. Nelson, J. N. Nicholls and M. D. Vargas, J. Organomet. Chem., 1983, 249, 255–272 CrossRef CAS.
- M. Tachikawa and E. L. Muetterties, Prog. Inorg. Chem., 1981, 28, 203–238 CAS.
- Y. Xiao, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2016, 4, 10379–10393 RSC.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098–16114 CrossRef CAS.
- J. Sun, J. Zhao, Z. Huang, K. Yan, X. Shen, J. Xing, Y. Gao, Y. Jian, H. Yang and B. Li, Nano-Micro Lett., 2020, 12, 13–49 CrossRef CAS PubMed.
- K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, Science, 2011, 334, 974–977 CrossRef CAS PubMed.
- J. A. Wiig, C. C. Lee, Y. Hu and M. W. Ribbe, J. Am. Chem. Soc., 2013, 135, 4982–4983 CrossRef CAS PubMed.
- J. Grunenberg, Angew. Chem., Int. Ed., 2017, 56, 7288–7291 CrossRef CAS PubMed.
- B. F. G. Johnson, R. D. Johnson and J. Lewis, J. Chem. Soc. A, 1968, 2865–2868 RSC.
- F. Scherbaum, A. Grohmann, B. Huber, C. Krüger and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1988, 27, 1544–1546 CrossRef.
- F. Scherbaum, A. Grohmann, G. Müller and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1989, 28, 463–465 CrossRef.
- N. T. Daugherty, T. J. Robilotto, J. Bacsa, T. G. Gray and J. P. Sadighi, Polyhedron, 2020, 181, 114464–114470 CrossRef CAS.
- C. J. Cassady and S. W. McElvany, J. Am. Chem. Soc., 1990, 112, 4788–4797 CrossRef CAS.
- R. Zhang, A. Dinca, K. J. Fisher, D. R. Smith and G. D. Willett, J. Phys. Chem. A, 2005, 109, 157–164 CrossRef CAS PubMed.
- S. Sabor, A. T. Benjelloun, M. M. A. Mogren and M. Mochlaf, Phys. Chem. Chem. Phys., 2014, 16, 21356–21362 RSC.
- R. G. Carlson, M. A. Gile, J. A. Heppert, M. H. Mason, D. R. Powell, D. V. Velde and J. M. Vilain, J. Am. Chem. Soc., 2002, 124, 1580–1581 CrossRef CAS PubMed.
- A. Hejl, T. M. Trnka, M. W. Day and R. H. Grubbs, Chem. Commun., 2002, 2524–2525 RSC.
- T. J. Morsing, A. Reinholdt, S. P. A. Sauer and J. Bendix, Organometallics, 2016, 35, 100–105 CrossRef CAS.
- M. H. Stewart, M. J. A. Johnson and J. W. Kampf, Organometallics, 2007, 26, 5102–5110 CrossRef CAS.
- S. H. Hong, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2004, 126, 7414–7415 CrossRef CAS PubMed.
- A. Reinholdt, J. E. Vibenholt, T. J. Morsing, M. Schau-Magnussen, N. E. A. Reeler and J. Bendix, Chem. Sci., 2015, 6, 5815–5823 RSC.
- A. Reinholdt, K. Herbst and J. Bendix, Chem. Commun., 2016, 52, 2015–2018 RSC.
- A. Reinholdt and J. Bendix, Inorg. Chem., 2017, 56, 12492–12497 CrossRef CAS PubMed.
- A. Reinholdt, A. F. Hill and J. Bendix, Chem. Commun., 2018, 54, 5708–5711 RSC.
- J. C. Peters, A. L. Odom and C. C. Cummins, Chem. Commun., 1997, 1995–1996 RSC.
- S. L. Latesky and J. P. Selegue, J. Am. Chem. Soc., 1987, 109, 4731–4733 CrossRef CAS.
- M. Etienne, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 1991, 113, 2324–2325 CrossRef CAS.
- I. A. Cade, A. F. Hill and C. M. A. McQueen, Organometallics, 2009, 28, 6639–6641 CrossRef CAS.
- B. J. Frogley and A. F. Hill, Chem. Commun., 2019, 55, 12400–12403 RSC.
- L. K. Burt and A. F. Hill, Dalton Trans., 2020, 49, 8143–8161 RSC.
- D. Mansuy, J. P. Lecomte, J. C. Chottard and J. F. Bartoli, Inorg. Chem., 1981, 20, 3119–3121 CrossRef CAS.
- A. Kienast, C. Bruhn and H. Homborg, Z. Anorg. Allg. Chem., 1997, 623, 967–972 CrossRef CAS.
- L. Galich, A. Kienast, H. Hüchstädt and H. Homborg, Z. Anorg. Allg. Chem., 1998, 624, 1235–1242 CrossRef CAS.
- C. Colomban, E. V. Kudrik, D. V. Tyurin, F. Albrieux, S. E. Nefedov, P. Afanasiev and A. B. Sorokin, Dalton Trans., 2015, 44, 2240–2251 RSC.
- R. D. Young, A. F. Hill, G. E. Cavigliasso and R. Stranger, Angew. Chem., Int. Ed., 2013, 52, 3699–3702 CrossRef CAS PubMed.
- S. I. Kalläne, T. Braun, M. Teltewskoi, N. Braun, R. Herrmann and R. Laubenstein, Chem. Commun., 2015, 51, 14613–14616 RSC.
- H. J. Barnett and A. F. Hill, Chem. Commun., 2020, 56, 7738–7740 RSC.
- H. J. Barnett and A. F. Hill, Angew. Chem., Int. Ed., 2020, 59, 4274–4277 CrossRef CAS PubMed.
- S. Takemoto, J. Ohata, K. Umetani, M. Yamaguchi and H. Matsuzaka, J. Am. Chem. Soc., 2014, 136, 15889–15892 CrossRef CAS PubMed.
- S. Takemoto, H. Morita, K. Karitani, H. Fujiwara and H. Matsuzaka, J. Am. Chem. Soc., 2009, 131, 18026–18027 CrossRef CAS PubMed.
- S. Takemoto, M. Tsujita and H. Matsuzaka, Organometallics, 2017, 36, 3686–3691 CrossRef CAS.
- R. L. Cordiner, A. F. Hill and J. Wagler, Organometallics, 2008, 27, 5177–5179 CrossRef CAS.
- A. Caselli, E. Solari, R. Scopelliti and C. Floriani, J. Am. Chem. Soc., 2000, 122, 538–539 CrossRef CAS.
- R. L. Miller and P. T. Wolczanski, J. Am. Chem. Soc., 1993, 115, 10422–10423 CrossRef CAS.
- J. A. Buss and T. Agapie, Nature, 2016, 529, 72–75 CrossRef CAS PubMed.
- A. L. Colebatch and A. F. Hill, Organometallics, 2016, 35, 2249–2255 CrossRef CAS.
- A. F. Hill and R. Shang, Organometallics, 2012, 31, 4635–4638 CrossRef CAS.
- A. L. Colebatch, A. F. Hill, R. Shang and A. C. Wills, Organometallics, 2010, 29, 6482–6487 CrossRef CAS.
- A. L. Colebatch, B. J. Frogley, A. F. Hill and C. S. Onn, Chem.–Eur. J., 2021, 27, 5322–5343 CrossRef CAS PubMed.
- R. Baati, D. K. Barma, J. R. Falck and C. Mioskowski, Tetrahedron Lett., 2002, 43, 2179–2181 CrossRef CAS.
- T. Kurogi, K. Irifune, T. Enoki and K. Takai, Chem. Commun., 2021, 57, 5199–5202 RSC.
- E. J. Corey and P. L. A. Fuchs, Tetrahedron Lett., 1972, 13, 3769–3772 CrossRef.
- D. G. Holah and J. P. Fackler, Jr, Inorg. Synth., 1967, 10, 26–33 CAS.
- See the ESI†.
- Y. Lan, P. Fan, X.-W. Liu, F.-F. Meng, T. Ahmad, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2017, 53, 12353–12356 RSC.
- G. S. Lee and S. H. Hong, Chem. Sci., 2018, 9, 5810–5815 RSC.
- P. J. Jones, A. L. Hale, W. Levason and F. P. McCullough, Jr, Inorg. Chem., 1983, 22, 2642–2644 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- A. Krapp, K. K. Pandey and G. Frenking, J. Am. Chem. Soc., 2007, 129, 7596–7610 CrossRef CAS PubMed.
- F. A. L. Anet and E. Leblanc, J. Am. Chem. Soc., 1957, 79, 2649–2650 CrossRef CAS.
- F. A. L. Anet, Can. J. Chem., 1959, 37, 58–61 CrossRef CAS.
- C. E. Castro and W. C. Kray, Jr, J. Am. Chem. Soc., 1963, 85, 2768–2773 CrossRef CAS.
- J. K. Kochi and D. D. Davis, J. Am. Chem. Soc., 1964, 86, 5264–5271 CrossRef CAS.
- C. E. Castro and W. C. Kray, Jr, J. Am. Chem. Soc., 1966, 88, 4447–4455 CrossRef CAS.
- Preliminary studies of the first steps of Paths A and B, [2 + 2]-cycloaddition of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O to CrC and insertion of aldehyde into Cr–C, respectively, with the help of DFT calculations showed that the intermediate formed by the first insertion reaction in Path B (+12.2 kcal mol−1) was much more stable than the [2 + 2]-cycloaddition product in Path A (+33.6 kcal mol−1) compared with ΔG(sol) values from 4-thf and PhCHO of 0 kcal mol−1. However, disproportionation of chromium species can also be considered during the reaction with aldehydes as observed in the reaction of 4-thf with alkynes, and it hampered mechanistic studies of these two reaction pathways.
O to CrC and insertion of aldehyde into Cr–C, respectively, with the help of DFT calculations showed that the intermediate formed by the first insertion reaction in Path B (+12.2 kcal mol−1) was much more stable than the [2 + 2]-cycloaddition product in Path A (+33.6 kcal mol−1) compared with ΔG(sol) values from 4-thf and PhCHO of 0 kcal mol−1. However, disproportionation of chromium species can also be considered during the reaction with aldehydes as observed in the reaction of 4-thf with alkynes, and it hampered mechanistic studies of these two reaction pathways.
- B. Twamley, R. Zehnder and P. J. Shapiro, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 51, m80–m81 CrossRef.
- H.-P. Guan, M. B. Ksebati, Y.-C. Cheng, J. C. Drach, E. R. Kern and J. Zemlicka, J. Org. Chem., 2000, 65, 1280–1290 CrossRef CAS PubMed.
- J. Donahue, G. L. Humphrey and V. Schomaker, J. Am. Chem. Soc., 1945, 67, 332–335 CrossRef.
- M. J. Murray and E. H. Stevenson, J. Am. Chem. Soc., 1944, 66, 314 CrossRef CAS.
- M. J. Murray and E. H. Stevenson, J. Am. Chem. Soc., 1944, 66, 812–816 CrossRef CAS.
- D. E. Applequist, G. F. Fanta and B. W. Henrikson, J. Org. Chem., 1958, 23, 1715–1716 CrossRef CAS.
- P. S. Skell and R. R. Engel, J. Am. Chem. Soc., 1965, 87, 1135 Search PubMed.
- P. S. Skell and R. R. Enge, J. Am. Chem. Soc., 1966, 88, 3749–3758 CrossRef CAS.
- F. P. Malan, E. Singleton, P. H. van Rooyen, J. Conradie and M. Landman, New J. Chem., 2018, 42, 19193–19204 RSC.
- K. A. Kreisel, G. P. A. Yap and K. H. Theopold, Organometallics, 2006, 25, 4670–4679 CrossRef CAS.
- D. S. McGuinness, V. C. Gibson, D. F. Wass and J. W. Steed, J. Am. Chem. Soc., 2003, 125, 12716–12717 CrossRef CAS PubMed.
- K. Öfele, Angew. Chem., Int. Ed., 1968, 7, 950 CrossRef.
- G. Huttner, S. Schelle and O. S. Mills, Angew. Chem., Int. Ed., 1969, 8, 515 CrossRef CAS.
- K. N. Juneau, L. S. Hegedus and F. W. Roepke, J. Am. Chem. Soc., 1989, 111, 4762–4765 CrossRef CAS.
- V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2006, 312, 722–724 CrossRef CAS PubMed.
- D. Holschumacher, C. G. Hrib, P. G. Jones and M. Tamm, Chem. Commun., 2007, 3661–3663 RSC.
- S. R. Caskey, M. H. Stewart, M. J. A. Johnson and J. W. Kampf, Angew. Chem., Int. Ed., 2006, 45, 7422–7424 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2093315 and 2093316. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04910k |
| ‡ Present address: Department of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawaoiwake-cho, Sakyo-ku, Kyoto 606-8502, Japan. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Keiichi
Irifune
*,
Keiichi
Irifune