
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation of a planar 1,2-dilithio-disilene and its conversion to a Si–B hybrid 2π-electron system and a planar tetraboryldisilene†
Miao
Tian
 ,
Jianying
Zhang
,
Lulu
Guo
and
Chunming
Cui
*
,
Jianying
Zhang
,
Lulu
Guo
and
Chunming
Cui
*
State Key Laboratory of Elemento-organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: cmcui@nankai.edu.cn
First published on 18th October 2021
Abstract
Lithium reagents have long played important roles in synthetic chemistry. However, unsaturated organosilicon lithium reagents are few in number. Herein, we describe the first isolation of a 1,2-dilithiodisilene: [(boryl)SiLi]2 (2) was prepared in 73% yield by the reduction of (boryl)tribromosilane (1, boryl = (HCArN)2B, Ar = 2,6-iPr2C6H3) with lithium in Et2O. The salt elimination reaction of 2 with dihaloboranes RBX2 afforded disilaborirenes [(boryl)Si]2BR (3a–c), whereas the reaction with two equivalents of B-bromocatecholborane ((cat)BBr) yielded the first tetraboryldisilene [(boryl)(cat)BSi]2 (4). X-ray diffraction analysis and density functional theory calculations indicated that the disilene 2 and tetraboryldisilene 4 feature an almost planar geometry and disilaborirenes 3a–c are aromatic with a silicon–boron hybrid 2π-electron delocalized structure. The results indicate that 1,2-dilithiodisilene 2 is a powerful synthetic reagent for the construction of novel silicon multiply bonded species with unique electronic structures and that the boryl substituents have significant electronic effects on the structure of silicon multiple bonding.
Introduction
There is great interest in the synthesis and reactivity of heavier main group multiply bonded species due to their unusual electronic structures and reaction patterns.1 A number of alkene analogues of silicon kinetically stabilized by bulky alkyl, aryl and silyl substituents have been isolated (A, Scheme 1).2 In light of the diverse bonding modes in disilenes, the corresponding anionic systems are expected to be useful as synthetic reagents for novel disilenes that otherwise could not be obtained.3 In addition, the anionic multiple bonding systems may exhibit cooperative effects in the activation of small molecules. However, anionic multiply bonded silicon species are much less common because of their synthetic challenges and extremely high reactivity. Nevertheless, several silicon analogues of vinyl anions (B, Scheme 1) that have been reported and their synthetic applications for new types of silicon species and unique reaction patterns to small molecules have been demonstrated.2a,b,3,4 It is envisioned that 1,2-alkali-metal-substituted disilenes (C, Scheme 1) could be versatile reagents for access to diverse silicon multiply bonded species and activation of inert molecules.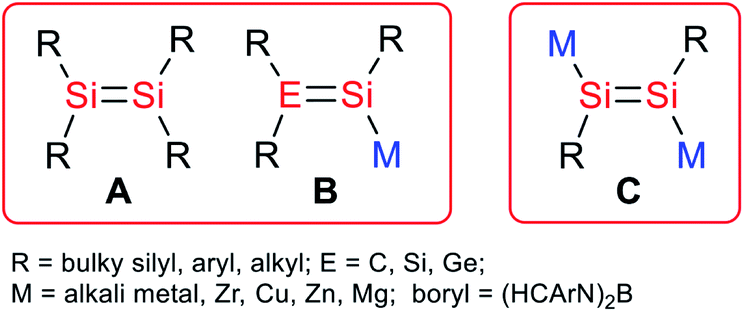 | ||
| Scheme 1 Structural motifs of the reported disilenes (A), silicon analogues of vinyl anions (B) and this work (C). | ||
1,2-Dialkali-metal substituted disilenes are also inherently related to disilynes, the silicon analogues of alkynes, because they could be viewed as doubly reduced disilynes. Disilynes have been of considerable interest because of their fundamental importance. By the employment of very bulky terphenyl, alkyl and silyl substituents, a few examples of disilynes have been isolated.5 However, their doubly reduced species have not been reported so far. It has been shown that the reduction of a silyl-substituted disilyne yielded the corresponding radical anion, and no further reduction products were disclosed.6 It is noted that the doubly reduced alkyne analogues have been isolated for germanium and tin by Power and Aldridge et al.7 However, to the best of our knowledge 1,2-alkali metal substituted disilenes (disilyne dianions) are still elusive. Very recently, our group reported the synthesis of a disilene and 1-magnesium-2,3-disilacyclopropene stabilized by a bulky boryl substituent.8 The strong σ electronic effects of the boryl ligand prompted us to investigate the synthetic probability of 1,2-dialkali-metal substituted disilenes.
Herein, we report the first synthesis of a dialkali metal-substituted disilene, the dilithio-diboryl-disilene 2 (Scheme 2), and its reactivity to haloboranes. Remarkably, in contrast to the trans-bent geometry observed for most of the disilenes, compound 2 adopts an almost planar geometry. Furthermore, a novel 2π aromatic B–Si–Si three-membered ring system with a 2e–3c bond and the first tetraboryldisilene was obtained from the reaction of 2 with haloboranes.
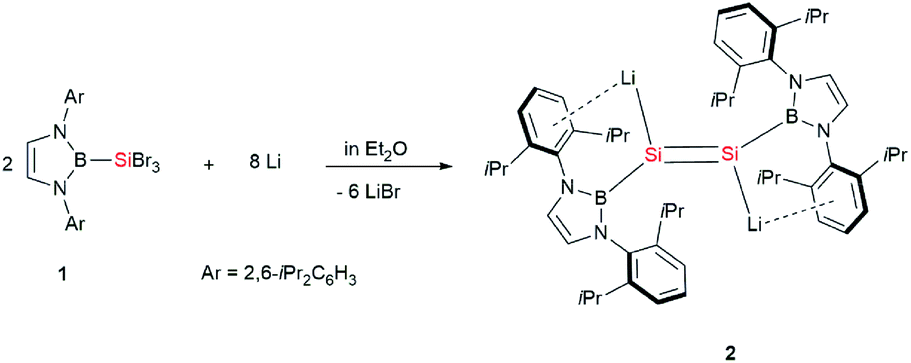 | ||
| Scheme 2 Synthesis of 2. | ||
Results and discussion
The dilithiodisilene [(boryl)SiLi]2 (2) was prepared by the reduction of 1 (ref. 7) with four equivalents of lithium powder in diethyl ether and was isolated as a red powder in 73% yield (Scheme 2). The structure of 2 has been characterized by multinuclear NMR spectroscopy, UV-vis spectroscopy and X-ray single-crystal analysis. The 29Si NMR spectrum of 2 in C6D6 shows a resonance at 297.0 ppm, which is down-field shifted in comparison to that reported for 1-magnesium-2,3-disilacyclopropene (204.1 ppm).8b The UV-vis spectrum of 2 in toluene shows the longest wavelength absorption maximum at 450.0 nm, which is slightly red-shifted compared with the maximum reported for 1-magnesium-2,3-disilacyclopropene (431.0 nm) (Fig. S2†).8b This value is very close to the value calculated for the π–π* transition of 2 (508.9 nm; HOMO − 1 → LUMO + 4, f = 0.2785) (Fig. S3†). The dilithiodisilene 2 is slightly soluble in n-hexane but is well dissolved in benzene and toluene. It is noted that compound 2 is not stable in THF and quickly decomposes into a complicated mixture.Dark red crystals of 2 suitable for X-ray diffraction studies were obtained from n-hexane at 0 °C. There are two independent molecules in the unit cell, and each of them is crystallographically C2 symmetric. The geometries of the two molecules are similar and thus only one is shown in Fig. 1. The structural analysis of 2 indicated an almost planar Si–Si geometry as indicated by the sum of the bond angles of 359.96° around the Si1 and Si1′ atoms and the six almost coplanar atoms B1, Li1, Si1, Si1′, Li′, B1′ (the largest out-of-plane deviation = 0.06 Å). This is in contrast to the nonplanar, trans-bent geometry of the majority of known disilenes. The planarity of 2 could be attributed to the electronic effects of the electropositive boryl and lithium substituents.9 The Si–Si bond length of 2.223(4) Å is same as that reported for 1-magnesium-2,3-disilacyclopropene (2.223(17) Å), while the Si–Li bond length of 2.482(12) Å is remarkably shorter than those observed in contact ion pairs of silenyl lithium species.4,10 The two five-membered diazaborolyl C2N2B planes are essentially coplanar with the BSiSi plane (the dihedral angle = 1.83°). These structural features indicate that the dilithio-diboryldisilene 2 features an ethylene-like structure.
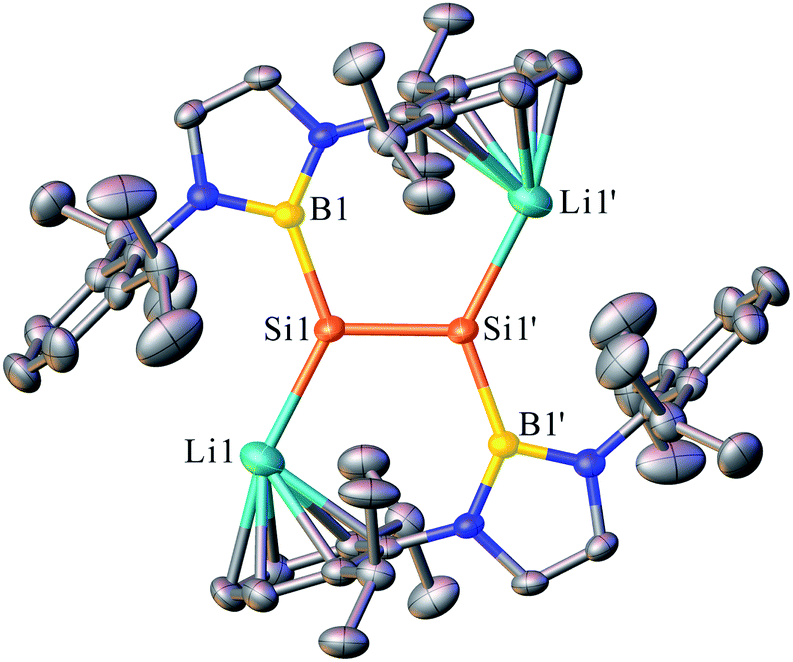 | ||
| Fig. 1 OLEX2 drawing of 2 with 50% ellipsoid probability. Hydrogen atoms have been omitted for clarity. | ||
To get insight into the electronic structure and bonding situations, density functional theory (DFT) calculations were performed for 2 at the B3LYP/6-31G(d,p) level (Fig. S4†). The calculated geometry is very close to that experimentally determined for 2. Natural bond orbital (NBO) analysis revealed highly filled σ (1.97e) and π (1.81e) bonds for the Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si double bonds. The Wiberg bond index (WBI, Table S2†) of 1.77 for the Si–Si bond is consistent with that for the double bond. Natural population analysis (NPA) charges (Si: −0.58; Li: +0.79) of 2 indicate the pronounced anionic character of the silicon atoms. The SiSi π bond orbital, which is perpendicular to the B–Si–Si–B plane, can be seen in the HOMO − 1 (Fig. 2). The HOMO of 2 is mainly composed of the silicon lone pair orbitals. The LUMO + 4 consists predominantly of the Si–Si π* bond with some contribution of the boron-centered p-orbitals.
Si double bonds. The Wiberg bond index (WBI, Table S2†) of 1.77 for the Si–Si bond is consistent with that for the double bond. Natural population analysis (NPA) charges (Si: −0.58; Li: +0.79) of 2 indicate the pronounced anionic character of the silicon atoms. The SiSi π bond orbital, which is perpendicular to the B–Si–Si–B plane, can be seen in the HOMO − 1 (Fig. 2). The HOMO of 2 is mainly composed of the silicon lone pair orbitals. The LUMO + 4 consists predominantly of the Si–Si π* bond with some contribution of the boron-centered p-orbitals.
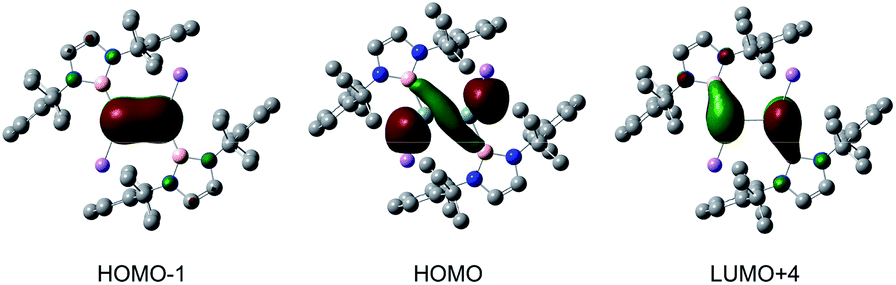 | ||
| Fig. 2 The calculated frontier orbitals of 2. | ||
Alkali metal-substituted disilenes may facilitate extensive studies on their reactions with halogenated reagents for the development of new reaction modes and access to novel multiply bonded silicon compounds.2a,b,3,4 Reduction of appropriately disubstituted dihalosilanes (R1R2SiX2) is the most general route to disilenes.2 However, in contrast to easy availability of alkyl, aryl and silyldihalosilanes, boryl-substituted dihalosilanes could not be easily prepared because of the lack of nucleophilic boron compounds. In view of the interesting electronic effects of boryl substituents, we explored the reaction of dilithiodisilene 2 with several types of haloboranes.
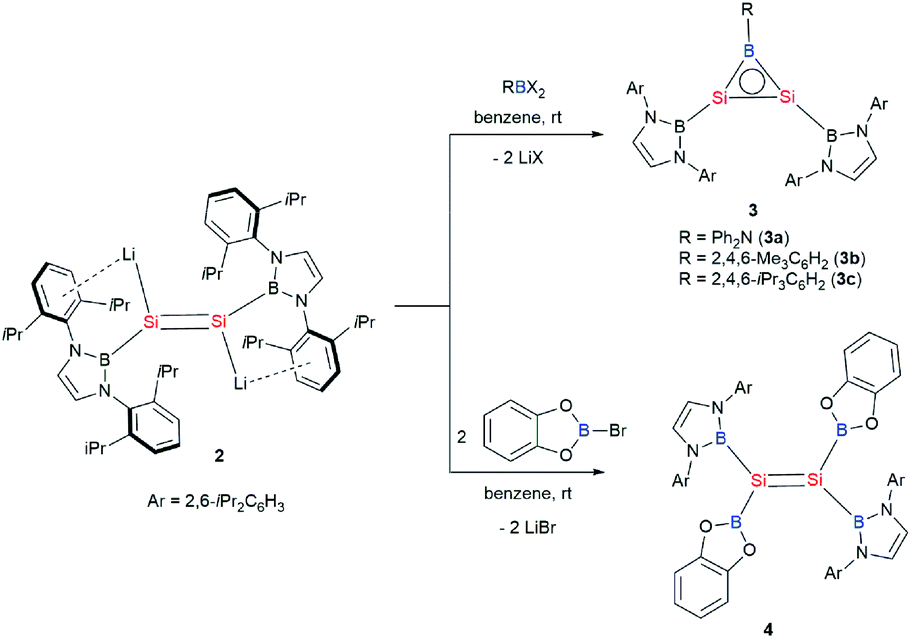
First, the reactions of 2 with dihaloboranes were explored with the expectation to generate a Si–Si–B three-membered ring system as it is isoelectronic with aromatic cyclopropenylium ions ([C3R3]+) and borirenes (R2C2BR). Thus, reactions of dilithiodisilene 2 with dihaloboranes RBX2 (R = Ph2N, Mes, Tip; X = Cl, Br; Mes = 2,4,6-Me3C6H2; Tip = 2,4,6-iPr3C6H2) have been conducted. Treatment of 2 with the haloboranes in benzene at room temperature afforded an orange yellow solution, from which disilaborirenes 3a–c could be obtained as light orange solids in 53%, 86% and 68% yields, respectively (Scheme 3). X-ray single-crystal analysis revealed that compounds 3a–c feature a planar B–Si–Si three-membered ring with both silicon and boron atoms being three-coordinate. The 29Si NMR spectra of compounds 3a–c exhibit a singlet signal at 112.2, 142.0 and 141.1 ppm, respectively. In the 11B NMR spectra of 3a–c, two broad signals are observed (δ 24.2 and 50.1 ppm for 3a, δ 23.5 and 72.9 ppm for 3b, and δ 23.4 and 73.3 ppm for 3c). The UV-vis spectra of compounds 3a–c in toluene show the longest wavelength absorption maximum at 380.0, 364.5 and 370.0 nm, respectively (Fig. S5†). The NMR and UV-vis data indicate that the amino substituent Ph2N on the boron atom in 3a has pronounced effects on the electronic structure of the B–Si–Si three-membered ring because of the π-interactions of the amino group with the boron atom.
 | ||
| Scheme 3 Reactivity of 2. | ||
Orange yellow crystals of 3a–c suitable for X-ray diffraction studies were obtained from n-pentane at −20 °C. The molecular structural analysis of 3a and 3c shown in Fig. 3 (for 3a–c, see Fig. S6–S8†) confirmed the B–Si–Si three-membered ring system. The Si–Si bond lengths of 2.1469(11), 2.133(2) and 2.1452(6) Å in 3a–c are in the range of that of a Si–Si double bond (2.12–2.25 Å)1e and slightly shorter than that determined for a cyclotrisilenylium cation (av. 2.217(3) Å).11 The B–Si bond lengths in the 3a–c BSi2 cycles ranging from 1.911(7) Å to 1.952(3) Å are close to those of 1.9190 and 1.9186(13) Å reported for L2Si2BR (D, Scheme 4) featuring the four-coordinate silicon atoms.12 The sums of the bond angles around Si1 and Si2 in 3a–c are close to 360°, indicating that the geometries of each silicon atom are essentially planar. A comparison of the B–Si bonds and internal angles of the Si2B ring in 3a–c reveals that the amino substituent Ph2N in 3a engages in a relatively weak π-interaction with the empty p orbital of the boron atom.
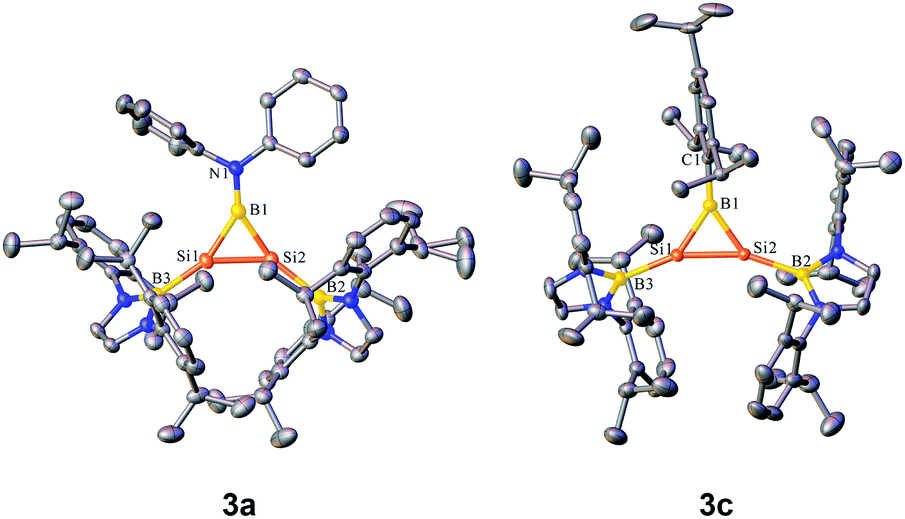 | ||
| Fig. 3 OLEX2 drawing of 3a and 3c with 50% ellipsoid probability. Hydrogen atoms have been omitted for clarity. | ||
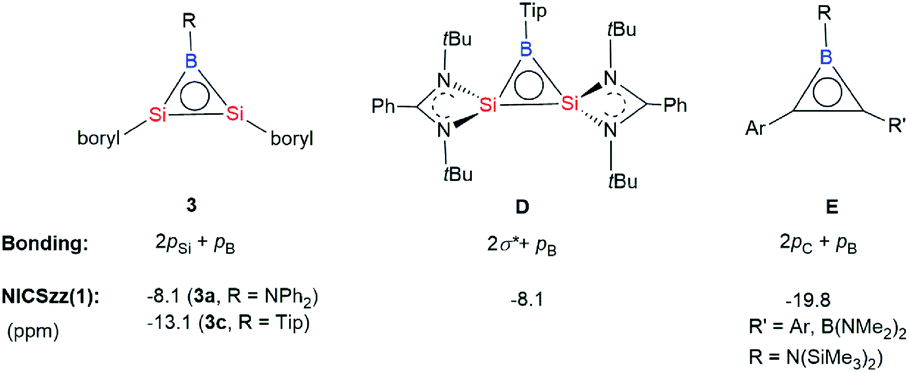 | ||
| Scheme 4 Comparison of the bonding and NICS values of disilaborirenes (3 and D) and borirenes (E). | ||
As borirenes are aromatic systems, we are interested in the aromaticity of the silicon analogues 3a–c for comparison.13 Thus, DFT calculations were carried out for amino-substituted 3a and the phenyl-substituted 3c at the B3LYP/6-31G(d,p) level. The optimized structures (Fig. S9 and S10†) are well correlated with the experimentally determined ones. The WBI values (1.25 and 1.28 for 3a and 3c) of the ring B–Si bonds indicate the partial double-bonds caused by 3c–2e bonding of the BSi2 unit in 3a (1.96e) and 3c (1.95e), as shown by NBO analysis (Tables S3–S6†). The calculated frontier orbitals for 3a and 3c are shown in Fig. 4. The HOMO of 3a consists of the SiSi π bonding and antiphase nitrogen lone pair, while the HOMO − 1 of 3c mainly corresponds to the delocalized Si2B π orbital (Fig. 4).
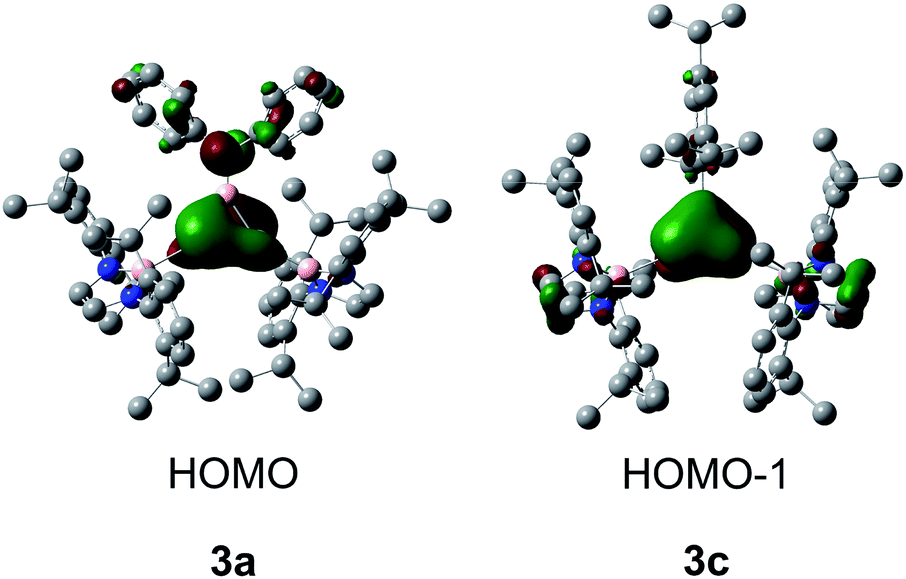 | ||
| Fig. 4 Frontier orbitals of 3a (left) and 3c (right). | ||
To determine the π aromaticity of disilaborirenes 3a and 3c, the nucleus independent chemical shift (NICSzz(1)) values were calculated. The NICS values of −8.1 and −13.1 ppm calculated for 3a and 3c indicate the aromaticity, but it is weaker than that (−19.8 ppm) reported for borirene E (Scheme 4).13 Although the aromaticity of 3a with R = Ph2N is similar to that reported for D (Scheme 4, Tip = 2,4,6-iPr3C6H2),12 that of 3c is much stronger than that of D. It is apparent that the degree of the π aromaticity of 3c with R = Tip is much greater than that of 3a with R = Ph2N because of the electron delocalization in the B–N bond in 3a. The aromaticity of 3a and 3c could be interpreted as the extensive 2π electron delocalization over the three-center bonding molecular orbitals from the p orbitals of B and Si atoms (Scheme 4). The bonding situation in the Si2B cycle is analogous to that of borirenes but distinct from that of D with four-coordinate Si atoms, in which the delocalized structure was formed by the σ* MOs of π-symmetry with the boron p orbital.12
Reaction of dilithiodisilene 2 with two equivalents of B-bromocatecholborane (cat)BBr resulted in the formation of the first tetraboryldisilene 4 (Scheme 3). Although a great number of disilenes have been synthesized, disilenes substituted by group 13 and 15 elements are still limited because of the lack of appropriate synthetic precursors.2b,3c,g,m,n,14 As the structure of a disilene is largely dependent on the substituents, the synthesis of boryl-substituents is of current interest. Tetraboryldisilene 4 was fully characterized by NMR spectroscopy, UV-vis spectroscopy and X-ray single-crystal analysis. The 29Si NMR spectrum shows a singlet at 105.6 ppm, which resides at a higher field in comparison to that of diboryldisilene (200.0 ppm). Consistent with the molecular structure of 4, the 11B NMR spectrum exhibits two broad signals at δ 26.2 and 37.3 ppm. The UV-vis spectra of 4 in toluene show the longest wavelength absorption maximum at 445.0 nm (Fig. S11†), which is same as the value calculated for the π–π* transition of 4 (445.0 nm; HOMO → LUMO, f = 0.4433) (Fig. S12†).
Yellow crystals of 4 suitable for X-ray diffraction studies were obtained from n-hexane at −20 °C. Analysis of the structure of 4 (Fig. 5 and S13†) revealed that the SiSi bond adopts an essentially planar geometry with a twist angle of 0° as well as a trans-bent angle of 1.86°, indicating the pronounced electronic effects (σ-donor) of the boryl substituents on the SiSi bond. The diazaborolyl C2N2B planes are almost orthogonal to the B–Si–Si planes with a dihedral angle of 86.58°. The structural features of 4 were also examined by DFT calculations at the B3LYP/6-31G(d,p) level. The HOMO and LUMO are mainly SiSi π and π* orbitals (Fig. S15†). On the basis of the results, compound 4 represents a planar disilene featuring the smallest trans-bent geometry and zero twist angle.15
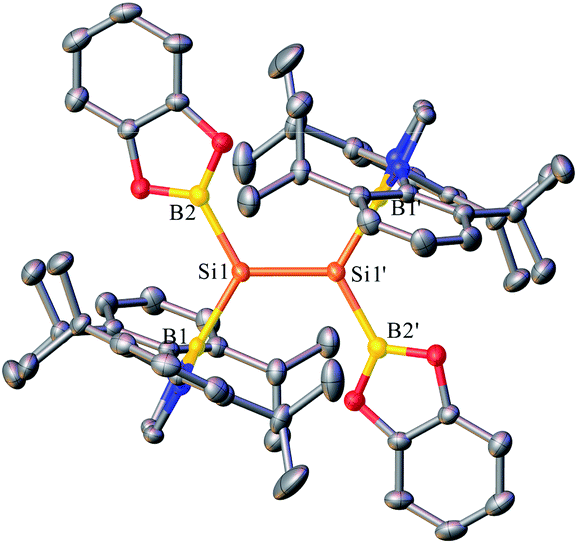 | ||
| Fig. 5 OLEX2 drawing of 4 with 50% ellipsoid probability. Hydrogen atoms have been omitted for clarity. | ||
Conclusions
In summary, dilithiodisilene 2 was prepared by the reduction of (boryl)SiBr3 with lithium in Et2O. Dilithiodisilene 2 represents one of the rare examples of ethylene-like disilenes. It turned out to be a valuable reagent for the construction of novel silicon multiply-bonded species as demonstrated by the first synthesis of a 3c-2π aromatic system 3a–c and a fully boryl-substituted disilene 4. The unique electronic effects of the boryl group allowed facile access to the dianonic species 2. DFT calculations predicted that the aromaticity of disilaborirenes 3a–c stems from the delocalization of the 2π electrons over the three-center bonding molecular orbital from the B and Si p atomic orbitals. Exploration of more challenging synthetic targets from dilithiodisilene 2 is currently underway in our laboratory.Data availability
All experimental procedures, spectroscopic data, results of the DFT calculations and crystallographic data can be found in the ESI.†Author contributions
M. Tian performed the major experimental work and wrote the original draft. J. Zhang performed the DFT calculations. L. Guo did the X-ray crystal analysis. C. Cui is the principal investigator and supervised for the project and finalized the draft of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grant No. 21632006) for generous support.Notes and references
- (a) For selected reviews, see: R. Borthakur and V. Chandrasekhar, Coord. Chem. Rev., 2021, 429, 213647–213678 CrossRef CAS; (b) P. P. Power, Organometallics, 2020, 39, 4127–4138 CrossRef CAS; (c) A. Agarwal and S. K. Bose, Chem.–Asian J., 2020, 15, 3784–3806 CrossRef CAS PubMed; (d) L. Zhao, S. Pan, N. Hozlmann, P. Schwerdtfeger and G. Frenking, Chem. Rev., 2019, 119, 8781–8845 CrossRef CAS PubMed; (e) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. Präsang and D. Scheschkewitz, Chem. Soc. Rev., 2016, 45, 900–921 RSC; (b) A. Rammo and D. Scheschkewitz, Chem.–Eur. J., 2018, 24, 6866–6885 CrossRef CAS PubMed; (c) A. Sekiguchi and V. Y. Lee, Chem. Rev., 2003, 103, 1429–1447 CrossRef CAS PubMed.
- (a) I. Bejan and D. Scheschkewitz, Angew. Chem., Int. Ed., 2007, 46, 5783–5786 CrossRef CAS PubMed; (b) K. Abersfelder and D. Scheschkewitz, J. Am. Chem. Soc., 2008, 130, 4114–4121 CrossRef CAS PubMed; (c) S. Inoue, M. Ichinohe and A. Sekiguchi, Chem. Lett., 2008, 37, 1044–1045 CrossRef CAS; (d) T. Iwamoto, M. Kobayashi, K. Uchiyama, S. Sasaki, S. Nagendran, H. Isobe and M. Kira, J. Am. Chem. Soc., 2009, 131, 3156–3157 CrossRef CAS PubMed; (e) J. Jeck, I. Bejan, A. J. P. White, D. Nied, F. Breher and D. Scheschkewitz, J. Am. Chem. Soc., 2010, 132, 17306–17315 CrossRef CAS PubMed; (f) M. Hartmanm, A. Haji-Abdi, K. Abersfelde, P. R. Haycock, A. J. P. White and D. Scheschkewitz, Dalton Trans., 2010, 39, 9288–9295 RSC; (g) P. Willmes, M. J. Cowley, M. Hartmann, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2014, 53, 2216–2220 CrossRef CAS PubMed; (h) T. Iwamoto, N. Akasaka and S. Ishida, Nat. Commun., 2014, 5, 5353–5359 CrossRef CAS PubMed; (i) P. Willmes, L. Junk, V. Huch, C. B. Yildiz and D. Scheschkewitz, Angew. Chem., Int. Ed., 2016, 55, 10913–10917 CrossRef CAS PubMed; (j) N. M. Obeid, L. Klemmer, D. Maus, M. Zimmer, J. Jeck, I. Bejan, A. J. P. White, V. Huch, G. Jung and D. Scheschkewitz, Dalton Trans., 2017, 46, 8839–8848 RSC; (k) T. Kosai, S. Ishida and T. Iwamoto, J. Am. Chem. Soc., 2017, 139, 99–102 CrossRef CAS PubMed; (l) T. Kosai, S. Ishida and T. Iwamoto, Dalton Trans., 2017, 46, 11271–11281 RSC; (m) T. Kosai and T. Iwamoto, J. Am. Chem. Soc., 2017, 139, 18146–18149 CrossRef CAS PubMed; (n) T. Kosai and T. Iwamoto, Chem.–Eur. J., 2018, 24, 7774–7780 CrossRef CAS PubMed; (o) Y. Heider, P. Willmes, D. Mühlhausen, L. Klemmer, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2019, 58, 1939–1944 CrossRef CAS PubMed.
- (a) D. Scheschkewitz, Angew. Chem., Int. Ed., 2004, 43, 2965–2967 CrossRef CAS PubMed; (b) M. Ichinohe, K. Sanuki, S. Inoue and A. Sekiguchi, Organometallics, 2004, 23, 3088–3090 CrossRef CAS; (c) S. Inoue, M. Ichinohe and A. Sekiguchi, Chem. Lett., 2005, 34, 1564–1565 CrossRef CAS; (d) K. Abersfelder, D. Güclü and D. Scheschkewitz, Angew. Chem., Int. Ed., 2006, 45, 1643–1645 CrossRef CAS PubMed; (e) M. Ichinohe, K. Sanuki, S. Inoue and A. Sekiguchi, Silicon Chem., 2006, 3, 111–116 CrossRef; (f) L. Zborovsky, R. Dobrovetsky, M. Botoshansky, D. Bravo-Zhivotovskii and Y. Apeloig, J. Am. Chem. Soc., 2012, 134, 18229–18232 CrossRef CAS PubMed; (g) D. Pinchuk, J. Mathew, A. Kaushansky, D. Bravo-Zhivotovskii and Y. Apeloig, Angew. Chem., Int. Ed., 2016, 55, 10258–10262 CrossRef CAS PubMed; (h) L. Zhu, J. Zhang, H. Yang and C. Cui, J. Am. Chem. Soc., 2019, 141, 19600–19604 CrossRef CAS PubMed; (i) I. Bejan, S. Inoue, M. Ichinohe, A. Sekiguchi and D. Scheschkewitz, Chem.–Eur. J., 2008, 14, 7119–7122 CrossRef CAS PubMed; (j) S. Inoue, M. Ichinohe and A. Sekiguchi, Angew. Chem., Int. Ed., 2007, 46, 3346–3348 CrossRef CAS PubMed; (k) I. Bejan, D. Güclü, S. Inoue, M. Ichinohe, A. Sekiguchi and D. Scheschkewitz, Angew. Chem., Int. Ed., 2007, 46, 3349–3352 CrossRef CAS PubMed; (l) A. Jana, M. Majumder, V. Huch, M. Zimmer and D. Scheschkewitz, Dalton Trans., 2014, 43, 5175–5181 RSC; (m) H. Tanaka, S. Inoue, M. Ichinohe and A. Sekiguchi, Organometallics, 2009, 28, 6625–6628 CrossRef CAS; (n) T. Kosai, S. Nishimura, N. Hayakawa, T. Matsuo and T. Iwamoto, Chem. Lett., 2019, 48, 1168–1170 CrossRef CAS; (o) A. Jana, V. Huch, M. Repisky, R. J. F. Berger and D. Scheschkewitz, Angew. Chem., Int. Ed., 2014, 53, 3514–3518 CrossRef CAS PubMed; (p) K. Abersfelder, A. J. P. White, H. S. Rzepa and D. Scheschkewitz, Science, 2010, 327, 564–566 CrossRef CAS PubMed; (q) M. J. Cowley, K. Abersfelder, A. J. P. White, M. Majumdar and D. Scheschkewitz, Chem. Commun., 2012, 48, 6595–6597 RSC; (r) T. Nguyen and D. Scheschkewitz, J. Am. Chem. Soc., 2005, 127, 10174–10175 CrossRef CAS PubMed; (s) N. Akasaka, K. Fujieda, E. Garoni, K. Kamada, H. Matsui, M. Nakano and T. Iwamoto, Organometallics, 2018, 37, 172–175 CrossRef CAS; (t) P. K. Majhi, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2021, 60, 242–246 CrossRef CAS PubMed.
- (a) A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755–1757 CrossRef CAS PubMed; (b) T. Sasamori, K. Hironaka, Y. Sugiyama, N. Takagi, S. Nagase, Y. Hosoi, Y. Furukawa and N. Tokitoh, J. Am. Chem. Soc., 2008, 130, 13856–13857 CrossRef CAS PubMed; (c) S. Ishida, R. Sugawara, Y. Misawa and T. Iwamoto, Angew. Chem., Int. Ed., 2013, 52, 12869–12873 CrossRef CAS PubMed; (d) M. Asay and A. Sekiguchi, Bull. Chem. Soc. Jpn., 2012, 85, 1245–1261 CrossRef CAS.
- R. Kinjo, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2007, 129, 26–27 CrossRef CAS PubMed.
- (a) L. Pu, M. O. Senge, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1998, 120, 12682–12683 CrossRef CAS; (b) L. Pu, A. D. Philips, A. F. Richards, M. Stender, R. S. Simons, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2003, 125, 11626–11636 CrossRef CAS PubMed; (c) A. F. Richards, M. Brynda and P. P. Power, Chem. Commun., 2004, 1592–1593 RSC; (d) A. Rit, J. Campos, H. Niu and S. Aldridge, Nat. Chem., 2016, 8, 1022–1026 CrossRef CAS PubMed.
- (a) Z. Liu, J. Zhang, H. Yang and C. Cui, Organometallics, 2020, 39, 4164–4168 CrossRef CAS; (b) M. Tian, J. Zhang, H. Yang and C. Cui, J. Am. Chem. Soc., 2020, 142, 4131–4135 CrossRef CAS PubMed.
- M. Karni and Y. Apeloig, J. Am. Chem. Soc., 1990, 112, 8590–8592 CrossRef.
- (a) L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2019, 58, 12007–12010 CrossRef CAS PubMed; (b) Y. Heider, P. Willmes, V. Huch, M. Zimmer and D. Scheschkewitz, J. Am. Chem. Soc., 2019, 141, 19498–19504 CrossRef CAS PubMed.
- M. Ichinohe, M. Igarashi, K. Sanuki and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 9978–9979 CrossRef CAS PubMed.
- S. K. Sarkar, R. Chaliha, M. M. Siddiqui, S. Banerjee, A. Münch, R. Herbst-Irmer, D. Stalke, E. D. Jemmis and H. W. Roesky, Angew. Chem., Int. Ed., 2020, 59, 23015–23019 CrossRef CAS PubMed.
- (a) J. J. Eisch, B. Shafii, J. D. Odom and A. L. Rheingold, J. Am. Chem. Soc., 1990, 112, 1847–1853 CrossRef CAS; (b) H. Braunschweig, H. Thomas and D. R. F. Seeler, Angew. Chem., 2005, 117, 7627–7629 CrossRef; (c) H. Braunschweig, A. Damme, R. D. Dewhurst, S. Ghosh, T. Kramer, B. Pfaffinger, K. Radacki and A. Vargas, J. Am. Chem. Soc., 2013, 135, 1903–1911 CrossRef CAS PubMed; (d) H. Braunschweig, R. D. Dewhurst, K. Radacki, C. W. Tate and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 6263–6266 CrossRef CAS PubMed.
- (a) K. Takeuchi, M. Ikoshi, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2010, 132, 930–931 CrossRef CAS PubMed; (b) K. Takeuchi, M. Ikoshi, M. Ichinohe and A. Sekiguchi, J. Organomet. Chem., 2011, 696, 1156–1162 CrossRef CAS; (c) P. Jutzi, A. Mix, B. Neumann, B. Rummel, W. W. Schoeller, H.-G. Stammler and A. B. Rozhenko, J. Am. Chem. Soc., 2009, 131, 12137–12143 CrossRef CAS PubMed; (d) S. Khan, S. S. Sen, H. W. Roesky, D. Kratzert, R. Michel and D. Stalke, Inorg. Chem., 2010, 49, 9689–9693 CrossRef CAS PubMed; (e) K. Izod, P. Evans and P. G. Waddell, Angew. Chem., Int. Ed., 2017, 56, 5593–5597 CrossRef CAS PubMed.
- (a) H. Watanabe, K. Takeuchi, N. Fukawa, M. Kato, M. Goto and Y. Nagai, Chem. Lett., 1987, 16, 1341–1344 CrossRef; (b) A. Fukazawa, Y. Li, S. Yamaguchi, H. Tsuji and K. Tamao, J. Am. Chem. Soc., 2007, 129, 14164–14165 CrossRef CAS PubMed; (c) M. Kobayashi, T. Matsuo, T. Fukunaga, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, J. Am. Chem. Soc., 2010, 132, 15162–15163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2081430, 2081431, 2088661, 2088665 and 2104803. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05125c |
| This journal is © The Royal Society of Chemistry 2021 |