
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioorthogonal regulation of DNA circuits for smart intracellular microRNA imaging†
Yingying
Chen
a,
Xue
Gong
a,
Yuhui
Gao
a,
Yu
Shang
a,
Jinhua
Shang
a,
Shanshan
Yu
a,
Ruomeng
Li
a,
Shizhen
He
a,
Xiaoqing
Liu
 ab and
Fuan
Wang
*ab
ab and
Fuan
Wang
*ab
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, P. R. China
bResearch Institute of Shenzhen, Wuhan University, Shenzhen 518057, P. R. China. E-mail: fuanwang@whu.edu.cn
First published on 8th November 2021
Abstract
Catalytic DNA circuits represent a versatile toolbox for tracking intracellular biomarkers yet are constrained with low anti-interference capacity originating from their severe off-site activation. Herein, by introducing an unprecedented endogenous DNA repairing enzyme-powered pre-selection strategy, we develop a sequential and specific on-site activated catalytic DNA circuit for achieving the cancer cell-selective imaging of microRNA with high anti-interference capacity. Initially, the circuitry reactant is firmly caged by an elongated stabilizing duplex segment with a recognition/cleavage site of a cell-specific DNA repairing enzyme, which can prevent undesired signal leakage prior to its exposure to target cells. Then, the intrinsic DNA repairing enzyme of target cells can liberate the DNA probe for efficient intracellular microRNA imaging via the multiply guaranteed molecular recognition/activation procedures. This bioorthogonal regulated DNA circuit presents a modular and programmable amplification strategy for highly reliable assays of intracellular biomarkers, and provides a pivotal molecular toolbox for living systems.
Introduction
The in-depth understanding of exquisite biotransformations requires the development of a robust toolbox for in situ monitoring intracellular biomolecules, especially disease-related biomarkers, and shows great potential for clinical diagnostics and therapeutics.1–4 Artificial biochemical circuits have thus been developed to explicate these complicated biological systems and further regulate biochemical reactions. Because of the intrinsic predictability and programmability of nucleic acids, DNA biocomputing circuits have been facilely engineered as intelligent artificial biochemical circuits and represent an indispensable tool for live cell imaging.5,6 The construction of robust, modular and compact artificial DNA circuits facilitates the exploration of the essential principles of complex biological systems, and it paves a facile path for information transmissions in molecular biosensing and biocomputing fields. Particularly, DNA circuits that can effectively integrate various functional modules of signal amplification hold great potential for cell imaging of low-abundance biomarkers.7–12 As a typical isothermal DNA circuit, the hybridization chain reaction (HCR) has already been proved for various biosensing applications.13–15 For HCR, the analyte-triggered cascade hybridization results in high-molecular-weight dsDNA copolymers. The low cellular diffusibility feature of the HCR product makes it suitable for intracellular imaging and colocalization of various analytes.16,17 However, the analyte, as the single input, directly triggers the autonomous HCR, which is prone to cause unreliable readouts for cell imaging.18,19 Even more concerningly, the metastable DNA probes may nonspecifically respond to surrounding complicated interferences encountered during the cellular delivery process, thus leading to compromised specificity and sensitivity for intracellular sensing. Thus, it is highly desirable to engineer endogenously programmed DNA circuits that can proceed via cell-specific exposure and activation to achieve highly reliable intracellular imaging with anti-interference capacity.Currently, a plethora of exogenous and endogenous activatable approaches have been exploited to regulate DNA circuits, to facilitate control over their structures and functions via specific activation.20 For exogenous regulation, photo activation is a representative method by virtue of its high spatiotemporal control property.21–23 However, its application in living systems is significantly constrained, due to the cumbersome chemical synthesis of photolabile groups on nucleobases, the limited penetration depth of light and the potential phototoxicity. As a result, endogenous cues, such as small molecules,24,25 RNAs,26,27 or enzymes,28,29 represent an alternative way to regulate DNA circuits. For the small molecules-activated strategy, e.g., ATP,24 their corresponding aptamers are needed to inter-correlate with DNA circuits. However, the available aptamers are always insufficient, and affinity is sometimes unwarrantable between small molecules and their aptamers. We have reported the implementation of multiple endogenous microRNA (miRNA)-activated DNA circuits for cell discrimination.27 Nevertheless, these miRNA-involved DNA circuits suffer from complicated designs and a lack of sufficient blocking means to eliminate off-site activation or nonspecific signal leakage, which are especially inadaptable in living systems. Among these endogenously activatable tools, enzymes represent superior candidates, owing to their high substrate specificity, excellent catalytic activity and biosafety. Endogenous enzymatic stimuli are thus introduced for developing various enzyme-responsive sensors and drug release systems.30–32 Especially, the DNA repairing enzyme represents a more appealing choice, considering the versatile and specific DNA substrate. Moreover, the DNA repairing enzyme is overexpressed in cancer cells because of the ubiquitous upregulation of DNA replication,33,34 which is of great interest in designing enzyme-regulated DNA circuitry with high activation efficiency, favorable cell-specificity, and minimal toxicity.
Herein, by introducing an endogenous DNA repairing enzyme-mediated sequential activation strategy, we developed an on-site bioorthogonally regulated DNA circuit for achieving specific microRNA imaging with high anti-interference ability. As illustrated in Scheme 1, the HCR circuitry system was selected for realizing cancer cell-selective amplified miRNA imaging. The stem region of one HCR reactant (hairpin H2) is elongated with a long duplex region to generate a HCR-blunt DNA pre-probe (H2-P). The elongated duplex segment can not only stabilize the DNA pre-probe by blocking the toehold domain of the HCR reactant but can also introduce the enzyme-specific recognition site. The introduction of the sequential and specific enzyme-controlled DNA pre-probe facilitates the on-site execution of an HCR circuit via two successive steps of specific enzyme regulation and amplified target sensing, thus ensuring selective and sensitive miRNA imaging. By virtue of the overexpressed DNA repairing enzyme in cancer cells, the DNA pre-probe is specifically cleaved to expose the circuitry reactant, which can then participate in target miRNA-triggered HCR for achieving amplified intracellular miRNA imaging by Förster resonance energy transfer (FRET) transduction. In contrast, the DNA pre-probe remains inactive and fails to motivate the HCR-powered miRNA sensing in normal cells because of the insufficient enzymes therein. Thus, based on the programmable and rational design of DNA probes, the precise manipulation of DNA circuits is available through endogenous enzymatic regulation in cancer-specific cells, which may provide a facile and effective tool for complicated intracellular applications.
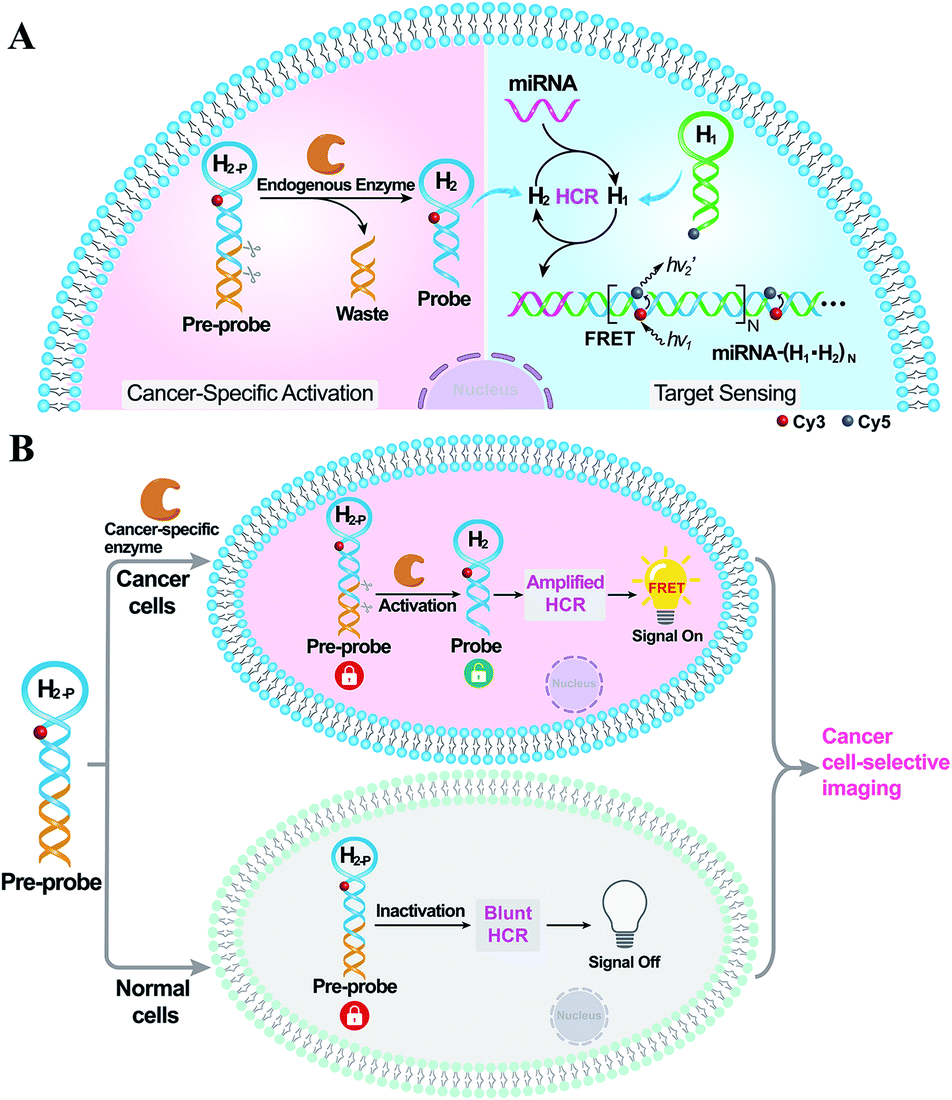 | ||
| Scheme 1 (A) Schematic of the on-site and sequential activation of the enzyme-guaranteed HCR amplification circuit for specific miRNA imaging in live cells. (B) Illustration of cancer cell-selective miRNA imaging by the on-site bioorthogonal activatable HCR system. | ||
Results and discussion
The principle of the sequential and specifically activated HCR amplifier for miRNA detection is illustrated in Scheme 1A (for the detailed reaction process, see Scheme S1†). As a proof of concept, the typical DNA repairing enzyme, human apurinic/apyrimidinic endonuclease 1 (APE1), which primarily resided in the cytoplasm of diverse cancer cells, was employed to activate the HCR pre-probe with abasic sites.34 Also, microRNA-21 (miR-21), a significant diagnostic cancer biomarker,35 was elected as the target analyte to prove the principle of our engineered system. The HCR amplification circuit was composed of two hairpins, H1 and H2-P. Herein, a given miR-21 initiator (I) was recognized by H1via a toehold-mediated strand-displacement mechanism, generating hybrids I–H1. However, hairpin H2-P was unable to hybridize with the sequences d–c in hybrids I–H1, as its toehold domain c*–d* was caged tightly by the additionally elongated duplex e–e* region, resulting in a temporarily blocked “inactive” H2-P pre-probe and the deactivated HCR circuit. This “inactive” H2-P pre-probe could be specifically activated into the “active” probe H2 by an endogenous enzyme APE1-mediated de-caging procedure, by which the duplex e–e* region of H2-P was removed. Then, the liberated H2 could hybridize with sequence d–c in hybrids I–H1, yielding complex I–H1·H2 to trigger the cyclic cross-opening of H1 and H2 and eventually generating HCR-assembled dsDNA products. H1 was modified with a fluorophore acceptor (Cy5) at its 5′-end, while H2-P was intermediately labeled with a fluorophore donor (Cy3) in the stem region. These two fluorophores (Cy3 and Cy5) were brought into close proximity through the autonomous cross-hybridization process, leading to an amplified FRET readout. During the miRNA-sensing process, the blocked DNA pre-probe H2-P was specifically exposed by endogenous enzyme APE1; then, the target could motivate the HCR amplifier. Therefore, the specifically activatable DNA pre-probe played an indispensable role to avoid nonspecific signal leakage, enabling the selective and sensitive bioimaging in live cells.The optimum dosage and incubation duration of APE1 enzyme were firstly tested by denatured polyacrylamide gel electrophoresis (PAGE) analysis (Fig. S1†). By incubating the HCR pre-probe H2-II-P with different concentrations of APE1 for 5 h, the substrate cleavage efficiency was quantified to be higher than 90% with 5 U mL−1 of APE1. The incubation duration was further explored by treating H2-II-P with 5 U mL−1 of APE1 for varied time-intervals, and up to 90% cleavage efficiency was achieved under the optimum duration of 5 h. Thus, 5 U mL−1 of APE1 and 5 h of incubation were selected as the optimum H2-P cleavage conditions for the following assays.
All DNA hairpins were theoretically (by Mfold software) and experimentally optimized for highly efficient miRNA sensing. The specifically APE1-recognized DNA pre-probe H2-P was designed with two different structures (H2-I-P and H2-II-P) (Fig. S2†). Here, H2-I-P was grafted with an additional loop region f between regions e/e* and a/d* as compared with H2-II-P. This symmetric organization could lead to a more stabilized configuration for APE1 activation, due to a more synergistic stabilization effect. However, the symmetric H2-I may impede the highly efficient execution of HCR on account of the steric effect; thus, the asymmetric H2-II was chosen as the optimized reactant to be recognized and regulated by APE1 enzyme. Native PAGE was also employed to evaluate the optimal structure of DNA pre-probe for APE1 recognition. As expected, the H2-I-P- or H2-II-P-containing HCR circuitry failed to proceed without the initiator miR-21 regardless of the presence or absence of APE1 (lanes 6 and 7, Fig. S3†), indicating that the pre-probes H2-I-P and H2-II-P were tightly blocked by the elongated duplex region. Moreover, the initiator miR-21 failed to trigger the HCR process in the absence of APE1 (lane 8, Fig. S3†), indicating that APE1 cleavage played a key role in unlocking the miR-21-triggered HCR circuit. For the APE1-guaranteed HCR, new HCR product bands with much lower electrophoretic mobility emerged obviously in the H2-II-P-involved HCR system after 3 h (lane 9, Fig. S3†), indicating that a higher HCR amplification efficiency was achieved by H2-II-P under the same reaction conditions. In consequence, the H2-II-P pre-probe was selected as the functional HCR reactant for further study. Furthermore, more DNA products were assembled with a prolonged 5 h of reaction duration (lane 10, Fig. S3†), demonstrating the autonomous and continuous assembly of the HCR circuit.
The APE1-controlled HCR system was further explored by a fluorescence experiment under the optimized hairpin dosage (Fig. S4A†). The feasibility demonstration of the APE1-regulated circuitry system is illustrated in Fig. 1A. In the absence of APE1 regulation, almost no fluorescence change was observed with or without the target miR-21 (curves c and d, respectively, Fig. 1B). Also, the mixture of H1 and APE1-exposed H2-II-P pre-probe generated very weak fluorescence response without the target analyte (curve a, Fig. 1B). Only in the presence of APE1 could the target miR-21 trigger the cascade hybridization of HCR reactants to generate an increasingly amplified FRET readout signal (curve b, Fig. 1B). The fluorescence feasibility demonstration was consistent with the native PAGE experiment (Fig. 1C; for details, see the ESI†). Thus, for this APE1-regulated miRNA-targeting HCR circuit, the DNA pre-probe was well blocked and stayed in a temporary dormant state without obvious signal leakage by avoiding the undesired spontaneous hybridization chain reaction. To further verify the APE1-specific recognition and cleavage of the DNA pre-probe, an additional mutant hairpin H2-III-P without the abasic site was introduced as an important control (Fig. 1D and S4B†). Extremely weak fluorescence was generated without miR-21, illustrating that the sensing activity of our HCR system was blocked appropriately by the afore-engineered DNA pre-probes. In the presence of miR-21, the intact H1 + H2-II-P mixture and the APE1-treated mutant H1 + H2-III-P mixture both displayed no obvious fluorescence change. Meanwhile, the APE1-regulated H1 + H2-II-P mixture elicited a significant fluorescence response to miR-21, indicating the crucial role of APE1 exposure for restoring the sensing property of the HCR amplification circuit. Subsequently, live cell imaging was performed to confirm the activatable miRNA sensing ability by choosing MCF-7 cells in which both APE1 and miR-21 were overexpressed (Fig. S4C†). By incubating MCF-7 cells with H1 and the APE1-active H2-II-P (with the abasic site) for 4 h, a dramatic fluorescence signal was observed. To ensure that the intracellular fluorescence signal indeed resulted from the enzyme-programmed DNA pre-probe, H1 and the APE1-blunt H2-III-P (without abasic site) were transfected as a negative control. As expected, the APE1-blunt H1 + H2-III-P group elicited a much lower intracellular fluorescence response under the same conditions, thus demonstrating the sequential APE1-specific recognition of the miR-21-activated HCR amplifier.
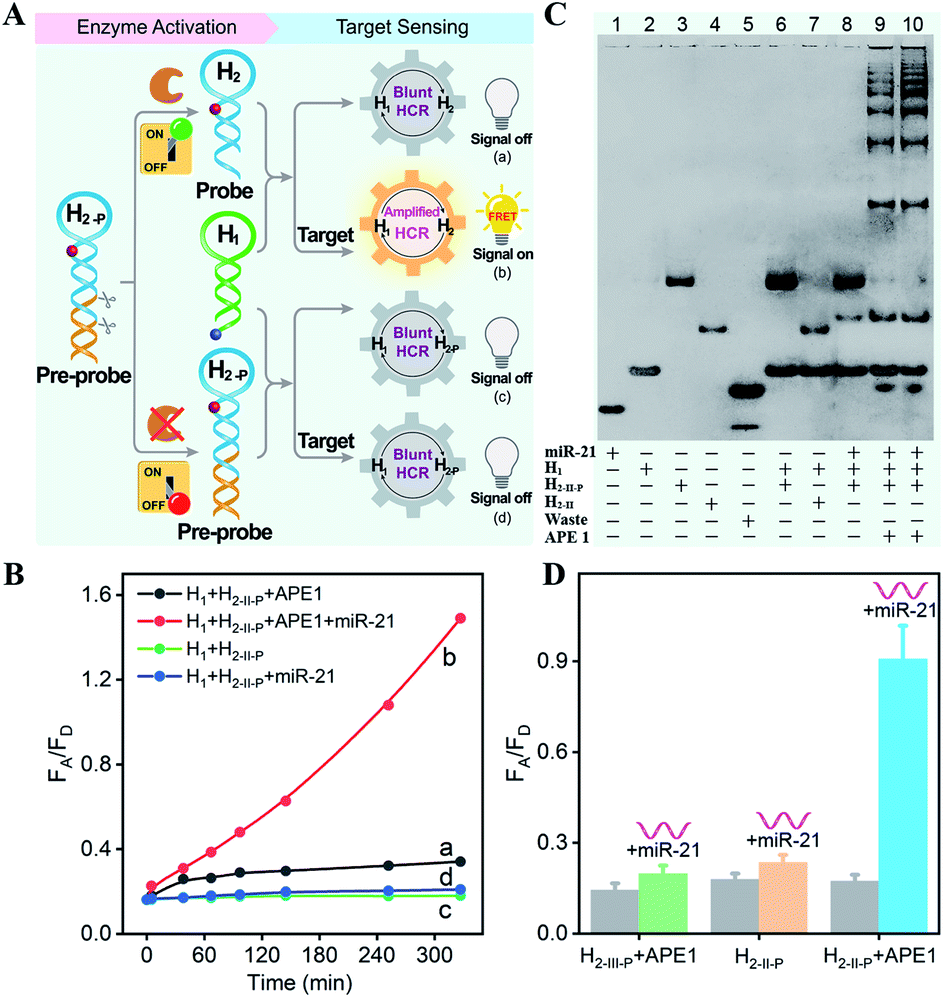 | ||
| Fig. 1 (A) Schematic of the feasibility of the sequential specific activated HCR amplification system. (B) Time-dependent FRET response of the APE1-liberated HCR system in the absence and presence of target. (C) Native gel electrophoresis characterization of the bioorthogonal activation HCR system. The “+” and “−” refer to the presence and absence of the corresponding components, respectively. (D) Evaluation of the specific regulation of HCR amplifier by fluorescence experiments based on the quantification of the fluorescence emission ratio of acceptor to donor (FA/FD). APE1-blunt groups: H1 + H2-III-P + APE1 and miR-21 + H1 + H2-III-P + APE1; intact groups (without APE1): H1 + H2-II-P and miR-21 + H1 + H2-II-P; APE1-regulated groups: H1 + H2-II-P + APE1 and miR-21 + H1 + H2-II-P + APE1. | ||
Having confirmed the feasibility of our sequentially and specifically activated HCR amplifier, its miR-21 sensing performance was assessed in order to provide a useful tool for sensitive imaging of miR-21 in cancer cells (Fig. 2A and S5A†). The summarized calibration curve of the fluorescence emission ratio of the acceptor to the donor (FA/FD) revealed a concentration-dependent tendency (Fig. 2B). From the derived linear relationship at a concentration range of 0–10 nM (R2 = 0.9997) (Fig. 2B inset), the limit of detection was calculated to be 66 pM according to the 3σ calculation method. For comparison, the sensing performance of the intact HCR system was also tested without enzymatic cleavage (Fig. S5B†). From the derived calibration curve (Fig. 2C), the limit of detection was calculated to be 2.15 nM from the linear relationship ranging from 0 to 20 nM (R2 = 0.9878) (Fig. 2C inset). It was apparent that the DNA circuitry was blocked efficiently with negligible signal leakage. The APE1 enzyme specifically recognized and exposed the active HCR probe from the inactive DNA pre-probe, thus facilitating the subsequent HCR-mediated miRNA sensing procedure. The specific APE1-modulated HCR amplifier enables the sensitive detection of miRNA and is expected to realize intracellular miRNA imaging, considering that the endogenous APE1 could guarantee the on-site activation of DNA circuity in live cells.
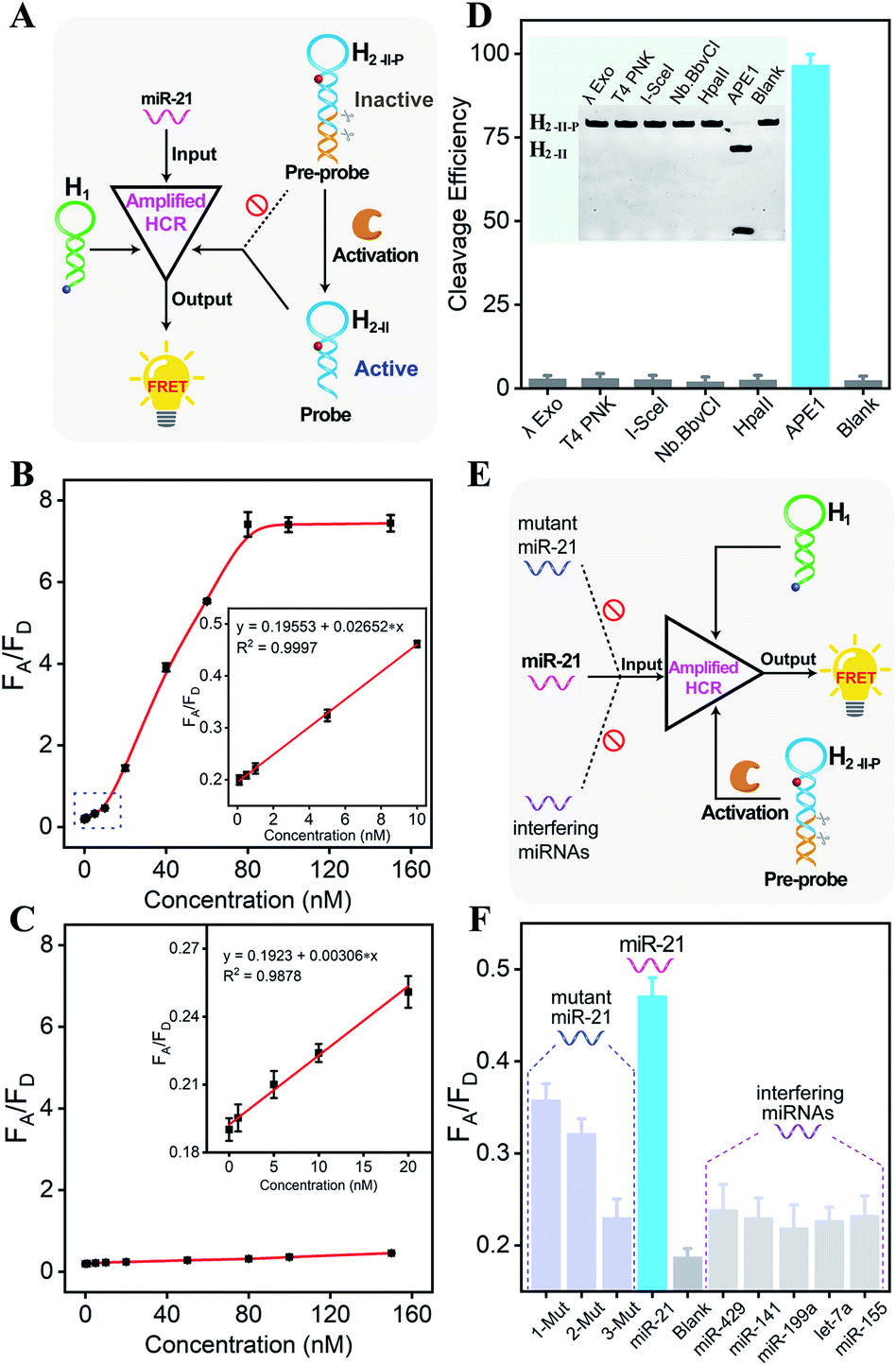 | ||
| Fig. 2 (A) Schematic of the comparison of different signal amplification properties of the APE1-regulated or intact HCR system. (B) Fluorescence transduction of the APE1-regulated HCR system in response to different concentrations of miR-21. Inset: the corresponding calibration curve. (C) Fluorescence transduction of the intact HCR system (without APE1 involvement) in response to different concentrations of miR-21. Inset: the corresponding calibration curve. (D) Denatured PAGE analysis of the specific recognition capability of APE1 (for detailed information, see the experimental section). The substrate cleavage efficiency was calculated using ImageJ software. (E) Schematic of the selectivity and specificity exploration of the HCR-amplified miR-21 detection platform. (F) Selectivity evaluation upon analyzing 10 nM miR-21, one-base mutant (1-Mut), two-base mutant (2-Mut), three-base mutant (3-Mut), and blank (no analyte). Specificity evaluation upon analyzing miR-21 and several interfering miRNAs (10 nM). Inset: Data are means ± SD (n = 3). All fluorescence intensities are referred to the fluorescence emission ratio of acceptor to donor (FA/FD). | ||
Because the aberrant expression and localization of APE1 enzyme are related to the development of various cancers, APE1 has attracted increasing attention as a significant biomarker in cancer prediction or prognostic applications.34,36 Considering the intricate biological matrixes during cellular delivery, the HCR-sensing platform could be specifically activated on-site by the overexpressed APE1 in cancer cells, which was expected to substantially improve the accuracy of miRNA detection in specific live cells. The APE1-specific activatable DNA circuitry was testified by both PAGE analysis (Fig. 2D) and a fluorescence assay (Fig. S6†). The initial PAGE analysis revealed almost complete exposure of the HCR pre-probe by APE1 enzyme (Fig. 2D), thus providing a solid basis for the subsequent HCR-amplified miRNA assay (Fig. S6†). Indeed, the fluorescence signal of the APE1-involved HCR differs greatly from that of other nuclease-treated systems, which is close to the background signal of the APE1-absent system. Therefore, the designed DNA circuit could be specifically cleaved by APE1, in sharp contrast to other possibly coexisting nucleases.
Then, the selectivity of the APE1-guaranteed HCR amplifier was assessed by introducing one-, two-, or three-base mutant miR-21 target (1-Mut, 2-Mut, and 3-Mut, respectively), as depicted in Fig. 2E. Interestingly, the fluorescence readout of miR-21 could be easily discriminated from these of mutant strands (Fig. 2F and S7†). The specificity was also evaluated by comparing the fluorescence readout of miR-21 with a series of interfering miRNAs, including miR-429, miR-141, miR-199a, let-7a, and miR-155. In contrast with the target miR-21, a much lower FRET readout was obtained from these interfering miRNAs, demonstrating the remarkable specificity of the APE1-regulated miRNA-sensing DNA circuit. To investigate the robustness of the present miRNA-sensing system, the miR-21 analysis system was also conducted in buffer containing 5% and 10% fetal bovine serum (Fig. S8†). The fluorescence response in diluted serum was comparable to that in ideal buffer, indicating the robustness of our DNA circuitry in complex biological fluids. In view of the promising selectivity, specificity, and anti-interference capability, the endogenous APE1-controlled HCR amplifier, as a versatile tool, shows great potential for facilitating more reliable and robust intracellular imaging applications.
Based on the above studies, the applicability of the APE1-guaranteed HCR amplifier for miR-21 imaging in living cells was further investigated by confocal laser scanning microscopy (CLSM). The fluorescence emission ratio of acceptor to donor (FA/FD) was adapted as a reliable visualization readout for improving the anti-interference ability of our miRNA-sensing system against the complex intracellular environment. The intracellular sensing capability of the sequentially and specifically activated HCR amplifier was explored in three kinds of cells with different APE1 and miR-21 expression levels in the cytoplasm, human breast cancer cells (MCF-7) with overexpressed APE1 and miR-21 profiles, human cervical cancer cells (HeLa) with moderate APE1 and miR-21 expression profiles, and human breast normal cells (MCF-10A) with negligible APE1 and miR-21 expression profiles.37–42 Herein, these functional HCR reactants were transfected into cells via a commercial transfection agent, lipofectamine 3000. The time-dependent increased fluorescence intensity confirmed the signal amplification process in live cells (Fig. S9†). The fluorescence intensity reached a saturated value after 4 h, which was thus chosen as the optimized incubation duration for live cells. On account of the aberrant APE1 expression in cancer cells, the endogenous APE1-modulated HCR amplifier was used for more reliable miRNA imaging, aiming to discriminate cancer cells from normal cells (Fig. 3A). As shown in Fig. 3B (for details, see Fig. S10†), a strong fluorescence signal was observed for cancer cells (MCF-7 and HeLa) transfected with the APE1-regulated HCR system (samples a and b, respectively). Meanwhile, the fluorescence readout was tremendously weak for normal cells (MCF-10A), with almost negligible expression of APE1 and miR-21 (sample c). Additionally, the APE1-blunt HCR system was similarly incubated with the different cancer cells (MCF-7 and HeLa) and normal cells (MCF-10A). In contrast, a visibly weak intracellular fluorescence signal was obtained in both cancer cells (samples d and e, respectively) and normal cells (sample f). These fluorescence results were further quantified and were summarized in Fig. 3C and D. Thus, the DNA circuit could only be specifically liberated by APE1, which is overexpressed in cancer cells, but remained inactive in normal cells. The pivotal role of the intrinsic endogenous APE1-programmed activation strategy was demonstrated for realizing cancer cell-selective miRNA imaging by our engineered DNA circuit.
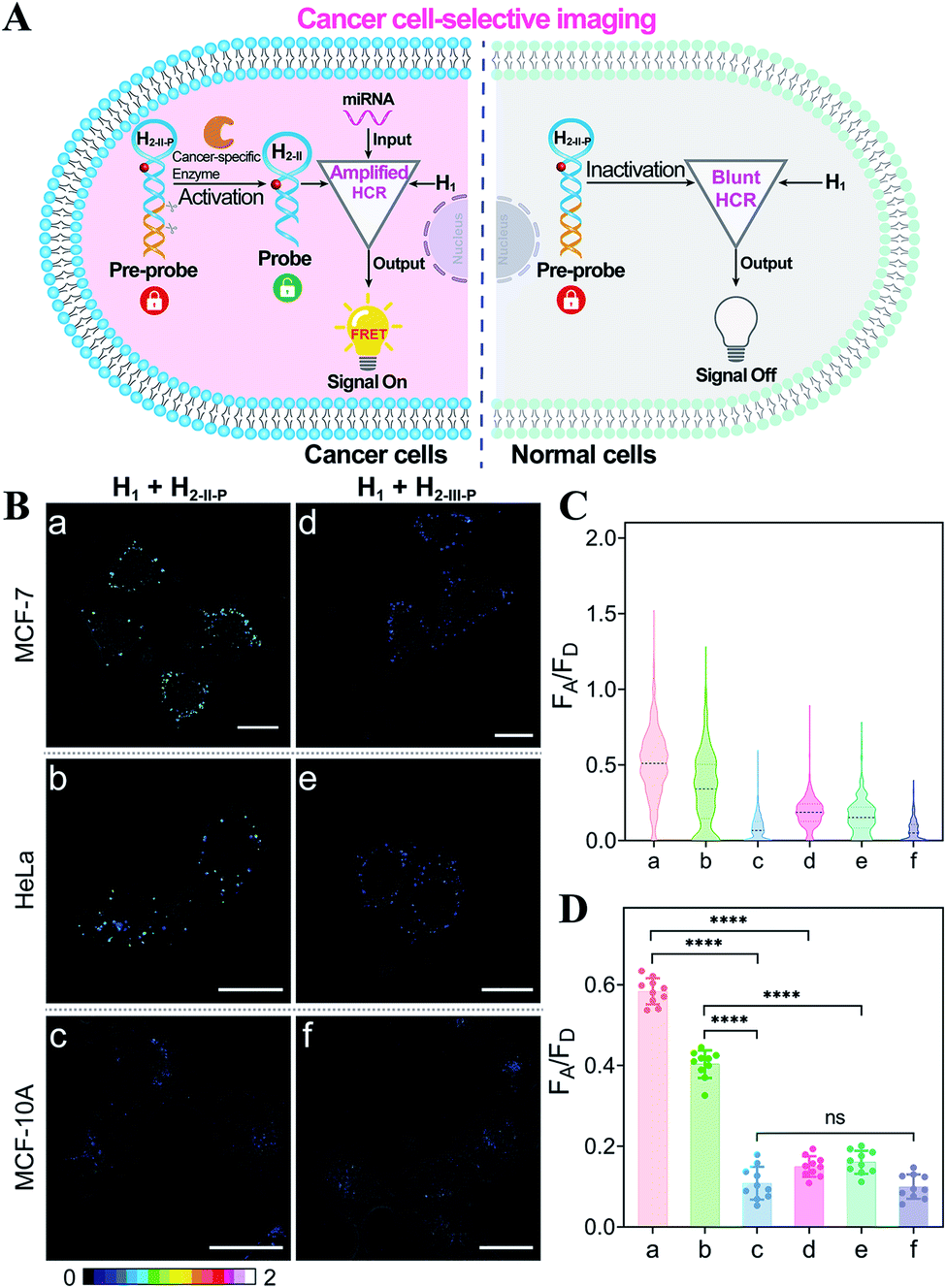 | ||
| Fig. 3 (A) Illustration of the endogenous APE1-guaranteed HCR-amplified miRNA imaging for discriminating cancer cells from normal cells. (B) Selective miR-21 imaging in living cells. Hairpin H2-II-P with an abasic site can be specifically recognized and liberated by APE1 for efficient reactant exposure, while hairpin H2-III-P without the abasic site cannot be recognized for reactant exposure. All scale bars correspond to 20 μm. (C) The relative fluorescence intensity distributions of the different cell samples in (B). (D) Statistical histogram analysis of the fluorescence readout (FA/FD) from numerous cell samples. ****p <0.0001, ns, not statistically significant (one-way ANOVA followed by Tukey's multiple comparisons test). | ||
The endogenous APE1-regulated HCR system was further investigated for monitoring cancer cells with varied expressions of miRNA (Fig. 4A). In order to upregulate and downregulate the expression of miRNA, the MCF-7 model cells were respectively pretreated with the miR-21 mimic or anti-miR-21 oligonucleotide,43 and then these pretreated cells were incubated with our proposed DNA circuit (Fig. 4B; for details, see Fig. S11†). As compared with the intact cells (sample a), an increased intracellular fluorescence signal was acquired in miR-21 mimic-pretreated cells (sample b), while an obviously declined fluorescence signal was elicited in anti-miR-21-inhibitor-pretreated cells (sample c). All of these fluorescence signals were quantified and are summarized in Fig. 4C and D. Clearly, the specific miRNA-sensing performance of our endogenous enzyme-guaranteed DNA circuit was demonstrated to distinguish cells with varied miR-21 expression profiles.
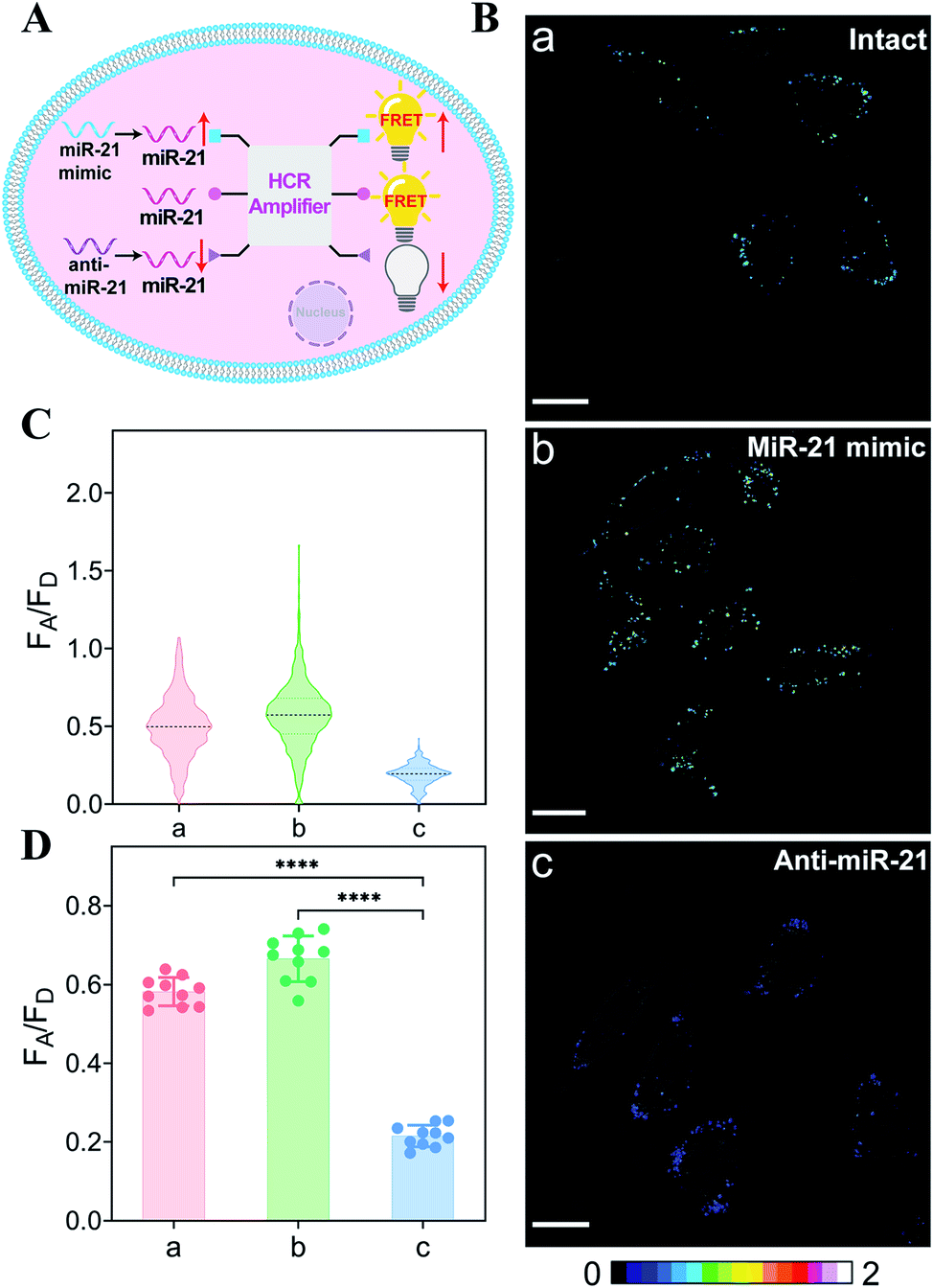 | ||
| Fig. 4 (A) Illustration of the endogenous APE1-modulated HCR-amplified miRNA imaging in MCF cells with different expressions of miR-21. (B) CLSM imaging of miR-21 in intact MCF-7 cells, the miR-21 mimic-pretreated MCF-7 cells (with upregulated miR-21), and the anti-miR-21-inhibitor-pretreated MCF-7 cells (with downregulated miR-21). All scale bars correspond to 20 μm. (C) The relative fluorescence intensity distributions of the different cell samples in (B). (D) Statistical histogram analysis of the fluorescence readout (FA/FD) from numerous cell samples. ****p <0.0001 (one-way ANOVA followed by Tukey's multiple comparisons test). | ||
The satisfying sensing performance of our on-site endogenous enzyme-controlled miRNA-imaging platform motivated us to explore its in vivo miR-21 imaging capacity in MCF-7 tumor-bearing mice, where the tumors were encoded with high expression of APE1 enzyme. Here, H1* was labeled with the Cy5/BHQ2 pair for whole-body fluorescence imaging (Fig. 5A). Moreover, the time-dependent in vivo visualization was monitored at different time points after the intratumoral injection of nude mice with lipo3000-loaded HCR mixture using an in vivo imaging system (IVIS). Liposomes represent a promising carrier for effectively delivering therapeutic agents.44 An increased fluorescence intensity was shown in the tumor sites that were treated with the bioorthogonal APE1-regulated HCR system (group a, Fig. 5B). The fluorescence signal intensified with prolonged incubation duration and reached the maximum value 8 h after injection. No significant fluorescence enhancement, however, was observed for tumor sites injected with the APE1-blunt HCR system (group b, Fig. 5B), thus highlighting the pivotal role of APE1 exposure in the on-site miRNA imaging with high reliability. In addition, the miR-21 inhibitor was pre-injected into tumor sites for downregulating miR-21 expression. A rather low fluorescence readout was acquired for these miRNA-inhibitor-treated mice by these APE1-regulated and APE1-blunt HCR systems (groups c and d of Fig. 5B, respectively), indicating their miRNA-specific sensing ability. Further quantification of these intratumoral imaging experiments showed that the APE1-regulated circuitry system (curve a, Fig. 5C) achieved 2.57-fold higher fluorescence intensity than the APE1-blunt circuitry system (curve b, Fig. 5C), 3.12-fold higher fluorescence readout than the APE1-regulated circuitry system pretreated with miR-21 inhibitor (curve c, Fig. 5C), or 3.51-fold higher than the APE1-blunt circuitry system pretreated with miR-21 inhibitor (curve d, Fig. 5C) at 8 h post-injection. These results further demonstrated that the DNA circuitry was specifically controlled on-site by the overexpressed APE1 enzyme to efficiently expose the initially blocked circuity reactant for selective miRNA imaging.
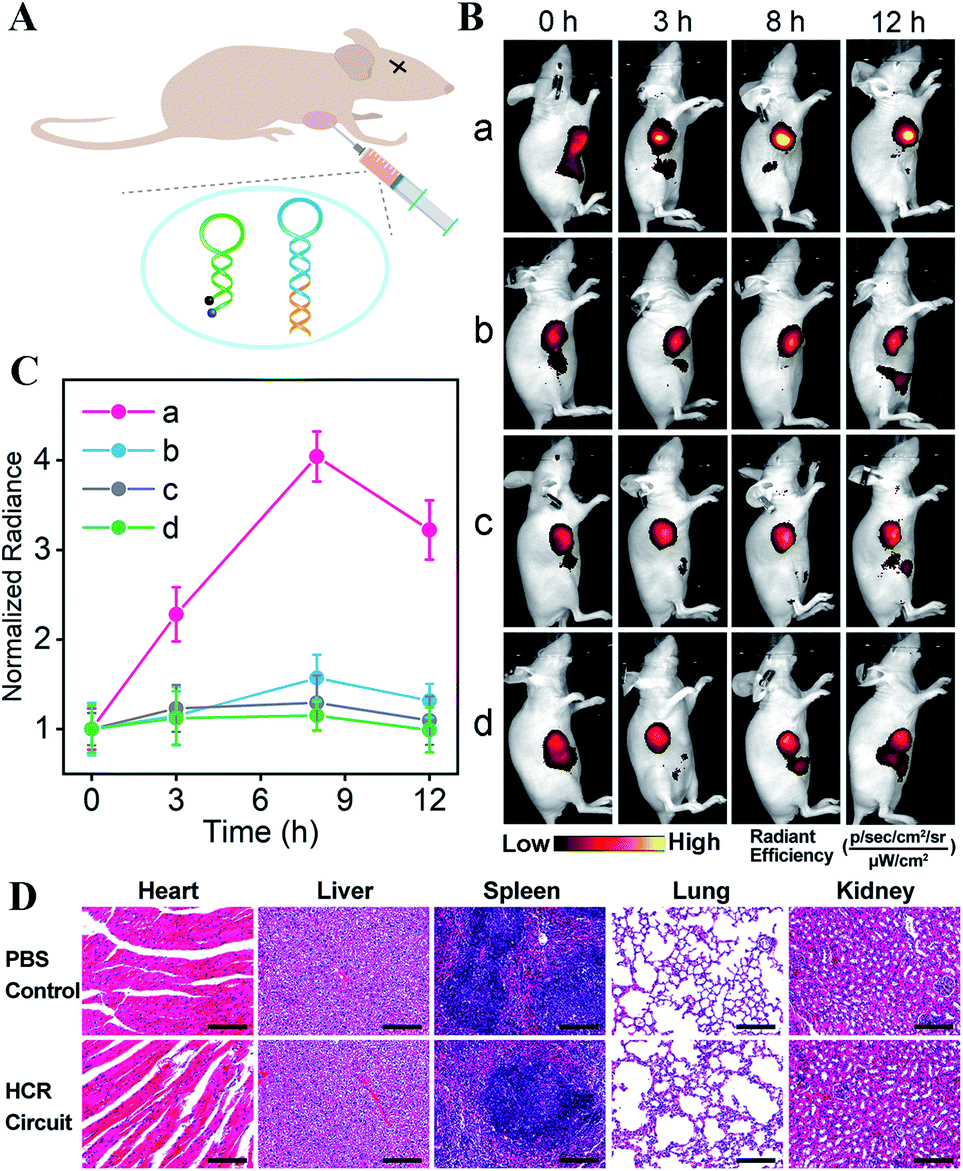 | ||
| Fig. 5 (A) Illustration of the on-site bioorthogonal APE1-regulated HCR circuitry system for in vivo miRNA imaging by intratumoral injection. (B) Whole-body fluorescence imaging of tumor-bearing mice at the indicated time points after injection of different systems: (a) the APE1-regulated HCR system, (b) the APE1-blunt HCR system, (c) the APE1-regulated HCR system after miR-21 inhibitor-pretreatment, (d) the APE1-blunt HCR system after miR-21 inhibitor-pretreatment. (C) Quantification of the intratumoral fluorescence readout signal in (B). Data are means ± SD (n = 3). (D) H&E staining analysis of the major organs harvested from nude mice administrated with the bioorthogonal APE1-guaranteed HCR circuitry system after 48 h; PBS was employed as the control group. All scale bars are 300 μm. | ||
These different HCR imaging systems were also systemically administrated into tumor-bearing nude mice through the tail vein to verify the in vivo enzyme-controlled imaging ability of our proposed functional DNA circuitry (Fig. S12A and B†). Quantitative analysis manifested that at the tumor site, the APE1-regulated circuitry system (curve a, Fig. S12C†) achieved 3.36-fold higher fluorescence intensity than the APE1-blunt circuitry system (curve b, Fig. S12C†), 3.54-fold higher fluorescence readout than the APE1-regulated circuitry system pretreated with miR-21 inhibitor (curve c, Fig. S12C†), and 3.56-fold higher fluorescence readout than the APE1-blunt circuitry system pretreated with miR-21 inhibitor (curve d, Fig. S12C†) at 8 h post-injection. The ex vivo imaging of the tumors and major organs was analyzed after 12 h of administration (Fig. S12D and E†). The tumor fluorescence intensity of the APE1-regulated HCR system was 2.80-fold higher than that of the APE1-blunt HCR system, 2.67-fold higher than that of the APE1-regulated HCR system pretreated with miR-21 inhibitor, and 3.33-fold higher than that of the APE1-blunt HCR system pretreated with miR-21 inhibitor. Moreover, almost no difference was observed in the normal organs for all these different systems, demonstrating the specific on-site activation of our functional circuitry. The high biocompatibility of our APE1-regulated DNA circuity system was further certified by H&E staining assay (Fig. 5D), hemolysis assay (Fig. S13†), and hematology and biochemical analysis (Fig. S14†). These results demonstrated the versatility and robustness of our proposed endogenous enzyme-guaranteed DNA circuit for realizing more challenging in vivo miRNA imaging applications.
The bioorthogonal regulation of DNA circuits provides a general and versatile toolbox to integrate with other different endogenous enzyme-guaranteed activation strategies to facilitate the on-site detection of various disease-specific biomarkers. Accordingly, a similar homing endonuclease, I-SceI enzyme, was also selected to activate the miR-21-sensing circuitry. I-SceI could induce minimal damage to most host genomes due to a special target recognition property.45 Here, the intracellular expression of I-SceI was achieved by transfecting the commercial plasmid pCBASceI into mammalian cells, as demonstrated by SDS-PAGE. Moreover, the transfection conditions were optimized to facilitate the following intracellular miRNA imaging experiments (Fig. S15†). The specifically I-SceI-regulated HCR circuitry system was constructed by introducing the corresponding DNA pre-probe (H2-III-P) encoded with the I-SceI-recognition site. The I-SceI-mediated circuitry regulation condition was firstly optimized (Fig. S16†), and the optimum catalytic conditions were chosen by incubating hairpin H2-III-P with 5 U mL−1 of I-SceI for 5 h at 37 °C, under which the substrate cleavage efficiency was higher than 90%. Then, the fluorescence experiment was performed to explore the miRNA-sensing performance by using the endogenously regulated DNA circuit (Fig. S17†). With I-SceI exposure, the time-dependent fluorescence profiles revealed that the HCR system proceeded rapidly to the miR-21 target (curve a), while a quite low fluorescence change was obtained in the absence of target (curve b). In contrast, without I-SceI, an ultralow fluorescence response was observed for the circuitry system regardless of the presence or absence of the target miR-21 (curves c and d, respectively). Thus, the functional circuitry was effectively restricted by the I-SceI-recognition site, and the I-SceI regulation was indispensable for motivating the efficient execution of the miR-21-sensing circuitry platform.
The miR-21-sensing performance of the present I-SceI-regulated HCR circuitry system was further evaluated. The miR-21-dependent fluorescence change was obtained (Fig. S18A†), and the limit of detection was determined to be 40.7 pM (3σ/slope) from the calibration curve (Fig. S18B†). Without I-SceI, however, the blocked DNA circuity displayed a poor response to the same miR-21 target (Fig. S18C†), and the limit of detection was calculated to be 2.43 nM (Fig. S18D†). Thus, the unexpected execution of our circuit was successfully restricted to avoid the potential signal leakage. Only with the assistance of I-SceI could the DNA circuit be liberated to sensitively detect the target miR-21, indicating promising applicability of the I-SceI-programmed DNA circuit for intracellular miRNA imaging. Subsequently, the I-SceI-modulated DNA circuitry system was similarly explored for miR-21 imaging in HeLa cells (Fig. S19A†). As expected, the generation of the fluorescence signal indeed proceeded via the control of I-SceI for only the I-SceI-expressed HeLa cells, which exhibited a highly distinct intracellular fluorescence readout (group a, Fig. S19B†), and not in the intact HeLa cells without I-SceI expression (group b, Fig. S19B†). Thus, the miRNA-sensing execution of the DNA circuit was indeed controlled by the endogenous I-SceI enzyme. In order to demonstrate the miR-21-specific imaging ability, the miR-21 inhibitor was similarly transfected into HeLa cells prior to the transfection of the DNA circuit. As expected, barely any detected fluorescence signal was observed in HeLa cells regardless of the presence or absence of I-SceI expression (groups c and d, respectively, Fig. S19B†). As a general and versatile sensing platform, our endogenous enzyme-programmed DNA circuitry showed great potential for realizing accurate and robust miR-21 imaging in complicated biological environments by eliminating the off-site leakage side effect.
Conclusion
In summary, we have engineered a bioorthogonal regulated DNA circuit for realizing on-site cancer cell-selective and amplified imaging of miRNA by means of the endogenous DNA repairing enzyme-controlled strategy. By ingeniously integrating the endogenous enzyme-recognizing duplex into a hairpin reactant, undesired signal leakage of the HCR circuit is substantially diminished via the additional endogenous enzymatic pre-selection procedure. Hence, this bioorthogonal regulation strategy provides a good opportunity to facilitate the application of DNA circuits in sensitive and selective live cell imaging. This endogenously activatable DNA circuit can be customized in disease diagnostics by introducing different enzyme-specific recognition sites. The sequential endogenous enzyme-preselected circuitry exposure and stringent target recognition provide a more versatile platform for in vivo bioimaging with high reliability and robustness. Additionally, the sequentially and specifically activated strategy could be similarly adapted to many other functional DNA circuits with improved anti-interference ability; thus, they hold great promise for clinical diagnosis and therapeutic evaluation.Data availability
All experimental data is available in the ESI.†Author contributions
F. Wang and Y. Chen conceived the study and designed the experiment. Y. Chen performed the main experiments and analysed the data. Y. Gao, Y. Shang, J. Shang and S. He assisted with cell experiments and modelling construction. S. Yu, R. Li and X. Gong assisted with the animal experiments and discussion. F. Wang and X. Liu revised and finalized the manuscript. All authors participated the writing – review and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The animal experiments were approved by the Animal Care and Use Committee of Wuhan University and complied with all relevant ethical regulations. This work is supported by National Natural Science Foundation of China (21874103 and 22074112) and the Central Funds Guiding the Local Science and Technology Development of Shenzhen (2021Szvup101).Notes and references
- K. Leung, K. Chakraborty, A. Saminathan and Y. Krishnan, Nat. Nanotechnol., 2019, 14, 176–183 CrossRef CAS PubMed.
- Z. Liu, F. Wang, X. Liu, Y. Sang, L. Zhang, J. Ren and X. Qu, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022769118 CrossRef CAS PubMed.
- W. Wei, X. He and N. Ma, Angew. Chem., Int. Ed., 2014, 53, 5573–5577 CrossRef CAS PubMed.
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901–986 CrossRef CAS PubMed.
- G. C. Gavins, K. Gröger, M. D. Bartoschek, P. Wolf, A. G. Beck-Sickinger, S. Bultmann and O. Seitz, Nat. Chem., 2021, 13, 15–23 CrossRef CAS PubMed.
- S. B. Ebrahimi, D. Samanta and C. A. Mirkin, J. Am. Chem. Soc., 2020, 142, 11343–11356 CrossRef CAS PubMed.
- Z. Qing, J. Xu, J. Hu, J. Zheng, L. He, Z. Zou, S. Yang, W. Tan and R. Yang, Angew. Chem., Int. Ed., 2019, 58, 11574–11585 CrossRef CAS PubMed.
- F. Wang, C.-H. Lu and I. Willner, Chem. Rev., 2014, 114, 2881–2941 CrossRef CAS PubMed.
- K. T. Kim, S. Angerani, D. Chang and N. Winssinger, J. Am. Chem. Soc., 2019, 141, 16288–16295 CrossRef CAS PubMed.
- J. Wei, H. Wang, X. Gong, Q. Wang, H. Wang, Y. Zhou and F. Wang, Nucleic Acids Res., 2020, 48, e60 CrossRef CAS PubMed.
- C.-P. Liang, P.-Q. Ma, H. Liu, X. Guo, B.-C. Yin and B.-C. Ye, Angew. Chem., Int. Ed., 2017, 56, 9077–9081 CrossRef CAS PubMed.
- C. Xue, S.-X. Zhang, C.-H. Ouyang, D. Chang, B. J. Salena, Y. Li and Z.-S. Wu, Angew. Chem., Int. Ed., 2018, 57, 9739–9743 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- Z. Cheglakov, T. M. Cronin, C. He and Y. Weizmann, J. Am. Chem. Soc., 2015, 137, 6116–6119 CrossRef CAS PubMed.
- H. Wu, T.-T. Chen, X.-N. Wang, Y. Ke and J.-H. Jiang, Chem. Sci., 2020, 11, 62–69 RSC.
- H. M. T. Choi, V. A. Beck and N. A. Pierce, ACS Nano, 2014, 8, 4284–4294 CrossRef CAS PubMed.
- J. Wei, X. Gong, Q. Wang, M. Pan, X. Liu, J. Liu, F. Xia and F. Wang, Chem. Sci., 2018, 9, 52–61 RSC.
- S. Bi, M. Chen, X. Jia, Y. Dong and Z. Wang, Angew. Chem., Int. Ed., 2015, 54, 8144–8148 CrossRef CAS PubMed.
- F. Xuan and I.-M. Hsing, J. Am. Chem. Soc., 2014, 136, 9810–9813 CrossRef CAS PubMed.
- L. Li, H. Xing, J. Zhang and Y. Lu, Acc. Chem. Res., 2019, 52, 2415–2426 CrossRef CAS PubMed.
- H. Chu, J. Zhao, Y. Mi, Y. Zhao and L. Li, Angew. Chem., Int. Ed., 2019, 58, 14877–14881 CrossRef CAS PubMed.
- F. Huang, M. Lin, R. Duan, X. Lou, F. Xia and I. Willner, Nano Lett., 2018, 18, 5116–5123 CrossRef CAS PubMed.
- Y. Shao, J. Zhao, J. Yuan, Y. Zhao and L. Li, Angew. Chem., Int. Ed., 2021, 60, 8923–8931 CrossRef CAS PubMed.
- X. Meng, H. Wang, M. Yang, J. Li, F. Yang, K. Zhang, H. Dong and X. Zhang, Anal. Chem., 2021, 93, 1693–1701 CrossRef CAS PubMed.
- Z. Zhang, Y. Jiao, Y. Wang and S. Zhang, Sci. Rep., 2016, 6, 29872 CrossRef CAS PubMed.
- J. Li, J. Wang, S. Liu, N. Xie, K. Quan, Y. Yang, X. Yang, J. Huang and K. Wang, Angew. Chem., Int. Ed., 2020, 59, 20104–20111 CrossRef CAS PubMed.
- X. Gong, J. Wei, J. Liu, R. Li, X. Liu and F. Wang, Chem. Sci., 2019, 10, 2989–2997 RSC.
- Y. Lin, Z. Yang, R. J. Lake, C. Zheng and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 17061–17067 CrossRef CAS PubMed.
- D. Yi, J. Zhao and L. Li, Angew. Chem., Int. Ed., 2021, 60, 6300–6304 CrossRef CAS PubMed.
- F. Chen, M. Bai, K. Cao, Y. Zhao, X. Cao, J. Wei, N. Wu, J. Li, L. Wang, C. Fan and Y. Zhao, ACS Nano, 2017, 11, 11908–11914 CrossRef CAS PubMed.
- J. Xue, F. Chen, M. Bai, X. Cao, P. Huang and Y. Zhao, Anal. Chem., 2019, 91, 4696–4701 CrossRef CAS PubMed.
- H. Zhang, S. Ba, J. Y. Lee, J. Xie, T.-P. Loh and T. Li, Nano Lett., 2020, 20, 8399–8407 CrossRef CAS PubMed.
- J. F. Alhmoud, J. F. Woolley, A.-E. A. Moustafa and M. I. Malki, Cancers, 2020, 12, 1050 CrossRef CAS PubMed.
- R. A. Caston, S. Gampala, L. Armstrong, R. A. Messmann, M. L. Fishel and M. R. Kelley, Drug Discovery Today, 2021, 26, 218–228 CrossRef CAS PubMed.
- J. Wei, H. Wang, Q. Wu, X. Gong, K. Ma, X. Liu and F. Wang, Angew. Chem., Int. Ed., 2020, 59, 5965–5971 CrossRef CAS PubMed.
- G. Tell, G. Damante, D. Caldwell and M. R. Kelley, Antioxid. Redox Signaling, 2005, 7, 367–384 CrossRef CAS PubMed.
- Y. Zhang, Y. Deng, C. Wang, L. Li, L. Xu, Y. Yu and X. Su, Chem. Sci., 2019, 10, 5959–5966 RSC.
- H. Zhang, S. Ba, Z. Yang, T. Wang, J. Y. Lee, T. Li and F. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 13634–13643 CrossRef CAS PubMed.
- F. Chen, M. Bai, K. Cao, Y. Zhao, J. Wei and Y. Zhao, Adv. Funct. Mater., 2017, 27, 1702748 CrossRef.
- S. Peng, Z. Tan, S. Chen, C. Lei and Z. Nie, Chem. Sci., 2020, 11, 7362–7368 RSC.
- K. Shi, B. Dou, C. Yang, Y. Chai, R. Yuan and Y. Xiang, Anal. Chem., 2015, 87, 8578–8583 CrossRef CAS PubMed.
- F. Yang, T.-T. Zhang, S.-S. Li, P. Song, K. Zhang, Q.-Y. Guan, B. Kang, J.-J. Xu and H.-Y. Chen, Anal. Chem., 2017, 89, 10239–10247 CrossRef CAS PubMed.
- L. Zhou, M. Gao, W. Fu, Y. Wang, D. Luo, K. Chang and M. Chen, Sci. Adv., 2020, 6, eabb0695 CrossRef CAS PubMed.
- P. B. Jeremy, B. Hanna, N. H. Margaret, G. Audrey, M. Marco, G. Dhanu, L. Taavi, V. Hadi, K. E. Elin, M. S. Molly and E.-A. Samir, Delivery of Oligonucleotide Therapeutics: Chemical Modifications, Lipid Nanoparticles, and Extracellular Vesicles, ACS Nano, 2021, 15, 13993–14021 CrossRef PubMed.
- J. B. Doyon, V. Pattanayak, C. B. Meyer and D. R. Liu, J. Am. Chem. Soc., 2006, 128, 2477–2484 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, experimental optimizations, extensive cellular imaging, and additional figures and schemes. See DOI: 10.1039/d1sc05214d |
| This journal is © The Royal Society of Chemistry 2021 |