
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural diversity of mixed polypnictogen complexes: dicationic E2E′2 (E ≠ E′ = P, As, Sb, Bi) chains, cycles and cages stabilized by transition metals†
Luis
Dütsch
,
Christoph
Riesinger
,
Gábor
Balázs
 ,
Michael
Seidl
and
Manfred
Scheer
*
,
Michael
Seidl
and
Manfred
Scheer
*
Department of Inorganic Chemistry, University of Regensburg, D-93040 Regensburg, Germany. E-mail: manfred.scheer@chemie.uni-regensburg.de
First published on 14th October 2021
Abstract
The reactivity of the tetrahedral dipnictogen complexes [{CpMo(CO)2}2(μ,η2:η2-EE′)] (E, E′ = P, As, Sb, Bi; “Mo2EE′”) towards different one-electron oxidation agents is reported. Oxidation with [Thia][TEF] (Thia+ = C12H8S2+; TEF− = Al{OC(CF3)3}4−) leads to the selective formation of the radical monocations [Mo2EE′]˙+, which immediately dimerize to the unprecedented dicationic E2E′2 ligand complexes [{CpMo(CO)2}4(μ4,η2:η2:η2:η2-E′EEE′)]2+via E–E bond formation. Single crystal X-ray diffraction revealed that, in the case of Mo2PAs and Mo2PSb, P–P bond formation occurs yielding zigzag E2P2 (E = As (1), Sb (2)) chains, whereas Mo2SbBi forms a Sb2Bi2 (5) cage, Mo2AsSb an unprecedented As2Sb2 unit representing an intermediate stage between a chain- and a cage-type structure, and Mo2AsBi a novel planar As2Bi2 (4a) cycle. Therefore, 1–5 bear the first substituent-free, dicationic hetero-E4 ligands, stabilized by transition metal fragments. Furthermore, in the case of Mo2AsSb, the exchange of the counterion causes changes in the molecular structure yielding an unusual, cyclic As2Sb2 ligand. The experimental results are corroborated by DFT calculations.
Introduction
The element carbon features the same affinity to both electropositive and electronegative elements, which makes it unique among all elements. Furthermore, this property is the basis of its infinite structural diversity.1 In particular, hydrocarbons form numerous chain- and cage-like as well as cyclic molecules or even combinations of these, which are important starting materials for organic syntheses and a large number of applications. For example, the isoprene molecule (2-methyl-1,3-butadiene) is the basic unit for the class of terpenes, which counts more than 8000 different molecules, and is widely used as e.g. flavours and fragrances.2 In contrast to carbon, the structural diversity of other p-block elements decreases strongly, which is caused by weaker covalent E–E bond energies. Thus, their chemistry is far less investigated. Since carbon and phosphorus are related to each other through the diagonal relationship and the isolobality between the {CH} fragment and the P atom (Scheme 1a),3 phosphorus is also capable of catenation. While numerous neutral and anionic polyphosphorus chains, cages and cycles have been known for a long time,4 the field of cationic representatives was only opened during the last two decades, mainly by the groups of Burford and Weigand.5 However, these compounds always carry organic substituents. Recently, we could show that polyphosphorus ligand complexes represent good starting materials for cationic polyphosphorus compounds upon oxidation. For example, oxidation of the hexaphosphabenzene6 complex [(Cp*Mo)2(μ,η6:η6-P6)] results in a bis-allylic distortion of the P6 ring.7 In contrast, oxidation of [Cp*Fe(η5-P5)] leads to dimerization via P–P bond formation yielding a formally neutral, bicyclic P10 ligand stabilized by two [Cp*Fe]+ fragments.8 The first substituent-free polyphosphorus cation, namely [P9]+ (I, Scheme 1b), was obtained by Krossing et al. via oxidation of P4 with [NO]+.9 This milestone in inorganic chemistry could only be accomplished with the help of weakly coordinating anions (WCAs),10 which are able to stabilize very labile and reactive cations due to their weak nucleophilic properties. Since the tetrahedrane derivative [{CpMo(CO)2}2(μ,η2:η2-P2)] (II; “Mo2P2”) is isolobal to P4, we carried out its oxidation, which leads to dimerization via P–P bond formation yielding the dicationic complex [{CpMo(CO)2}4(μ,η2:η2:η2:η2-P4)]2+ (VI) including an unique P4 chain free from organic substituents.11 In comparison to polyphosphorus compounds, representatives of the heavier group 15 elements such as arsenic, antimony as well as bismuth are considerably less known. Interestingly, we could transfer the reactivity of II towards oxidations to its heavier derivatives [{CpMo(CO)2}2(μ,η2:η2-E2)] (E = As (III), Sb (IV), Bi (V); “Mo2E2”), which yield similar dimerization products [{CpMo(CO)2}4(μ,η2:η2:η2:η2-E4)]2+ (E = As (VII), Sb (VIII), Bi (IX)) including an analogous dicationic As4 (VII) chain as well as unique dicationic Sb4 (VIII) and Bi4 (IX) “butterfly-like” cages, respectively, which are stabilized by transition metal fragments.11 Even rarer is the field of hetero-polypnictogen complexes, especially the ones containing As–Sb,12 As–Bi13 and Sb–Bi14 bonds, since the hetero-element bond energy decreases. Therefore, they have to be stabilized by bulky organic substituents, as for instance in the neutral hetero-tripnictogen chains tBu2EP(tBu)E′tBu2 (E, E′ = As, Sb; X)15 and tBu2PAs(tBu)EtBu2 (E = P, As, Sb, Bi; XI)16 (Scheme 1c). Otherwise, they tend to disproportionate by forming homonuclear bonds.12a The only example of a cationic hetero-polypnictogen complex is, to the best of our knowledge, the arsane-stabilized dicationic P4 butterfly compound [(AsPh3)2(μ,η1:η1-P4][AlCl4]2 (XII; Scheme 1d),17 whereas representatives of the heavier pnictogens are unknown, which might be caused by the lack of suitable precursors.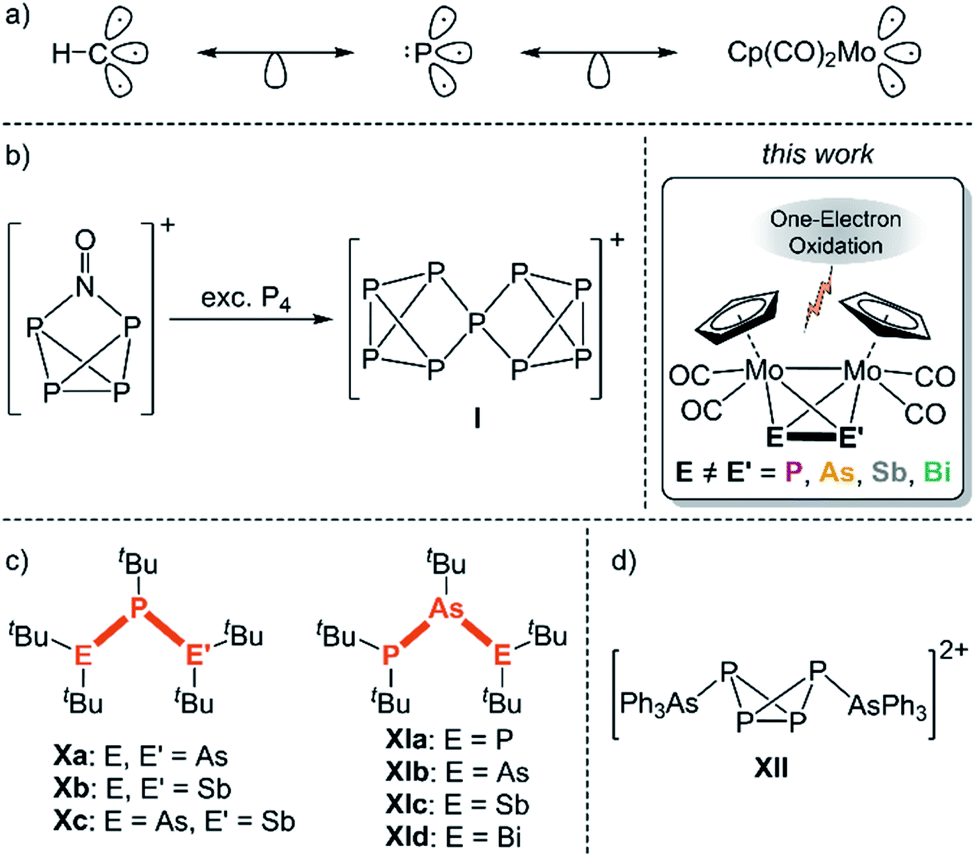 | ||
| Scheme 1 (a) Isolobal relation between the {CH} fragment, phosphorus and the 15 VE fragment {CpMo(CO)2}; (b) first substituent-free polyphosphorus cation P9+ (I); (c) selected examples of neutral hetero-polypnictogen complexes (X, XI); (d) dicationic hetero-polypnictogen compound XII; this work: one-electron oxidation of the hetero-dipnictogen complexes [{CpMo(CO)2}2(μ,η2:η2-EE′)] (EE′ = PAs (A), PSb (B), AsSb (C), AsBi (D), SbBi (E)). | ||
To target this, only very recently we were able to extend the class of tetrahedral Mo2E2 (II–V) compounds by their respective substituent-free hetero-dipnictogen congeners [{CpMo(CO)2}2(μ,η2:η2-EE′)] (EE′ = PAs (A), PSb (B), AsSb (C), AsBi (D), SbBi (E); Scheme 1). They are now accessible via a simple procedure in high yields, which makes further reactivity studies feasible.18C–E feature the very first covalent bonds between two different heavy pnictogen atoms that do not possess organic substituents. Hence, A–E should serve as excellent precursors for the formation of unprecedented extended hetero-polypnictogen frameworks upon oxidation. Thus, the question arises between which pnictogen atoms of the hetero-EE′ ligand the new bonds will be formed after oxidation and whether the ionisation potential of the pnictogen or the bond energy of the newly formed bond will determine the reaction outcome. Herein, we report on the reactivity of A–E towards salts of the strong one-electron oxidant thianthrenium ([Thia]˙+ = [C12H8S2]˙+) to form unprecedented hetero-pnictogen chain and cage moieties. Additionally, the influence of the stabilizing counterion on the reactivity and the solid-state structure was investigated, and a remarkable effect is shown.
Results and discussion
Cyclic voltammetry
The cyclic voltammograms (CV) of A–E (Fig. 1) reveal a chemically pseudo-reversible oxidation at +0.19 V (Mo2PAs = A), +0.08 V (Mo2PSb = B), +0.12 V (Mo2AsSb = C), −0.10 V (Mo2AsBi = D) and −0.07 V (Mo2SbBi = E) vs. Cp2Fe0/+ and the reductive back wave significantly shifted to −0.31 V (A), −0.43 V (B), −0.20 V (C), −0.36 V (D) and −0.44 V (E).19 Compared to Mo2P2 (IV, +0.28 V),11 the oxidation potential of Mo2PAs is considerably lower but almost equal to that of the heavier congener Mo2As2 (V, +0.19 V).11 The same is observed for Mo2PSb, where the oxidation potential equals the one of Mo2Sb2 (+0.05 V).11 However, the oxidation potentials of Mo2AsSb, Mo2AsBi and Mo2SbBi are in between the oxidation potentials of their respective homo-dipnictogen complexes, with the latter two being almost similar.11 Therefore, Mo2PSb steps out of line, as a higher or at least similar oxidation potential compared to Mo2AsSb is expected. Mo2SbBi also shows an additional small oxidation wave at +0.05 V, which can be attributed to small amounts of Mo2Sb2, which are formed as trace impurities during its synthesis.18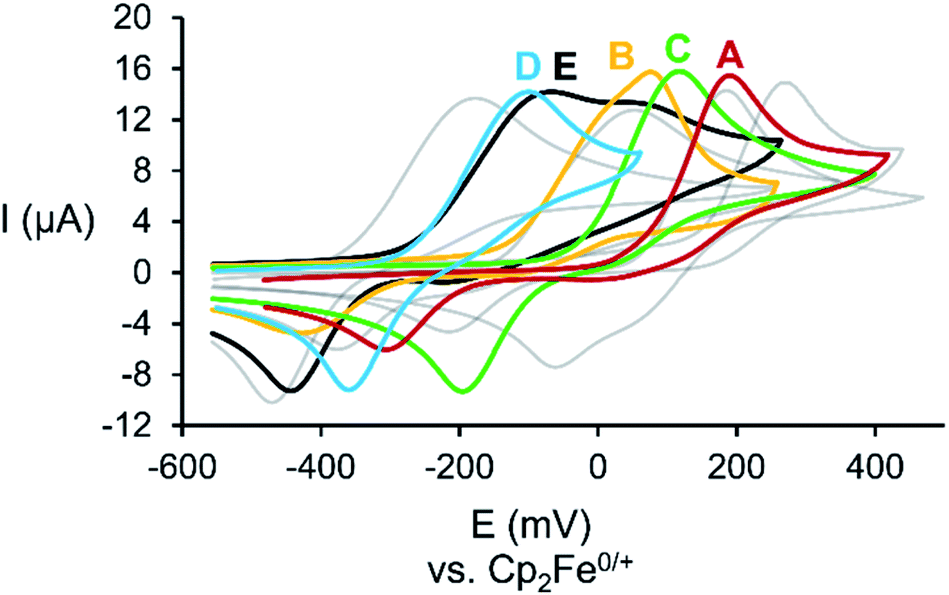 | ||
| Fig. 1 Cyclic voltammograms of the starting materials A–E (coloured) as well as their homo-dipnictogen congeners (grey; from right to left: II, III, IV and V) in CH2Cl2 solution (only the first oxidation and its respective back wave are shown); c([NBu4][PF6]) = 0.1 M. | ||
The CVs of A and B suggest that the heavier pnictogen atom (As in A and Sb in B) contributes more to the oxidation potential than the P atom and, therefore, a dimerization via As–As or Sb–Sb bond formation upon one-electron oxidation should be favoured over P–P bond formation. DFT calculations also show that the heavier pnictogen atom contributes more to the HOMO and that the pnictogen atomic orbital contribution increases with increasing atomic number, i.e. P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E (%) = 7:7, 10:12 and 13:18 for E = As, Sb and Bi, respectively. However, this contrasts with the experimental findings, which are discussed in the following.
:E (%) = 7:7, 10:12 and 13:18 for E = As, Sb and Bi, respectively. However, this contrasts with the experimental findings, which are discussed in the following.
One-electron oxidation of A–E
When an orange red solution of A or B is reacted with the very strong one-electron oxidant [Thia]+ (E = 0.86 V vs. Cp2Fe0/+)20 containing the WCA [Al{OC(CF3)3}4]− (=[TEF]−) in CH2Cl2, immediately dark greenish red solutions of the P–P coupled products [{CpMo(CO)2}4(μ4,η2:η2:η2:η2-EPPE)][TEF]2 (E = As (1), Sb (2)), featuring a P2E2 chain, are obtained selectively, and 1 and 2 can be isolated in 73% and 88% yields (Scheme 2). DFT calculations show that the formation of the isomers containing a P–P bond are energetically favoured compared to the possible isomers with E–E bonds (42 kJ mol−1 and 38 kJ mol−1 for 1 and 2, respectively). The next starting materials, the heavier analogues Mo2AsSb (C) and Mo2AsBi (D), represent very interesting compounds as their lighter homo-dipnictogen congener Mo2As2 (III) builds dicationic E4 chains upon oxidation, whereas their heavier homo-dipnictogen congeners Mo2Sb2 (IV) and Mo2Bi2 (V) form dicationic E4 cage-like ligand complexes. Therefore, the question arose which way C and D tend to follow upon one-electron oxidation. Their CVs (Fig. 1) indicate an oxidation behaviour, which is in between their homo-dipnictogen derivatives. Interestingly, the reaction of C and D with [Thia][TEF] selectively leads to the E′–E′ coupled, dicationic products [{CpMo(CO)2}4(μ4,η2:η2:η2:η2-EE′E′E)][TEF]2 (EE′ = AsSb (3a), AsBi (4a)) in excellent isolated yields of 92% and 89% (Scheme 2). 3a represents an astonishing intermediate stage between the chain- and the cage-type structures and 4a possess an unprecedented planarized As2Bi2 cyclic ligand which differs significantly from hitherto observed structures. Interestingly, in contrast to 1 and 2, in both cases, no bonds between the lighter pnictogen atoms (As in 3a and 4a) are formed. However, oxidation of E leads to an Sb–Sb coupled cage-like compound [{CpMo(CO)2}4(μ4,η2:η2:η2:η2-BiSbSbBi)][TEF]2 (5) in 84% crystalline yield (Scheme 2), which exhibits an Sb2Bi2 ligand with a butterfly-like structure.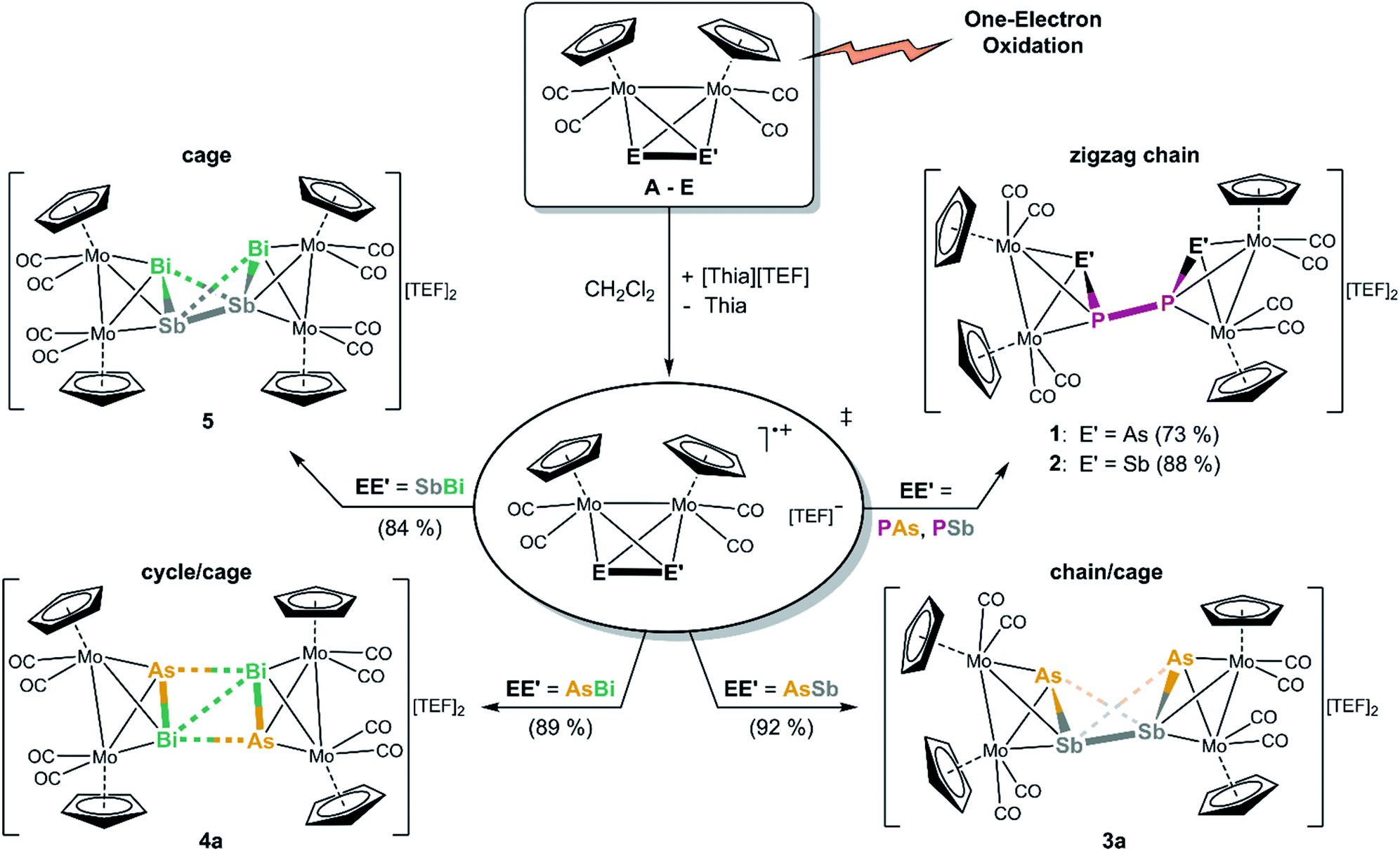 | ||
| Scheme 2 Oxidation of the tetrahedral hetero-dipnictogen complexes [{CpMo(CO)2}2(μ,η2:η2-EE′)] (EE′ = PAs (A), PSb (B), AsSb (C), AsBi (D), SbBi (E) resulting in dimerization reactions leading to dicationic E2E′2 chains, cages and cycles. Isolated yields are given in parentheses. | ||
In each case, the potentially first formed radical cations [A]˙+, [B]˙+, [C]˙+, [D]˙+ or [E]˙+, respectively, immediately dimerize and do not dissociate in solution since no signals can be observed in the respective X-band EPR spectra (vide infra). This is also supported by DFT calculations which show that the dimerization of the radical cations [A]˙+–[E]˙+ is exothermic (A: 147 kJ mol−1, B: 158 kJ mol−1, C: 143 kJ mol−1, D: 144 kJ mol−1 and E: 166 kJ mol−1).
Structural characterization of 1–5
Analytically pure crystals of 1 and 3a–5 suitable for single crystal X-ray diffraction are received as dark red (1, 3a) or black (4a, 5) blocks or plates after precipitation with n-hexane, washing with toluene and recrystallization from CH2Cl2/n-hexane or o-difluorobenzene/n-hexane at 4 °C. Despite several attempts, 2 could only be crystallized as thin plates which allowed to yield a weak X-ray dataset revealing just a first insight into the heavy-atom framework of the molecular structure and, therefore, no detailed structural data of 2 are discussed in the following.21The cationic moieties of 1–5 (Fig. 2a–d) each consist of two molecules of the oxidized starting materials [A]˙+, [B]˙+, [C]˙+, [D]˙+ or [E]˙+, respectively, whose Mo2EE′ tetrahedra are linked together via a newly formed E–E or E′–E′ bond. The received central structural motifs in 1 and 2 are an asymmetrical AsPPAs or SbPPSb zigzag chain, respectively, with a gauche conformation (dihedral angle in 1: 134.77(1)°). Hence, they are related to their all-phosphorus and all-arsenic derivatives VI and VII.11 The P–As distances in 1 are only slightly longer than in free A (2.232(1) Å),22 but still slightly shorter than a P–As single bond (2.32 Å).23 The newly formed central P–P bond (2.2163(1) Å) matches well with an anticipated classical single bond and with the corresponding distance in the DFT-optimized geometry (2.198 Å). The respective P–P distance in the DFT-optimized geometry of 2 is calculated to 2.201 Å.
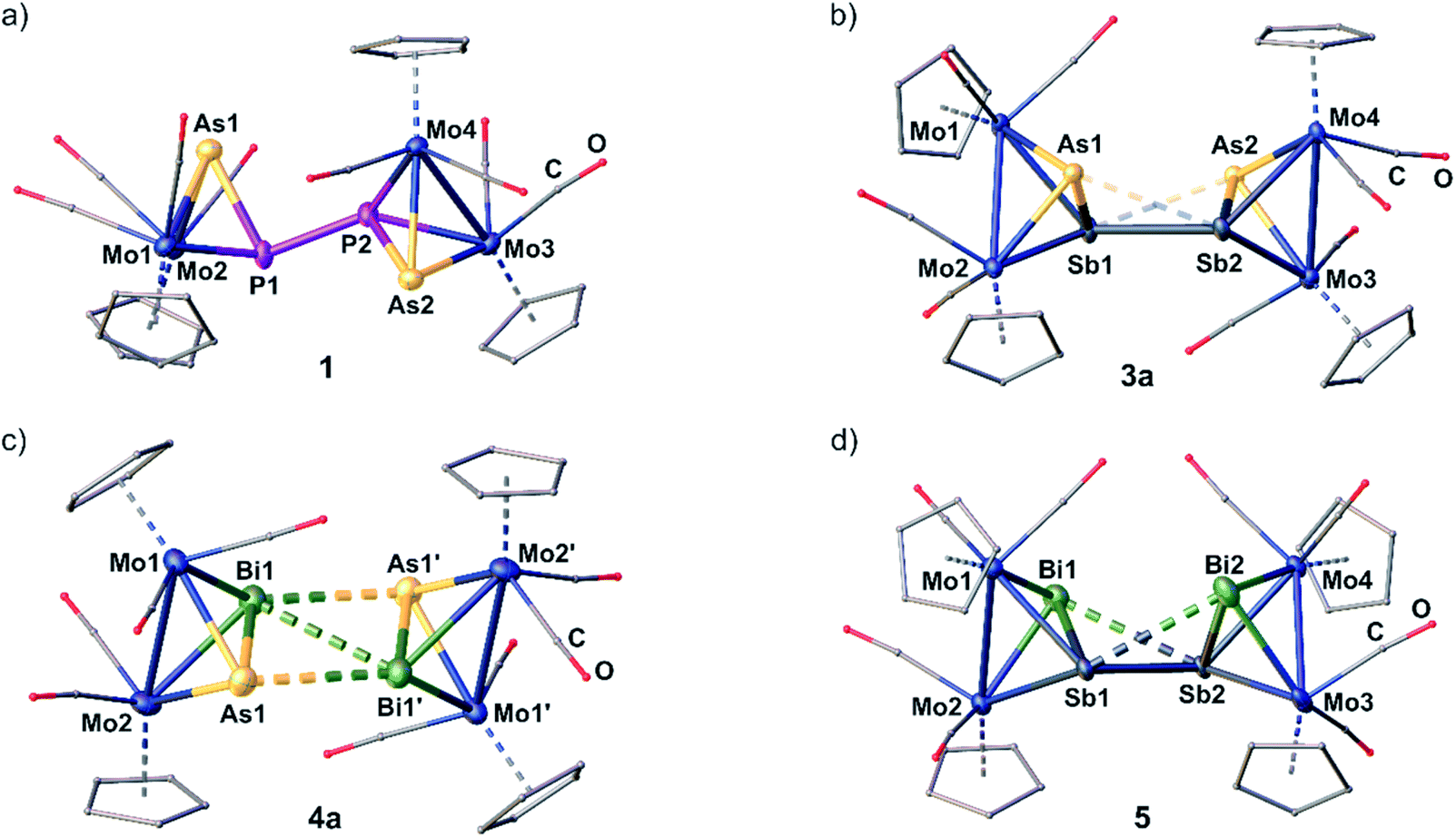 | ||
| Fig. 2 Molecular structure of the dicationic parts of 1 (a), 3a (b), 4a (c), and 5 (d). Anisotropic displacement is set to the 50% probability level. H atoms are omitted and C as well as O atoms are drawn as small spheres for clarity. Short E⋯E and E⋯E′ contacts are drawn as dashed bonds and long E⋯E′ contacts as translucent dashed bonds. Selected bond lengths [Å] and angles [°]: 1: P1–As1 2.3099(1), P1–P2 2.2163(1), P2–As2 2.2490(1), Mo1–Mo2 3.1853(1), Mo3–Mo4 3.1559(1), As1–P1–P2 96.79(1), P1–P2–As2 103.86(1), As1–P1–P2–As2 134.77(1); 3a: As1–Sb1 2.6874(1), Sb1–Sb2 3.0024(1), Sb2–As2 2.6795(1), As1–Sb2 3. 3.3214 (1), As2–Sb1 3. 2730(1), As1–Sb1–Sb2 71.18(1), Sb1–Sb2–As2 70.08(1), As1–Sb1–Sb2–As2 128.98(1); 4a: As1–Bi1 2.8261(1), Bi1–Bi1′ 3.2725(1), As1–Bi1′ 3.2577(1), Mo1–Mo2 3.1922(1), As1–Bi1–Bi1′ 64.09(1), As1–Bi1–Bi1′–As1′ 180.00(1); 5: Sb1–Bi1 2.9760(1), Sb1–Sb2 3.2237(1), Sb2–Bi2 2.9851(1), Sb1–Bi2 3.3236(1), Sb2–Bi1 3.3048(1), Mo1–Mo2 3.2172(1), Mo3–Mo4 3.2205(1), Bi1–Sb1–Sb2 64.69(1), Sb1–Sb2–Bi2 64.18(1), Bi1–Sb1–Sb2–Bi2 112.70(1). | ||
In 3a–5, hetero-tetrapnictogen ligands (AsSbSbAs (3a), AsBiBiAs (4a) and BiSbSbBi (5)) are observed, which, however, differ from those of the P–P and As–As coupled derivatives 1, 2, VI and VII and reveal cage-like structural motifs. Thereby, the intra-tetrahedral E–E′ bond lengths are elongated by ∼0.2 Å compared to the respective starting materials but are all just slightly longer than the respective single bonds.23 In contrast, the newly formed E–E or E′–E′ bonds, respectively, are comparably longer and exceed the respective single bonds by 0.20 Å (3a), 0.25 Å (4a) and 0.42 Å (5). Interestingly, 3a, 4a and 5 exhibit two further short E⋯E′ contacts (As⋯Sb: 3.2730(1)–3.3214(1) Å; As⋯Bi: 3.2577(1) Å; Sb⋯Bi: 3.3048(1)–3.3236(1) Å).24 In 4a and 5, they exceed their respective single bonds by 0.4–0.5 Å, whereas, in 3a, they are elongated even more by 0.7 Å. But all of them are still far below the sum of their van der Waals radii (As–Sb/Bi: Σ = 3.91/3.92 Å, Sb–Bi: Σ = 4.13 Å).25 Thus, 5 exhibits a cage-like central Sb2Bi2 core, which can be described as a distorted “butterfly-like” (bicyclo[1.1.0]butane) framework stabilized by four [CpMo(CO)2] fragments. This is the first example of a mixed polypnictogen butterfly-type compound. So far, only similar metal-coordinated Sb4 and Bi4 complexes have been reported either as dicationic (VIII and IX)11 or as neutral species.26 In contrast, 3a and 4a exhibit central cage-like As2Sb2 and As2Bi2 cores, respectively, which differ from the hitherto discussed structures. The structure in 3a reveals to be a very remarkable intermediate stage between the zigzag E4 and E2E′2 chains in 1, 2, VI and VII on the one hand, and the distorted “butterfly-like” E4 and Sb2Bi2 cages in 5, VIII and IX on the other hand.11 This based on the very long distances of the additional E⋯E′ contacts, the arrangement of the Cp ligands (Fig. 2 and ESI†) and the observed angles within the AsSbSbAs unit (vide infra). Moreover, compound 4a represents an entirely unprecedented structure, where the dication is symmetrical and contains a completely planar, central As2Bi2 cage. Therefore, it can rather be described as a dicationic As2Bi2 cycle or as a planarized, distorted As2Bi22+ “butterfly-like” (bicyclo[1.1.0]butane) framework stabilized by four [CpMo(CO)2] fragments (for natural charge distribution see ESI†). DFT calculations, though, suggest a “butterfly-like” geometry similar to 5. The fact that the build-up of As–Bi and Bi–Bi interactions, respectively, is favoured over an As–As bond formation is also very remarkable.
The transition from a chain-like (1) to a more cage-like structural motif in 3a–5 is also reflected by the angles within the E′EEE′ chains. While the As1–P1–P2 and P1–P2–As2 angles in 1 are close to 100°, the respective angles in 3a–5 decrease considerably to 64° (4a, 5) and 71° (3a). Also, the dihedral angles ∢(E′–E–E–E′) change from 135° in 1 to 113° in 5 and 180° in the planar As2Bi2 cycle of 4a, while the same angle is just slightly decreased to 128° in 3a illustrating again that it represents an intermediate stage between a chain and a cage type structure. In each of the compounds 1 and 3a–5, the Mo–Mo bonds are elongated by 0.1–0.2 Å compared to their respective starting materials, while the Mo–E and Mo–E′ bonds slightly decrease in length. DFT calculations for the gas phase reproduce well the experimental geometric parameters of 1 and 2 in the solid state, while, for 3–5, cage-like geometries are predicted. The Mayer bond order for the central P–P bonds in 1 and 2 is 0.79 and 0.81, respectively, while the bond order of the central E–E bonds in the cage-like geometries of 3a, 4a and 5 lies between 0.42 and 0.52. However, they are supported by two additional E⋯E′ interactions with bond orders between 0.21 and 0.35 (cf. ESI†). Hence, the Mayer bond orders of the newly formed bonds and interactions for the compounds with a cage-like geometry add up to a bond order of nearly 1 (cf. ESI†).
In general, hetero-polypnictogen chains are almost unknown. While few examples for AsPPAs27 and SbPPSb27a chains and cycles have been reported, which, however, could only be stabilized by organic substituents or were only obtained as an inseparable product mixture,28 heavier hetero-polypnictogen chains without phosphorus have, to the best of our knowledge, not been observed yet (except for a tetrabismuth-substituted diarsane As2(BiClR)4 (R = CH(SiMe3)2).13 Therefore, 1 and 2 are the first E2P2 (E = As, Sb) ligands only stabilized by transition metal fragments, and 3a–5 the first E2E′2 ligands of the heavy pnictogen elements As, Sb and Bi in general. Additionally, the polypnictogen cages in 3a and 4a show geometries which have not been observed before for p-block elements.
DFT computations
DFT calculations19 show that the single occupied molecular orbital (SOMO) in the potentially first formed paramagnetic monocation [B]˙+ is delocalized over the molybdenum atoms as well as the PSb ligand and the CO units with major contributions from Mo, P and Sb (Fig. 3). The spin density is mainly localized on Mo (24 and 40%) and with smaller contributions from the pnictogen atoms (14% on P and 16% on Sb). Interestingly, although the spin density on Sb is slightly higher than on P, the dimerization of [B]˙+ occurs via P–P bond formation. The spin density on the EE′ unit in [A]˙+–[E]˙+ increases with increasing atomic number of E or E′ (cf. ESI†). Furthermore, DFT calculations consistently reproduce the experimentally observed effect of P–As and Mo–Mo bond elongations, although the absolute bond lengths are slightly overestimated.19 Additionally, the torsion angle in the dimerization product 1 comes close to 180° during the geometry optimization. Therefore, the E4 chains become planar. This suggests that the experimentally observed gauche arrangement, determined by single crystal X-ray diffraction of 1, may be caused by crystal packing effects.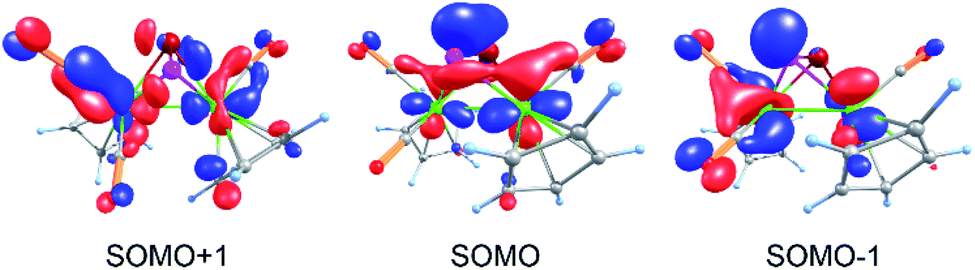 | ||
| Fig. 3 Frontier molecular orbitals (α spin) in [B]˙+, calculated at the TPSSh/def2-TZVP level of theory. | ||
Spectroscopic investigations
The 1H NMR spectra of 1–5 in CD2Cl2 solution only feature one sharp singlet at δ = 5.66 ppm (1), 5.61 ppm (2), 5.68 ppm (3a), 5.69 ppm (4a) and 5.72 ppm (5), respectively, for the Cp ligands. In the case of 5, also small singlets at δ = 5.64 and 5.78 ppm are detected, which can be attributed to trace impurities of VIII and IX. The latter are received by oxidation of IV and V, which are formed in the synthesis of E, and cannot be completely separated from each other. Likewise, one singlet is observed in the 13C{1H} NMR spectra for the Cp ligands indicating a highly dynamic behaviour of the Cp ligands in solution, which cannot be resolved on the NMR timescale. Characteristic signals for the [TEF]− anion and the CO ligands are observed in the 19F{1H} as well as the 13C{1H} NMR spectra.The 31P{1H} NMR spectrum of 1 at room temperature shows only one relatively sharp signal at δ = −28.8 ppm (ω1/2 = 11 Hz), which is shifted to higher field by 60 ppm compared to the starting material A (δ = 30.1 ppm).29 Upon cooling to 193 K, the signal moves farther to higher field (δ = −39.4 ppm) and undergoes broadening (ω1/2 ∼ 1700 Hz) suggesting that the fast dynamic processes in 1, which render all P atoms as well as Cp and CO ligands chemically equivalent on the NMR timescale, are constrained at lower temperatures. Below 253 K, two new signals at δ = −119.7 ppm and 21.4 ppm arise in addition to the broad singlet indicating the formation of a new, unidentified species. The 31P{1H} NMR spectrum of 2 also reveals a sole singlet at δ = 35.0 ppm, which again is shifted to higher field by 60 ppm in comparison to the starting material B (δ = 98.8 ppm).18 This verifies the suggestion that, analogously to 1, a P–P coupled dicationic product is formed (Scheme 2).
Solutions of 1, 2, 4a and 5 in CD2Cl2 or CH2Cl2 are all silent in the X-band EPR spectra at room temperature and at 77 K. This indicates that no dissociation of the dicationic species occurs, which is in good agreement with the calculated dissociation energies (vide supra). Likewise, 3a is EPR-silent at room temperature as well, but shows a very weak axial signal (giso = 1.954) upon cooling to 77 K. This suggests that very small amounts of the radical monocation [C]˙+ might be present in frozen solution at very low temperatures. In contrast, no dimeric products can be observed in the ESI mass spectra of 1–5 suggesting that, in the gas phase, solely the monocations [Mo2EE′]+ are present (only [Mo2PAs]22+ could be observed in very concentrated solutions of 1 in a minor ratio).
31P{1H} MAS NMR and IR spectra show that, in contrast to its lighter congener VI, 1 does not undergo reversible isomerisation.11 Furthermore, at least five CO bands are observed in the IR spectra supporting the asymmetrical molecular structure (Fig. 2a).
Influence of the counter ion on the solid-state structure of 3 and 4
The [TEF]− anion causes major problems during the refinement and solution of single crystal X-ray diffraction experiments (e.g. in 2 or VII)11 due to its high symmetry, weak coordination properties and the free rotation of the perfluorinated tert-butoxy groups, which can lead to a severe disorder. However, without the [TEF]− anion, the dicationic products are insoluble in all common solvents except for MeCN, MeNO2 and acetone, in which fast decomposition occurs even at low temperatures,11 or in liquid SO2, which complicates crystallization (due to its low boiling point (−10 °C) and its toxicity)30 or crystal mounting (due to gas evolution probably caused by embedded SO2 molecules).Therefore, we introduced a similar perhalogenated alkoxyaluminate anion [Al{OC(CCl3)(CF3)2}4]− (= [TEFCl]−), where one CF3 group on every tert-butoxy ligand is replaced by a CCl3 substituent.31 This lowers the symmetry of the anion and can lead to a decrease in disorder. Moreover, it was of interest to determine if small changes in the structure of the counterion can influence the outcome of the solid-state structure. However, the strong one-electron oxidant [Thia]+ is unknown with this counterion. Hence, a route for a high-yielding synthesis of [Thia][TEFCl] had to be developed. A simple one-step reaction of Li[TEFCl], NO[SbF6] and thianthrene gives the deep purple [Thia][TEFCl] in 89% yield [eqn (1)]. The reaction is performed in liquid SO2 to ensure that all starting materials are fully dissolved. [Thia][TEFCl] is highly soluble in CH2Cl2 even at lower temperatures and can be crystallized as dark purple blocks from CH2Cl2/n-hexane.19 Furthermore, the reaction can be carried out in a multigram scale.
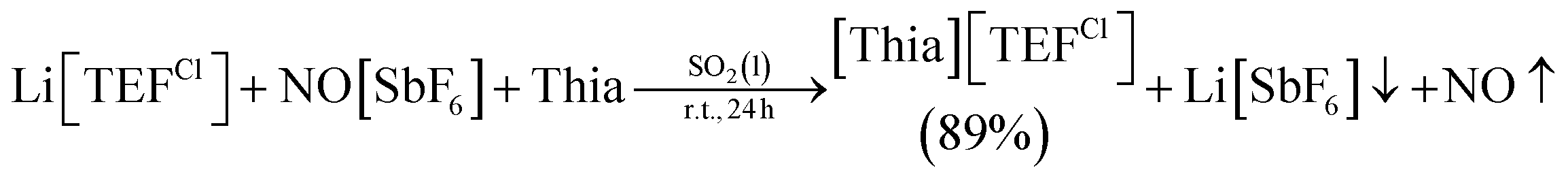 | (1) |
To gain a first insight into the influence of the counterion within the oxidation of tetrahedral dipnictogen complexes, [Thia][TEFCl] was reacted with solutions of C–E. It appears that the counter anion has no influence on the reactivity itself since again only the dimeric, dicationic E–E coupled products [{CpMo(CO)2}4(μ4,η2:η2:η2:η2-E2E′2)][TEFCl]2 (E2E′2 = As2Sb2 (3b), As2Bi2 (4b)) can be obtained (Scheme 3) in good crystalline yields of 77% and 81%, respectively. Despite several attempts, the oxidation product of E could not be crystallized due to the high solubility of the [TEFCl]− anion leading to oily products. However, the exchange of the counterion surprisingly has a dramatic impact on the molecular structure of 3b, which differs significantly from its [TEF]− derivative 3a (4b only shows slight deviations to 4a).
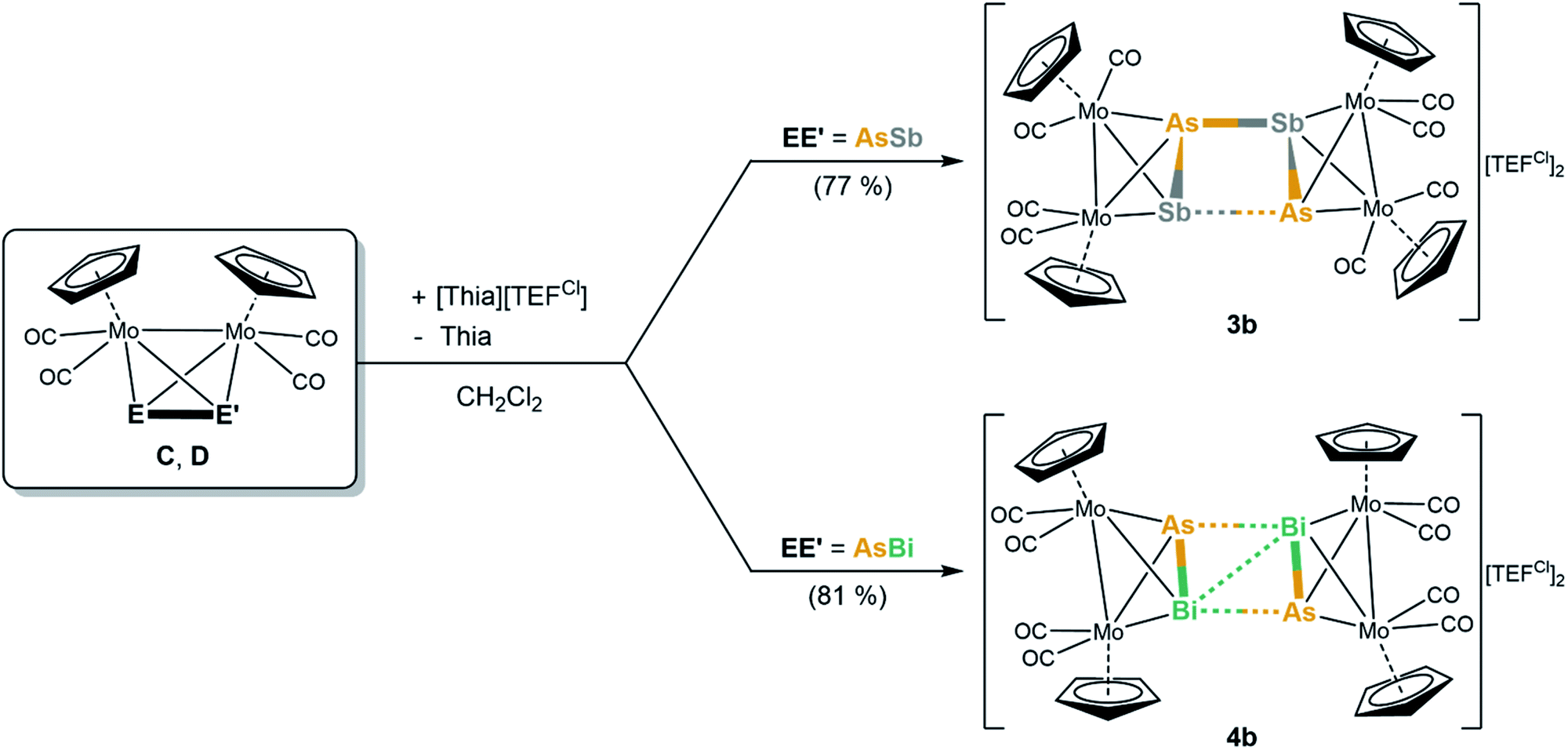 | ||
| Scheme 3 Oxidation of C and D with Thia[TEFCl] resulting in dimerization reactions yielding a dicationic, cyclic As2Sb2 ligand (3b) or a planarized, distorted butterfly-like As2Bi2 motif (4b), respectively. Isolated yields in parentheses. | ||
The dication in 3b builds up a completely unprecedented structural motif (Fig. 4a). It contains a central, cyclic As2Sb2 ligand. It is very remarkable that the arsenic and antimony atoms within the cycle are bound in an alternating fashion.19 The intratetrahedral As–Sb bonds are elongated compared to free C by 0.1 to 0.2 Å,18 but are still in the range of a single bond. The Mo–Mo bonds are widened up in the same manner. The Mo2AsSb tetrahedra are tilted against each other by approximately 13° leading to a dihedral angle of the As2Sb2 ring of 155.39(1)°. Furthermore, they are interconnected via two newly formed As–Sb bonds (As2–Sb1: 2.9108(1) Å; As1–Sb2: 3.0270(1) Å), with one of them being 0.11 Å longer than the other one, but even the shorter bond exceeds the sum of the covalent radii (Σ(As–Sb) = 2.62 Å)23 by 0.29 Å. Additionally, the As2Sb2 cycle reveals a very long diagonal As1–As2 (4.3525(1) Å) distance which exceeds the sum of the van der Waals radii25 by far excluding any further interactions. But more interestingly, it also exhibits a relatively short Sb1⋯Sb2 contact (3.4492(1) Å), which is 0.7 Å below the sum of the van der Waals radii (Sb–Sb: Σ = 4.12 Å).25 This leads to a slight distortion within the cycle with angles between 73.09(1)° and 100.37(1)°, with the smaller angles being at the arsenic atoms. Therefore, 3b can be regarded as an intermediate stage between the As2Sb2 cage in 3a and the As2Bi2 cycles in 4a and 4b, respectively. Overall, while cyclic As4 units are known as the heavier dianionic cyclo-butadiene analogues,32 the As2Sb2 cycle in 3b is the first example of its kind.
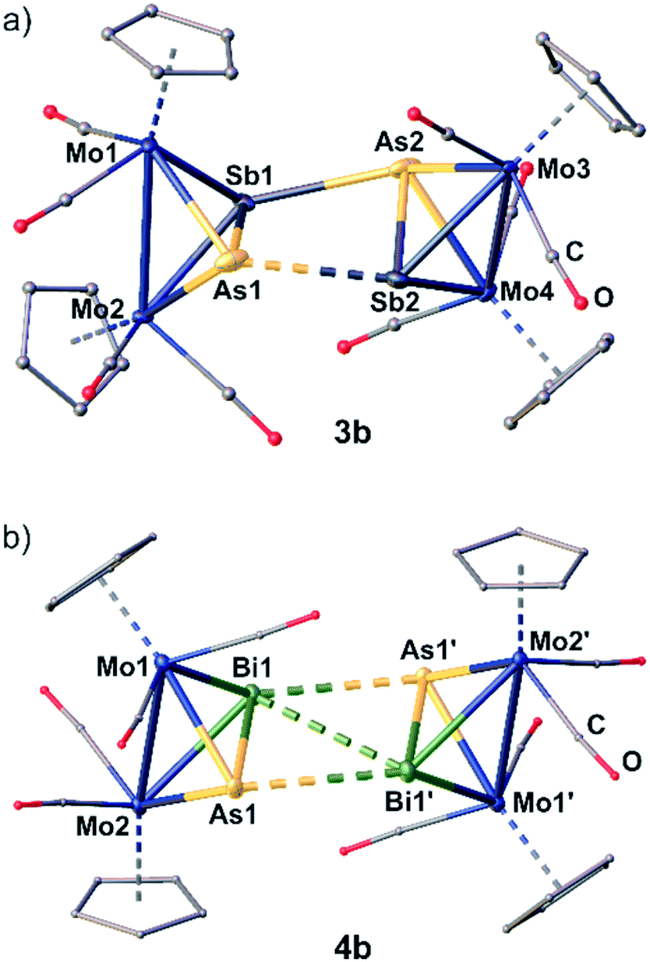 | ||
| Fig. 4 Molecular structure of the dicationic parts of 3b (a) and 4b (b). Anisotropic displacement is set to the 50% probability level. H atoms are omitted and C as well as O atoms are drawn as small spheres for clarity. Selected bond lengths [Å] and angles: 3b: As1–Sb1 2.7540(1), Sb1–As2 2.9108(1), As2–Sb2 2.6504(1), Sb2–As1 3.0270(1), Sb1–Sb2 3.4492(1), As1–Sb1–Sb2–As2 154.39(1); 4b: As1–Bi1 2.8377(1), Bi1–Bi1′ 3.4273(1), As1–Bi1′ 3.2184(1), Mo1–Mo2 3.1763(1), As1–Bi1–Bi1′ 60.96(1), As1–Bi1–Bi1′–As1′ 180.0(1). | ||
Geometry optimizations (TPSSh/def2-TZVP level) starting from the experimental geometries of 3a as well as 3b lead in both cases to a “cage-like” geometry similar to 5, indicating that the anion has a strong influence on the formed geometry in the solid state. In comparison to 3b, the [TEFCl]− counterion has no big influence on the molecular structure of 4. The cation in 4b (Fig. 4b) is similar to its [TEF]− congener 4a regarding all bond lengths and angles except for the Bi–Bi bond, which is elongated by 0.15 Å in 4b compared to 4a.
To investigate the influence of the counter ion towards a possible dissociation of the dications in solution, X-band EPR spectra of 4b were recorded which were silent both at room temperature and in frozen solution at 77 K. This indicates that no radical monocations [D]˙+ are present in solution just as in the case of 4a.
Conclusion
In summary, we have studied the one-electron oxidation chemistry of the tetrahedral hetero-dipnictogen complexes A–E. We successfully discovered the structural diversity of the rare class of hetero-polypnictogen compounds. The unique EE′ ligand complexes are readily oxidized by the organic radical cation [Thia]˙+. The initially formed radical monocations [A]˙+, [B]˙+, [C]˙+, [D]˙+ and [E]˙+, respectively, dimerize immediately in solution via E–E bond formation giving the novel dicationic products [{CpMo(CO)2}4(μ4,η2:η2:η2:η2-E′EEE′)][TEF]2 (EE′ = PAs (1), PSb (2), SbAs (3a), BiAs (4a), SbBi (5)), which reveal unprecedented four-membered hetero-pnictogen chains, free from organic substituents and are stabilized in the coordination sphere of transition metals. Remarkably, in 1, 2 and 5, the new bonds are formed between the respective lighter pnictogen atoms, whereas the aggregation in 3a and 4a takes place via the heavier pnictogen atoms. The products 1 and 2 bear unique, unsubstituted P2E2 chains in gauche conformation, while 5 exhibits a distorted “butterfly-like” (bicyclo[1.1.0]butane) Sb2Bi2 cage with two additional short Sb⋯Bi contacts. However, 3a represents a novel and very remarkable intermediate stage between those two structural motifs, in which the additional As⋯Sb contacts are considerably longer and also the bond angles and the arrangement of the Cp substituents differ. 4a even shows an entirely unprecedented structure exhibiting a planar As2Bi2 cycle, which can be interpreted as a planarized “butterfly-like” core. Moreover, 1 and 2 contain the first unsubstituted E2P2 (E = As, Sb) ligands that are only stabilized in the coordination sphere of transition metal fragments, and 3a–5 exhibit the first E2E′2 ligands of the heavy pnictogen elements As, Sb and Bi in general. The exchange of the counterion (using [TEFCl]−) has no effect on the molecular structure of 4. However, in 3b, the [TEFCl]− anion causes cyclization of the As2Sb2 ligand yielding a unique, cyclic As2Sb2 ligand in which the As and Sb atoms are bound in an alternating fashion. The influence of the counterion on the molecular structure of dicationic E4 and E2E′2 compounds will be a topic of future research. Overall, it could be proved that the oxidation of hetero-polypnictogen ligand complexes is a useful synthetic tool to gain access to the class of unsubstituted, cationic hetero-polypnictogen frameworks stabilized in the coordination sphere of transition metals, which are not obtained by other ways.Data availability
All experimental procedures, spectroscopic data, information on the theoretical calculations and crystallographic data can be found in the ESI.†Author contributions
L. D. performed the experimental work (except for 4a) and wrote the original draft. C. R. performed the preparation and characterization of 4a. G. Balázs performed the DFT calculations and M. Seidl the X-ray structural analysis. M. Scheer supervised and acquired funding for the project and finalized the draft of the manuscript. All authors contributed in preparing the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the project Sche 384/36-1. C. R. is grateful to the Studienstiftung des Deutschen Volkes for a PhD fellowship. This paper is dedicated to Prof. Holger Braunschweig on the occasion of his 60th birthday.Notes and references
- A. F. Holleman and E. Wiberg, Inorganic Chemistry, Walter de Gruyter, Berlin, 2001, vol. 101, p. 778 Search PubMed.
- M. Eggersdorfer, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000, vol. 36, pp. 29–45 Search PubMed.
- R. Hoffmann, Angew. Chem., Int. Ed., 1982, 21, 711–724 CrossRef.
- (a) M. Baudler and K. Glinka, Chem. Rev., 1993, 93, 1623–1667 CrossRef CAS; (b) M. Baudler and K. Glinka, Chem. Rev., 1994, 94, 1273–1297 CrossRef CAS.
- (a) C. A. Dyker and N. Burford, Chem.–Asian J., 2008, 3, 28–36 CrossRef CAS PubMed; (b) A. P. M. Robertson, P. A. Gray and N. Burford, Angew. Chem., Int. Ed., 2014, 53, 6050–6069 CrossRef CAS PubMed; (c) M. Donath, F. Hennersdorf and J. J. Weigand, Chem. Soc. Rev., 2016, 45, 1145–1172 RSC.
- O. J. Scherer, H. Sitzmann and G. Wolmershäuser, Angew. Chem., Int. Ed., 1985, 24, 351–353 CrossRef.
- M. Fleischmann, F. Dielmann, L. J. Gregoriades, E. V. Peresypkina, A. V. Virovets, S. Huber, A. Y. Timoshkin, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13110–13115 CrossRef CAS PubMed.
- (a) R. F. Winter and W. E. Geiger, Organometallics, 1999, 18, 1827–1833 CrossRef CAS; (b) M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 2972–2976 CrossRef CAS PubMed.
- (a) T. Köchner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529–6531 CrossRef PubMed; (b) T. Köchner, S. Riedel, A. J. Lehner, H. Scherer, I. Raabe, T. A. Engesser, F. W. Scholz, U. Gellrich, P. Eiden, R. A. P. Schmitt, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 8139–8143 CrossRef PubMed.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- L. Dütsch, M. Fleischmann, S. Welsch, G. Balázs, W. Kremer and M. Scheer, Angew. Chem., Int. Ed., 2018, 57, 3256–3261 CrossRef PubMed.
- (a) A. J. Ashe and E. G. Ludwig, J. Organomet. Chem., 1986, 303, 197–204 CrossRef CAS; (b) D. Nikolova and C. von Hänisch, Eur. J. Inorg. Chem., 2005, 378–382 CrossRef CAS; (c) J. G. Stevens, J. M. Trooster, H. F. Martens and H. A. Meinema, Inorg. Chim. Acta, 1986, 115, 197–201 CrossRef CAS.
- S. Traut, A. P. Hähnel and C. von Hänisch, Dalton Trans., 2011, 40, 1365–1371 RSC.
- (a) K. M. Marczenko and S. S. Chitnis, Chem. Commun., 2020, 56, 8015–8018 RSC; (b) T. Sasamori, N. Takeda and N. Tokitoh, Chem. Commun., 2000, 1353–1354 RSC.
- B. Ringler, M. Müller and C. von Hänisch, Eur. J. Inorg. Chem., 2018, 640–646 CrossRef CAS.
- C. Ritter, N. Michel, A. Rinow, B. Ringler and C. von Hänisch, Eur. J. Inorg. Chem., 2021, 2514–2522 CrossRef CAS.
- M. Donath, E. Conrad, P. Jerabek, G. Frenking, R. Fröhlich, N. Burford and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 2964–2967 CrossRef CAS PubMed.
- L. Dütsch, C. Riesinger, G. Balázs and M. Scheer, Chem.–Eur. J., 2021, 27, 8804–8810 CrossRef PubMed.
- For details see ESI†.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- Independent on numerous attempts the obtained datasets of the single crystal X-ray diffraction experiments of 2 are very weak since 2 only crystallizes as thin plates or forms an oil. Therefore, only a first insight into the geometry of the heavy-atom framework of the molecular structure can be given revealing a P-P coupled SbPPSb chain as central unit. No bond lengths or angles can be discussed. The presence of a P-P coupled product containing an SbPPSb chain is supported by 31P NMR. For further details, see the ESI†.
- (a) J. E. Davies, L. C. Kerr, M. J. Mays, P. R. Raithby, P. K. Tompkin and A. D. Woods, Angew. Chem., Int. Ed., 1998, 37, 1428–1429 CrossRef CAS; (b) J. E. Davies, M. J. Mays, P. R. Raithby, G. P. Shields, P. K. Tompkin and A. D. Woods, J. Chem. Soc., Dalton Trans., 2000, 1925–1930 RSC.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- Two independent molecules in the asymmetric unit of 4a are observed. The given bond lengths and angles are the ones of one dication. The second molecule is very similar.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- (a) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657–10661 CrossRef CAS PubMed; (b) L. Tuscher, C. Helling, C. Ganesamoorthy, J. Krüger, C. Wölper, W. Frank, A. Nizovtsev and S. Schulz, Chem.–Eur. J., 2017, 23, 12297–12304 CrossRef CAS PubMed; (c) J. Krüger, C. Wölper and S. Schulz, Inorg. Chem., 2020, 59, 11142–11151 CrossRef PubMed.
- (a) E. Conrad, N. Burford, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2009, 131, 5066–5067 CrossRef CAS PubMed; (b) M. Mehta, J. E. McGrady and J. M. Goicoechea, Chem.–Eur. J., 2019, 25, 5445–5450 CrossRef CAS PubMed; (c) J. Bresien, A. Hinz, A. Schulz and A. Villinger, Eur. J. Inorg. Chem., 2018, 1679–1682 CrossRef CAS; (d) L. Weber, D. Bungardt and R. Boese, Z. Anorg. Allg. Chem., 1989, 578, 205–224 CrossRef CAS.
- C. Schwarzmaier, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 290–293 CrossRef CAS PubMed.
- P. W. Roesky, N. Reinfandt, C. Schoo, L. Dütsch, R. Köppe, S. N. Konchenko and M. Scheer, Chem.–Eur. J., 2021, 27, 3974–3978 CrossRef PubMed.
- A. F. Holleman and E. Wiberg, Inorganic Chemistry, Walter de Gruyter, Berlin, 2001, vol. 101, pp. 531–532 Search PubMed.
- X. Zheng, Z. Zhang, G. Tan and X. Wang, Inorg. Chem., 2016, 55, 1008–1010 CrossRef CAS PubMed.
- (a) N. Reinfandt, C. Schoo, L. Dütsch, R. Köppe, S. N. Konchenko, M. Scheer and P. W. Roesky, Chem.–Eur. J., 2021, 27, 3974–3978 CrossRef CAS PubMed; (b) F. Spitzer, G. Balázs, C. Graßl and M. Scheer, Chem. Commun., 2020, 56, 13209–13212 RSC; (c) F. Spitzer, G. Balázs, C. Graßl, M. Keilwerth, K. Meyer and M. Scheer, Angew. Chem., Int. Ed., 2018, 57, 8760–8764 CrossRef CAS PubMed; (d) O. J. Scherer, J. Vondung and G. Wolmershäuser, J. Organomet. Chem., 1989, 376, C35–C38 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2105248–2105253. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04924k |
| This journal is © The Royal Society of Chemistry 2021 |