
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-trapped exciton emission and piezochromism in conventional 3D lead bromide perovskite nanocrystals under high pressure†
Yue
Shi
,
Wenya
Zhao
,
Zhiwei
Ma
,
Guanjun
Xiao
 * and
Bo
Zou
* and
Bo
Zou
State Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, China. E-mail: xguanjun@jlu.edu.cn
First published on 21st October 2021
Abstract
Developing single-component materials with bright-white emission is required for energy-saving applications. Self-trapped exciton (STE) emission is regarded as a robust way to generate intrinsic white light in halide perovskites. However, STE emission usually occurs in low-dimensional perovskites whereby a lower level of structural connectivity reduces the conductivity. Enabling conventional three-dimensional (3D) perovskites to produce STEs to elicit competitive white emission is challenging. Here, we first achieved STEs-related emission of white light with outstanding chromaticity coordinates of (0.330, 0.325) in typical 3D perovskites, Mn-doped CsPbBr3 nanocrystals (NCs), through pressure processing. Remarkable piezochromism from red to blue was also realized in compressed Mn-doped CsPbBr3 NCs. Doping engineering by size-mismatched Mn dopants could give rise to the formation of localized carriers. Hence, high pressure could further induce octahedra distortion to accommodate the STEs, which has never occurred in pure 3D perovskites. Our study not only offers deep insights into the photophysical nature of perovskites, it also provides a promising strategy towards high-quality, stable white-light emission.
Introduction
Lighting equipment is an important part of daily life. Recently, single-component materials with white emission typically originating from self-trapped excitons (STEs) have become appealing options for solid-state lighting applications. This is because they have a simplified device structure, and avoid the self-absorption and color instability seen in traditional multiple emitters.1 The radiative recombination of STEs in low-dimensional metal halide perovskites (LMHPs) has been investigated widely, but the lower level of structural connectivity of metal halide octahedra in LMHPs reduces conductivity, which seriously limits their practical application in optoelectric devices.2,3 The three-dimensional (3D) perovskites CsPbX3 (X = Cl, Br, I) have excellent charge-transport properties. They are regarded as deal candidates for high-efficiency light-emitting diodes because of high quantum efficiency, high color purity, and easy tunability of wavelengths.4–8 However, realization of white emission based on STEs in 3D perovskite materials is challenging.High-pressure studies on metal halide perovskite (MHP) materials have revealed increases in optical tuning,2,9–11 bandgap optimization,12,13 morphological control6,14–17 and dramatic increases in quantum yields.18–21 In particular, a new concept of pressure induced emission (PIE), whereby a non-luminescent material exhibits emission upon compression, was proposed in the research of compressed zero-dimensional (0D) perovskite Cs4PbBr6 nanocrystals (NCs).22 Thus, PIE provides distinct advantages for light-emitting applications achievable by the design of structural distortion.23–26 Localization of carriers resulting from a low electronic dimension and seriously distorted metal halide octahedra enhance the optical activity and binding energy of STEs. Therefore, the radiative recombination of STEs may also be activated in doped 3D perovskite materials with local carriers by continuously tuning the structural distortion with the assistance of a high-pressure method.
The dopant Mn2+ has been studied extensively in CsPbBr3 NCs. Its excited carriers within the host perovskite excitation are trapped into Mn2+, leading to the formation of local electrons via energy transfer.27–30 Doping Mn2+ in CsPbBr3 induces the local distortion of the octahedral framework considerably, in contrast to the case of CsPbCl3 NCs.31 This phenomenon occurs because of the greater disparity of M–Br (M = Pb, Mn) bond-dissociation energies between PbBr2 (249 kJ mol−1) and MnBr2 (314 kJ mol−1) compared with PbCl2 (301 kJ mol−1) and MnCl2 (338 kJ mol−1) precursors.32 Motivated by such results, we were encouraged to systematically investigate the pressure response of replacing Mn2+ with Pb2+ within CsPbBr3 NCs. We documented significant enhancement of broadband emission in doped 3D all-inorganic halide perovskites, CsPbxMn1−xBr3 NCs, under 7.62 GPa, where the emissive color transferred to expected white light from the initial orange. An isostructural phase transformation at ∼2.00 GPa was demonstrated by in situ high-pressure angle dispersive synchrotron X-ray diffraction (ADXRD) and optical absorption. The large uneven lattice deformation around the Mn dopants accommodates the STEs, and exciton self-trapping never occurs in pure CsPbBr3. We not only discovered an effective approach to enhance the broadband emission originating from STEs in doped 3D all-inorganic perovskites CsPbxMn1−xBr3 NCs, we also provide insights into the microscopic mechanisms that could guide future designs for materials with light-emitting applications.
Results and discussion
The morphology and structure of the synthesized Mn-doped CsPbBr3 NCs were investigated using transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) characterizations. As shown in Fig. S1a–c,† the samples before compression exhibited a nanocube morphology with good crystallinity. The Mn-doped CsPbBr3 NCs had an average diameter of 9.28 nm with a standard deviation of 1.07 nm. Under an ambient condition, the photoluminescence (PL) spectrum (Fig. 1a) revealed dual emission peaks centered at ∼439 nm (CsPbxMn1−xBr3 band-edge emission) and 613 nm (Mn-related emission), which are consistent with previous reports.32In situ high-pressure PL measurement of CsPbxMn1−xBr3 NCs was carried out to investigate optical properties up to 19 GPa (Fig. 1a–c and S2†). With an increase in pressure, the intensity of the band-edge PL peak experienced weakening and vanished completely at 2.05 GPa, whereas the Mn-related emission declined slightly before 0.90 GPa. Upon further compression to 2.05 GPa, the intensity of Mn-related emission strengthened sharply. Both peaks showed a red shift below 2.05 GPa. When the applied pressure exceeded 2.05 GPa, a new emission appeared and experienced persistent enhancement in its intensity with increasing pressure. This new emission could be clearly fitted by two sub-bands denoted as Peak I and Peak II in Fig. S3,† which are located around ∼448 and ∼508 nm, respectively. Peak I and Peak II first exhibited a red shift with increasing pressure until 5.16 GPa, then the peak position of two emissions was virtually unchanged. Detailed information about the new emission is shown in Table S1 and Fig. S4.† Interestingly, the new emission exhibited excellent stability, and suppressed the general emission quenching at high pressure. Even up to 19 GPa, the new emission had reasonable emissive properties. Furthermore, the CIE chromaticity coordinates of Mn-doped CsPbBr3 NCs could be controlled effectively by pressure across a wide range from red to blue, which was corroborated clearly from the change of emission color throughout compression (Fig. 1d, e and Table 1). High-quality white-light emission with chromaticity coordinates of (0.330, 0.325) were obtained through pressure processing. The ability to achieve white emission in 3D perovskites with tunable chromaticity indicated a prospective strategy for creating colorful solar designs using strain/pressure engineering with reasonable chemical regulation or device construction. When the pressure was released completely, the PL spectra of pressure-treated samples showed a distinct difference compared with that in its initial state (Fig. S5†), which exhibited Mn2+-dominated emission. To provide insights into this phenomenon, we further investigated the morphology and structure of Mn-doped CsPbBr3 NCs after decompression using TEM and HRTEM characterizations. The quenched samples exhibited deformation to some extent, with relatively poor crystallinity (Fig. S6†), which led to an increase in defects. Therefore, the defects produced after pressure processing would result in suppression of band-edge emission.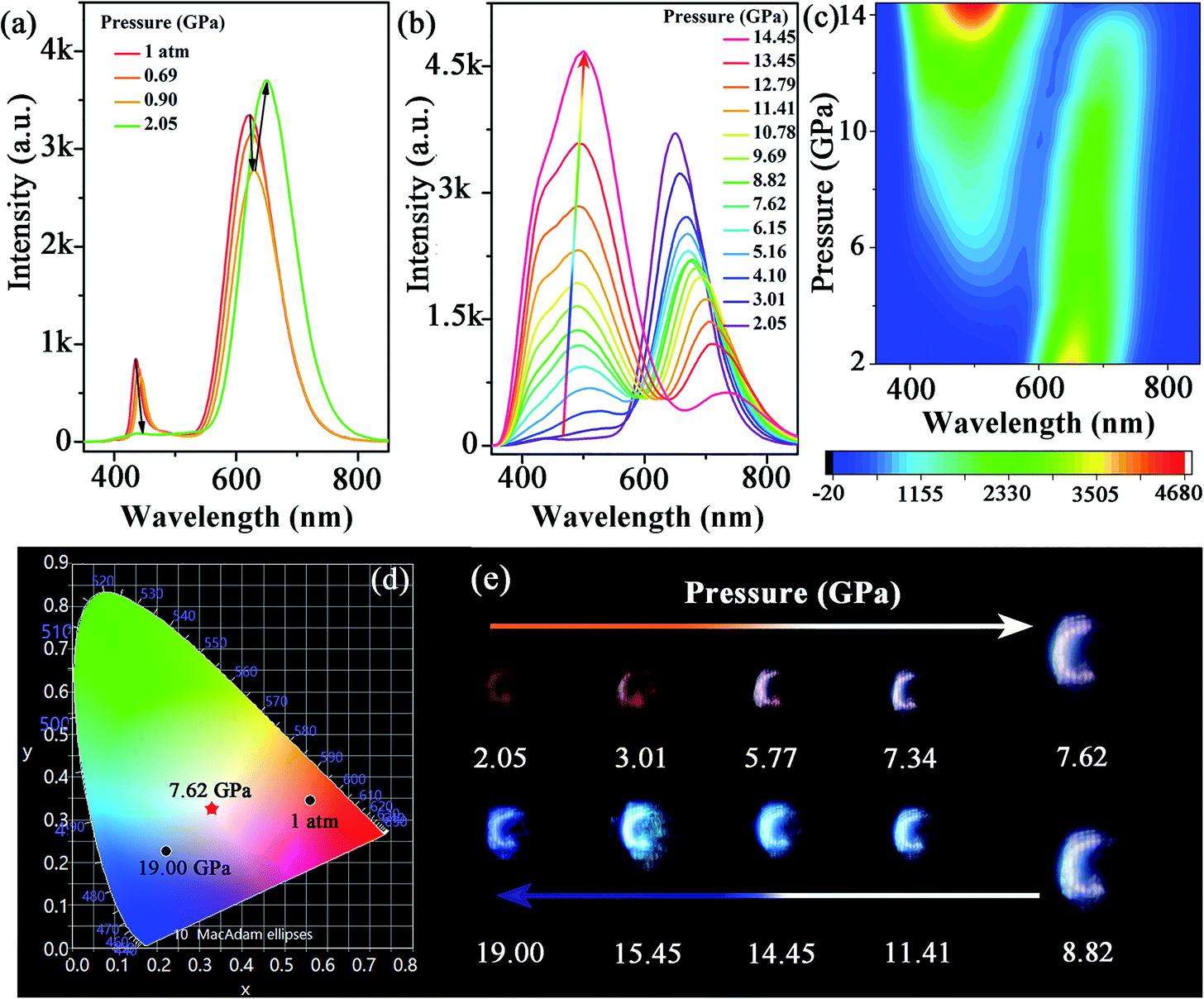 | ||
| Fig. 1 (a–c) PL spectra of Mn-doped CsPbBr3 NCs under high pressure. (d) Pressure-dependent chromaticity coordinates of the emissions. (e) PL micrographs upon compression. | ||
| Pressure (GPa) | CIE x | CIE y | Pressure (GPa) | CIE x | CIE y |
|---|---|---|---|---|---|
| 1 atm | 0.561 | 0.345 | 10.78 | 0.260 | 0.309 |
| 2.05 | 0.626 | 0.338 | 11.41 | 0.241 | 0.302 |
| 4.10 | 0.504 | 0.358 | 13.45 | 0.229 | 0.309 |
| 5.16 | 0.432 | 0.356 | 14.45 | 0.232 | 0.328 |
| 6.64 | 0.366 | 0.338 | 17.00 | 0.234 | 0.275 |
| 7.62 | 0.330 | 0.325 | 18.00 | 0.229 | 0.252 |
| 8.82 | 0.307 | 0.317 | 19.00 | 0.222 | 0.226 |
| 9.69 | 0.276 | 0.310 |
We also undertook in situ high-pressure ultraviolet-visible (UV-Vis) absorption measurements up to ∼23.98 GPa (Fig. 2a). At the ambient condition, the absorption edge of CsPbxMn1−xBr3 NCs was ∼440 nm, a blue shift compared with perfect CsPbBr3 NCs.33 Upon compression, the absorption spectra showed a red shift before 1.83 GPa. After that, the bandgap increased suddenly, which was coincident with emergence of a new PL peak. The indirect bandgap of CsPbxMn1−xBr3 NCs was estimated by extrapolating the linear portion of (αhυ)1/2versus hυ in Tauc plots, where α is the absorption coefficient and hυ is the photon energy. The bandgap value (Eg) of CsPbxMn1−xBr3 NCs was ∼2.80 eV at the ambient condition (Fig. 2b). At a relatively low pressure of 1.50 GPa, Eg exhibited linear narrowing by 0.07 eV (Fig. 2c) that was related to lattice contraction.33 Upon further compression, the rate of photon energy increased sharply above 1.83 GPa (Fig. 2c and S7†). This sudden change in bandgap energy could be attributed to distortion of inorganic octahedra.34 Density functional theory (DFT) calculations revealed that the indirect bandgap of CsPbxMn1−xBr3 NCs was determined mainly by changes in the valence band maximum (VBM) and conduction band minimum (CBM) (Fig. 2d). The distinctly different evolution between emission Peak I, Peak II and absorption edge associated with free excitons ruled out a mechanism based on band-edge transition.35 Furthermore, the new emissions showed a gradual red shift, whereas the band edge showed a gradual blue shift, resulting in a large increase in the Stokes shift (Fig. S4a†), which could be ascribed to the enhanced strength of electron–phonon coupling. Likewise, the intensity of Peak I and Peak II at different pressures showed a linear dependence on the excitation power density up to 22 mW cm−2 (Fig. 2e–g), indicating that both emissions originated from STEs.36,37 Typically, the presence of STEs was manifested through broadband luminescence.38 Once electrons and holes are photogenerated, they are self-trapped rapidly to form STEs due to the strong electron–phonon coupling with an increase in pressure, which leads to the emission enhancement of STEs.22,39
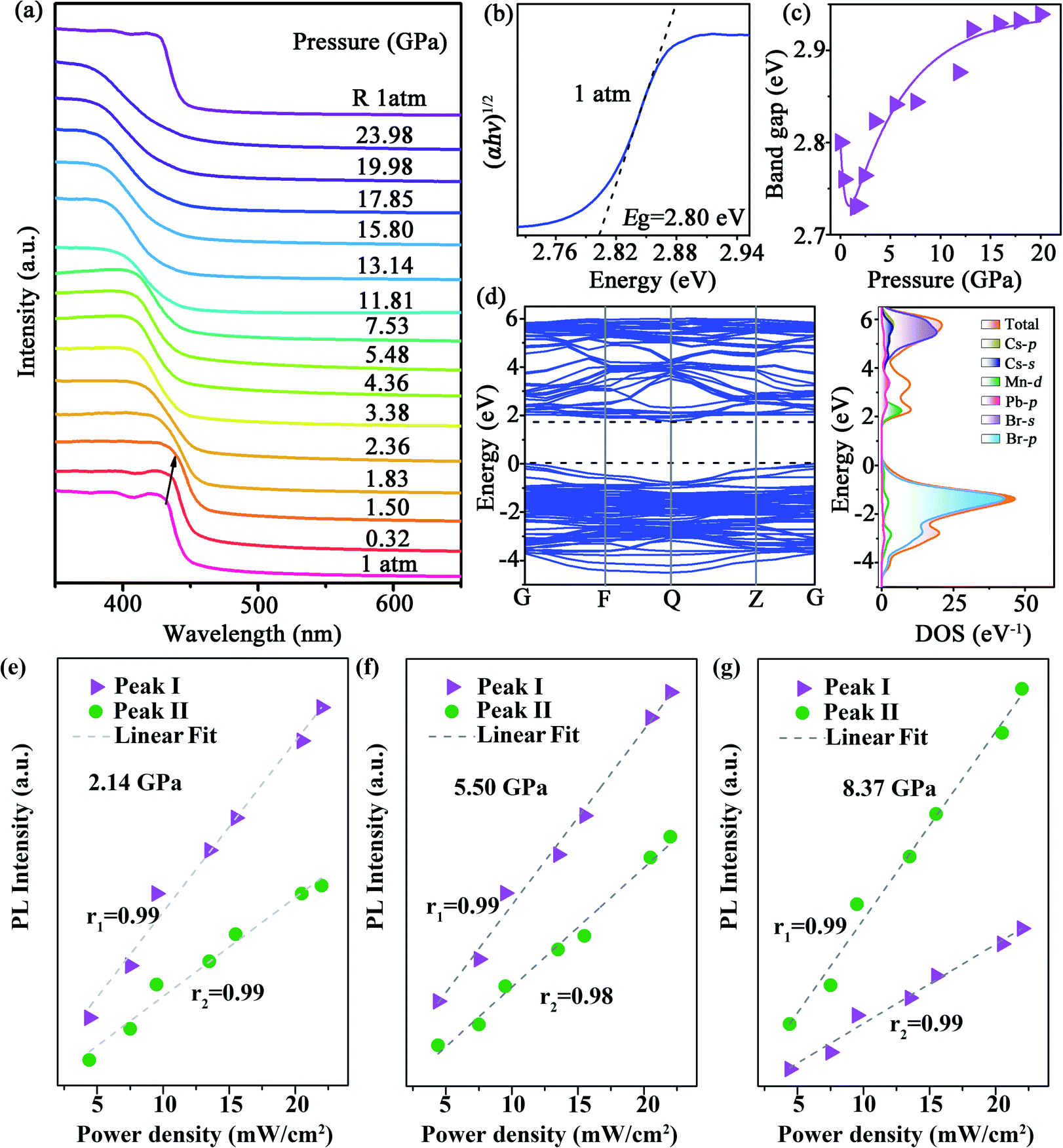 | ||
| Fig. 2 (a) Absorption spectra of CsPbxMn1−xBr3 NCs as a function of pressure. (b) Indirect bandgap Tauc plot for CsPbxMn1−xBr3 NCs at ambient pressure. (c) Bandgap evolution of CsPbxMn1−xBr3 NCs as a function of pressure. (d) Left: calculated electron-band structure of CsPbxMn1−xBr3 NCs at 1 atm. Right: total and partial density of states projected on the orbitals of different atoms. PL intensity of emission Peak I and Peak II at 2.14 GPa (e), 5.50 GPa (f) and 8.37 GPa (g) as a function of power density. The intensity of Peak I is denoted as triangular purple dots, whereas the intensity of Peak II is denoted as circular green dots. | ||
To verify the correlation between the optical properties and structural distortion of CsPbxMn1−xBr3 NCs, in situ high-pressure ADXRD experiments were undertaken. At the ambient condition, all diffraction peaks exhibited slight broadening, which could have been related to the uneven distortion among PbBr6 and MnBr6 octahedra. Upon compression, all diffraction peaks shifted to higher 2θ angles due to lattice contraction (Fig. 3a). Although the changes in PL and absorption spectra were obvious at 2.05 GPa, the ADXRD patterns over different pressures barely changed. This observation suggested isostructural phase transition in CsPbxMn1−xBr3 NCs, which are (in general) considered to be derived from electronic structural transitions (see ESI†). The pressure-induced rotation of PbBr6 and MnBr6 octahedra with an opposite direction and structural distortion were responsible for the isostructural phase transition. At higher pressures, CsPbxMn1−xBr3 NCs tended to be amorphous, and only a few broad diffraction peaks were detected. Comparison of the peak positions to those of pure CsPbBr3 revealed the ADXRD peaks of CsPbxMn1−xBr3 NCs to shift towards higher diffraction angles. This phenomenon indicated lattice contraction owing to the substitution of Pb2+ by smaller Mn2+ in host lattices (Fig. 3f). Refined ADXRD data at 1 atm agreed well with experimental data (Rwp = 0.61% and RP = 0.36%) (Fig. 3g). The lattice constants for different axes of CsPbxMn1−xBr3 NCs were collected as a function of pressure (Fig. 3b). The compressed rate of a and c axes experienced a turning point at >2.15 GPa, which matched closely with changes in PL and absorption. The discontinued evolution of lattice parameters at 2.15 GPa were ascribed to the isostructural phase transition. The bulk modulus (B0) of CsPbxMn1−xBr3 NCs was obtained by fitting the pressure-dependent unit-cell volumes based on the Birch–Murnaghan equation of state. Note that the B0 after isostructural phase transition was much larger than the initial value (14.91 GPa), indicating the less compressible nature of high-pressure structure (Fig. 3c). Moreover, the polar compressibility indicatrix in the ac and ab planes revealed obvious anisotropy within the structure (Fig. 3d and e).
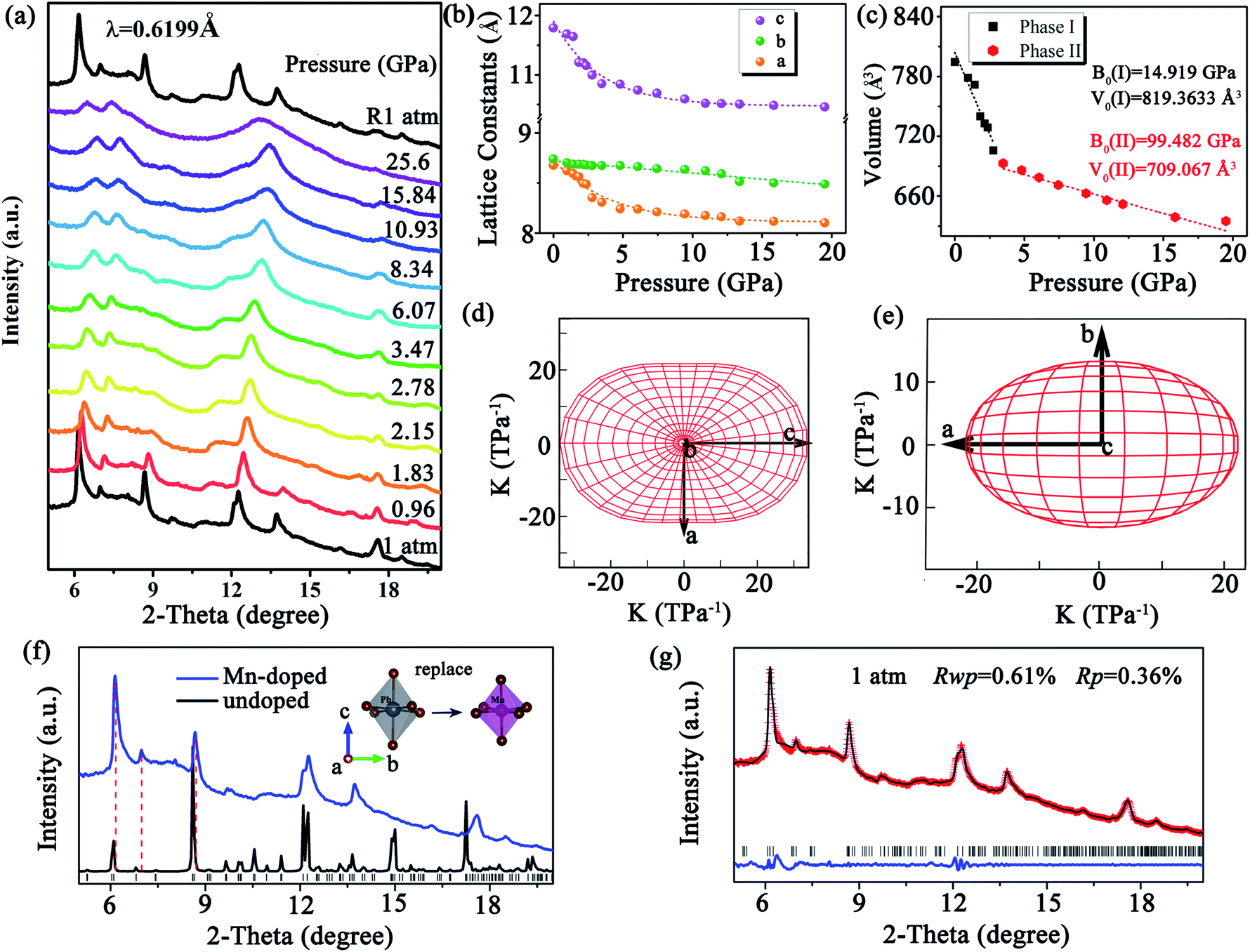 | ||
| Fig. 3 (a) Representative in situ high-pressure ADXRD patterns of CsPbxMn1−xBr3 NCs. (b) Pressure-dependent lattice constants of CsPbxMn1−xBr3 NCs. (c) Cell-volume evolution of CsPbxMn1−xBr3 NCs upon compression. (d and e) Polar compressibility indicatrix in the ab and ac planes of CsPbxMn1−xBr3 NCs. (f) ADXRD pattern of CsPbxMn1−xBr3 NCs, along with the reference ADXRD pattern for the orthorhombic phase of CsPbxMn1−xBr3. Inset shows the partial cation exchange in CsPbBr3 NCs (schematic). Therein, Pb2+ are partially replaced by Mn2+, thereby resulting in divalent-cation-doped CsPb1−xMnxBr3 NCs. (g) Refinements of experimental (red fork), simulated (black profile), and different (blue line) ADXRD patterns at 1 atm. Therein, black vertical markers indicate the corresponding Bragg reflections. | ||
We revealed the distortion of Mn–Br and Pb–Br octahedra within CsPbxMn1−xBr3 under pressure quantitatively (Fig. 4a and b). The degree of octahedral distortions was determined using the following parameters:11
 | ||
| Fig. 4 (a) δ2 values (variance in octahedral angle) and Δd values (distortion in bond length) of Pb octahedra within CsPbxMn1−xBr3 NCs under pressure. (b) δ2 values and Δd values of Mn octahedra within CsPbxMn1−xBr3 NCs under pressure. (c) Crystal structures viewed along the a axis of CsPbxMn1−xBr3 NCs before and after compression. Configuration coordinate models for CsPbxMn1−xBr3 NCs at ambient pressure (d) and high pressure (e). ST denotes self-trapped states. | ||
Distortion of bond length:
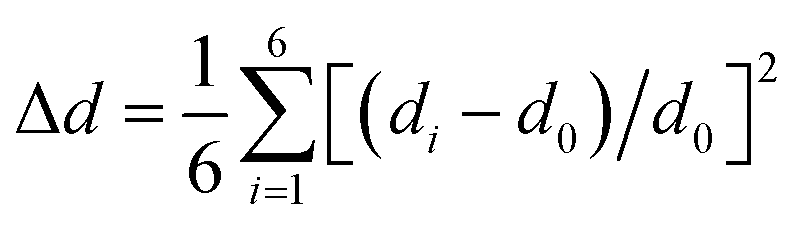 | (1) |
Variance in octahedral angle:
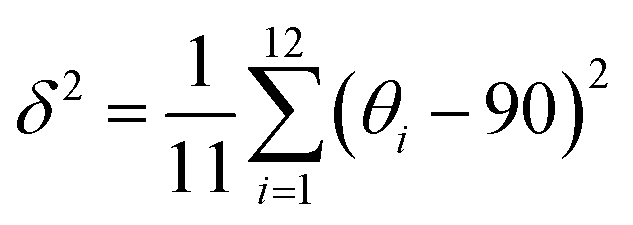 | (2) |
Conclusions
We realized, for the first time, high-quality white-light emission with chromaticity coordinates of (0.330, 0.325) by designing STEs formation in dense traditional 3D perovskite Mn-doped CsPbBr3 NCs. The size-mismatched 3D-perovskite structure resulting from the introduction of Mn dopants and the subsequent large distortion suppressed radiative recombination of free excitons and stabilized more STEs resulting, ultimately, in STE emission. Moreover, considerable piezochromism and intriguing PIE associated with STEs were discovered under high pressure. Our study not only deepens insight into PIE, but also provides guidelines to improve the designability of stable white-emission 3D perovskites for solid-state lighting and displays.Data availability
Data are available within the article or its ESI materials.† The data that cannot be found in the article or in ESI materials† are available on request from the authors.Author contributions
J. X. and B. Z. devised the method and conceived the project. Y. S. designed and performed the experiments. W. Z. and Z. M. performed the partial experiments and calculations. All authors analyzed the results and contributed to writing the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFE0120300), National Science Foundation of China (21725304, 22131006, 12174144 and 11774125), China Postdoctoral Science Foundation (2021M690065), and Fundamental Research Funds for the Central Universities. This work was undertaken mainly at BL15U1 at Shanghai Synchrotron Radiation Facility. Portions of this work were carried out at 4W2HP-Station within Beijing Synchrotron Radiation Facility.Notes and references
- S. Pathak, N. Sakai, F. Wisnivesky Rocca Rivarola, S. D. Stranks, J. Liu, G. E. Eperon, C. Ducati, K. Wojciechowski, J. T. Griffiths, A. A. Haghighirad, A. Pellaroque, R. H. Friend and H. J. Snaith, Chem. Mater., 2015, 27, 8066–8075 CrossRef CAS.
- S. Sun, C. K. Gan, A. G. del Águila, Y. Fang, J. Xing, T. T. H. Do, T. J. White, H. Li, W. Huang and Q. Xiong, Sci. Adv., 2019, 5, eaav9445 CrossRef PubMed.
- Z. Yang, C. Qin, Z. Ning, M. Yuan, J. Tang and L. Ding, Sci. Bull., 2020, 65, 1057–1060 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- Y. Nagaoka, K. Hills-Kimball, R. Tan, R. Li, Z. Wang and O. Chen, Adv. Mater., 2017, 29, 1606666 CrossRef PubMed.
- S. Guo, K. Bu, J. Li, Q. Hu, H. Luo, Y. He, Y. Wu, D. Zhang, Y. Zhao, W. Yang, M. G. Kanatzidis and X. Lu, J. Am. Chem. Soc., 2021, 143, 2545–2551 CrossRef CAS PubMed.
- Y. Liu, B. Dong, A. Hagfeldt, J. Luo and M. Graetzel, SmartMat, 2021, 2, 33–37 CrossRef.
- Q. Li, Z. Chen, M. Li, B. Xu, J. Han, Z. Luo, L. Tan, Z. Xia and Z. Quan, Angew. Chem., Int. Ed., 2020, 60, 2583–2587 CrossRef PubMed.
- Q. Li, Z. Chen, B. Yang, L. Tan, B. Xu, J. Han, Y. Zhao, J. Tang and Z. Quan, J. Am. Chem. Soc., 2020, 142, 1786–1791 CrossRef CAS PubMed.
- M. E. Sun, T. Geng, X. Yong, S. Lu, L. Ai, G. Xiao, J. Cai, B. Zou and S. Q. Zang, Adv. Sci., 2021, 8, 2004853 CrossRef CAS PubMed.
- Z. Ma, F. Li, D. Zhao, G. Xiao and B. Zou, CCS Chem., 2020, 2, 71–80 CrossRef CAS.
- Q. Li, Y. Wang, W. Pan, W. Yang, B. Zou, J. Tang and Z. Quan, Angew. Chem., Int. Ed., 2017, 56, 15969–15973 CrossRef CAS PubMed.
- Z. Wang and S. Saxena, Solid State Commun., 2002, 123, 195–200 CrossRef CAS.
- T. Yin, B. Liu, J. Yan, Y. Fang, M. Chen, W. K. Chong, S. Jiang, J. L. Kuo, J. Fang, P. Liang, S. H. Wei, K. P. Loh, T. C. Sum, T. J. White and Z. X. Shen, J. Am. Chem. Soc., 2018, 141, 1235–1241 CrossRef PubMed.
- L. Meng, J. M. D. Lane, L. Baca, J. Tafoya, T. Ao, B. Stoltzfus, M. Knudson, D. Morgan, K. Austin, C. Park, P. Chow, Y. Xiao, R. Li, Y. Qin and H. Fan, J. Am. Chem. Soc., 2020, 142, 6505–6510 CrossRef CAS PubMed.
- T. Wang, R. Li, Z. Quan, W. S. Loc, W. A. Bassett, H. Xu, Y. C. Cao, J. Fang and Z. Wang, Adv. Mater., 2015, 27, 4544–4549 CrossRef CAS PubMed.
- Y. Wang, S. Guo, H. Luo, C. Zhou, H. Lin, X. Ma, Q. Hu, M. H. Du, B. Ma, W. Yang and X. Lu, J. Am. Chem. Soc., 2020, 142, 16001–16006 CrossRef CAS PubMed.
- Y. Wang, H. Zhang, J. Zhu, X. Lu, S. Li, R. Zou and Y. Zhao, Adv. Mater., 2020, 32, 1905007 CrossRef CAS PubMed.
- Z. Wang, C. Schliehe, T. Wang, Y. Nagaoka, Y. C. Cao, W. A. Bassett, H. Wu, H. Fan and H. Weller, J. Am. Chem. Soc., 2011, 133, 14484–14487 CrossRef CAS PubMed.
- H. Luo, S. Guo, Y. Zhang, K. Bu, H. Lin, Y. Wang, Y. Yin, D. Zhang, S. Jin, W. Zhang, W. Yang, B. Ma and X. Lu, Adv. Sci., 2021, 8, 2100786 CrossRef CAS PubMed.
- Z. Ma, Z. Liu, S. Lu, L. Wang, X. Feng, D. Yang, K. Wang, G. Xiao, L. Zhang, S. A. T. Redfern and B. Zou, Nat. Commun., 2018, 9, 4506 CrossRef PubMed.
- Z. Ma, F. Li, L. Sui, Y. Shi, R. Fu, K. Yuan, G. Xiao and B. Zou, Adv. Opt. Mater., 2020, 8, 2000713 CrossRef CAS.
- X. Lü, C. Stoumpos, Q. Hu, X. Ma, D. Zhang, S. Guo, J. Hoffman, K. Bu, X. Guo, Y. Wang, C. Ji, H. Chen, H. Xu, Q. Jia, W. Yang, M. G. Kanatzidis and H.-K. Mao, Natl. Sci. Rev., 2021, nwaa288 CrossRef PubMed.
- S. Guo, Y. Zhao, K. Bu, Y. Fu, H. Luo, M. Chen, M. P. Hautzinger, Y. Wang, S. Jin, W. Yang and X. Lu, Angew. Chem., Int. Ed., 2020, 59, 17533–17539 CrossRef CAS PubMed.
- R. Fu, W. Zhao, L. Wang, Z. Ma, G. Xiao and B. Zou, Angew. Chem., Int. Ed., 2021, 60, 10082–10088 CrossRef CAS PubMed.
- D. Zhao, G. Xiao, Z. Liu, L. Sui, K. Yuan, Z. Ma and B. Zou, Adv. Mater., 2021, 33, 2100323 CrossRef CAS PubMed.
- D. Parobek, B. J. Roman, Y. Dong, H. Jin, E. Lee, M. Sheldon and D. H. Son, Nano Lett., 2016, 16, 7376–7380 CrossRef CAS PubMed.
- W. J. Mir, M. Jagadeeswararao, S. Das and A. Nag, ACS Energy Lett., 2017, 2, 537–543 CrossRef CAS.
- S. K. Mehetor, H. Ghosh, B. Hudait, N. S. Karan, A. Paul, S. Baitalik and N. Pradhan, ACS Energy Lett., 2019, 4, 2353–2359 CrossRef CAS.
- Y. Cao, G. Qi, L. Sui, Y. Shi, T. Geng, D. Zhao, K. Wang, K. Yuan, G. Wu, G. Xiao, S. Lu and B. Zou, ACS Mater. Lett., 2020, 2, 381–388 CrossRef CAS.
- D. Parobek, Y. Dong, T. Qiao and D. H. Son, Chem. Mater., 2018, 30, 2939–2944 CrossRef CAS.
- G. Xiao, Y. Cao, G. Qi, L. Wang, C. Liu, Z. Ma, X. Yang, Y. Sui, W. Zheng and B. Zou, J. Am. Chem. Soc., 2017, 139, 10087–10094 CrossRef CAS PubMed.
- F. Bai, K. Bian, X. Huang, Z. Wang and H. Fan, Chem. Rev., 2019, 119, 7673–7717 CrossRef CAS PubMed.
- Z. Ma, F. Li, G. Qi, L. Wang, C. Liu, K. Wang, G. Xiao and B. Zou, Nanoscale, 2019, 11, 820–825 RSC.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- N. Pradhan, F. Liu, T. Zhang, D. Mondal, S. Teng, Y. Zhang, K. Huang, D. Wang, W. Yang, P. Mahadevan, Y. S. Zhao and R. Xie, Angew. Chem., Int. Ed., 2021, 60, 13548–13553 CrossRef PubMed.
- J. Yu, J. Kong, W. Hao, X. Guo, H. He, W. R. Leow, Z. Liu, P. Cai, G. Qian, S. Li, X. Chen and X. Chen, Adv. Mater., 2018, 1806385 CrossRef PubMed.
- Y. Shi, Z. Ma, D. Zhao, Y. Chen, Y. Cao, K. Wang, G. Xiao and B. Zou, J. Am. Chem. Soc., 2019, 141, 6504–6508 CrossRef CAS PubMed.
- Y. Zhou, J. Chen, O. M. Bakr and H.-T. Sun, Chem. Mater., 2018, 30, 6589–6613 CrossRef CAS.
- W. Liu, Q. Lin, H. Li, K. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS PubMed.
- D. Rossi, D. Parobek, Y. Dong and D. H. Son, J. Phys. Chem. C, 2017, 121, 17143–17149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experiments, HRTEM images, high-pressure optical spectra, and ball and stick model. See DOI: 10.1039/d1sc04987a |
| This journal is © The Royal Society of Chemistry 2021 |