
Room-temperature conversion of the photoelectrochemical oxidation of methane into electricity at nanostructured TiO2†
Yanir
Kadosh
,
Eli
Korin
and
Armand
Bettelheim
 *
*
Department of Chemical Engineering, Ben-Gurion University of the Negev, Be'er Sheva, Israel. E-mail: armandb@bgu.ac.il
First published on 16th October 2020
Abstract
The energy potential of methane is restrained by the energy input required to break its C–H bond. Therefore, most of the energy conversion processes of methane use thermochemical activation which is highly endothermic. The present report demonstrates the effective photoelectrochemical activity of a TiO2 nanotube arrays photoanode towards methane oxidation in acidic electrolyte and ambient conditions. The examined photoanode exhibits a higher photocurrent density response in the presence of methane as compared to that obtained in its absence (0.54 vs. 0.27 mA cm−2, respectively). Products characterization reveals a relatively high faradaic efficiency towards the formation of CO2 and formic acid (72 and 16% at 0.3 V vs. RHE, respectively). These results are correlated to the role of the special surface architecture of the nanotube arrays in dictating the reaction pathways. The first time room-temperature operation of a solar driven fuel cell (photo-fuel cell), in which methane oxidation is converted to electricity, is also demonstrated. This device performing with an acidic electrolyte or as a gas phase photo-fuel cell exhibited output maximum power densities of 69 and 82 μW cm−2, respectively.
Introduction
Methane is known for its great potential as an energy source, it is abundant, chemically versatile, and has a small environmental footprint.1 However, methane is also known for its inert chemical nature. Its symmetrical chemical structure and the absence of polarity result in one of the highest C–H bond energies among hydrocarbons (434 kJ mol−1).2,3 Methane is also in a gaseous state at room temperature, which makes it harder for utilization. Therefore, extracting its chemical potential can be achieved by an energy conversion process. Such a process can either convert methane's chemical energy into a useful derivative, such as oxygenates and higher hydrocarbons, or into electricity in a fuel cell device.4–6Currently, steam reforming is the most viable way for the conversion of methane.7 In this process a mixture of CO and H2 (“syngas”) is produced from a reaction of methane with steam. The obtained mixture reacts in a further reaction step to produce valuable products, such as oxygenates or longer hydrocarbons. The major challenges involved in this process originate from the steam reforming reaction. This step is highly endothermic and necessitates high energy input (>650 °C).8
Shifting the conversion route from thermochemical to electrochemical is of a great interest. Electrochemical processes can drive catalytic reactions at moderate temperatures by reducing the activation energy barrier with applied potential.9 Particularly, photoelectrochemical (PEC) processes can dramatically reduce the activation barrier by the interaction with light.10 The use of a semiconductor photoanode in a PEC system can produce highly reactive species (holes) upon irradiation.11
The PEC oxidation of liquid fuels, such as methanol, has been thoroughly investigated,12 but only a very few reports aim for the activation of methane's C–H bond at moderate temperatures.13,14 Recently, the PEC room temperature homocoupling of methane to form ethane using a WO3 gas diffusion photoanode under an applied voltage of 1.2 V has been reported.14 Unlike the scarcity of PEC methane activation reports, photochemical processes of methane oxidation (in the absence of an external electric field) gained more attention in the literature and a variety of photocatalysts have been investigated. Dispersed oxides (such as Al2O3, Al2O3–TiO2, MgO/Al2O3 and Ga2O3)15–18 supported on silica and Zn+-modified zeolite19 were found to promote the photocatalytic non-oxidative coupling of methane to ethane. More works reported the partial photocatalytic oxidation of methane to oxygenates using Bi (Bi2WO6, BiVO4 and Bi2WO6/TiO2),20 Ti (FeOx/TiO2,21 TiO2), W (WO3)22 and Ni (NiO)22 based photocatalysts.
The present report deals with the investigation of the capability of a TiO2 nanotube arrays (TiO2NTA) photoanode to drive the PEC oxidation of methane. The unique architecture of the nanotube arrays (NTA) enables the facile diffusion of reactants to the TiO2 surface, inhibits recombination of holes and electrons, and the small wall thickness of the nanotubes reduces the resistance of charge carriers along the nanotubes.23–27 We show that these features are most probably responsible for the more complete ambient PEC oxidation of CH4 (CO2 as main product) than for such a process performed by TiO2 prepared by atomic layer deposition (ALD),13 or as demonstrated here, for the compact TiO2 layer remaining after the NTA removal. Encouraged by these results, this led us to consider examining the concept of the galvanic energy conversion of methane to electricity using a solar driven fuel cell (photo-fuel cell, PFC). Such devices use solar energy to produce electricity by oxidizing organic substances which serve as fuels and which are preferably oxidized to CO2.28 Although solid oxide fuel cells are known for their ability to consume methane as a fuel at high temperatures (650–1100 °C),29 we demonstrate here for the first time that it is possible to operate at room temperature a PFC which converts methane's chemical energy into electricity with significant power density and long term stability.
Experimental
Photoanode fabrication and characterization
The TiO2NTA photoanode was fabricated by anodization of a Ti foil. The anodization process was carried out in a mixture of ethylene glycol (anhydrous, 99.8%, Sigma Aldrich) and deionized water (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 vol%) with 0.3 wt% NH4F (ACS reagent, ≥98.0%, Sigma Aldrich). A titanium foil (0.25 mm, 99.7%, Sigma Aldrich) was first washed by a sonication in acetone, ethanol and deionized water, each for 15 min, and then dried in air. Then the titanium foil and a Pt mesh were connected to the anode and cathode ports, respectively, of a DC power supply (N5771A – KEYSIGHT) and kept 2 cm apart. A constant voltage of 30 V was applied between the electrodes for 1.5 hour while the solution was stirred by a magnetic stirrer. After the anodization process, the titanium foil was immersed in deionized water for 1 hour and was left to dry overnight. Subsequently, the anodized foil was annealed at 500 °C in air for 1 hour. The morphology and the nanostructure of the photoanode was examined by high resolution scanning electron microscope (HRSEM, FEI Verios) and transmission electron microscope (TEM, Tecnai 12G2 TWIN, 120 kV). The crystal structure was examined by PANalytical's Empyrean multipurpose diffractometer. Diffuse reflectance spectrum (DRS) of the photoanode was recorded by Ocean Optics UV-Vis spectrophotometer (DH-2000-BAL) using a reflectance spectroscopy accessory (QR400-7-UV/VIS). Calibration was performed with a Spectralon standard (SRS-99-010). To assess the impact of the presence of nanotube arrays on the process, photoanodes were also examined after the nanotubes were peeled off by an adhesive tape.30 The adhesive tape was applied to the top surface of the photoanode and then removed mechanically. Subsequently, the photoanode was washed with acetone and deionized water and was left to dry overnight.
:10 vol%) with 0.3 wt% NH4F (ACS reagent, ≥98.0%, Sigma Aldrich). A titanium foil (0.25 mm, 99.7%, Sigma Aldrich) was first washed by a sonication in acetone, ethanol and deionized water, each for 15 min, and then dried in air. Then the titanium foil and a Pt mesh were connected to the anode and cathode ports, respectively, of a DC power supply (N5771A – KEYSIGHT) and kept 2 cm apart. A constant voltage of 30 V was applied between the electrodes for 1.5 hour while the solution was stirred by a magnetic stirrer. After the anodization process, the titanium foil was immersed in deionized water for 1 hour and was left to dry overnight. Subsequently, the anodized foil was annealed at 500 °C in air for 1 hour. The morphology and the nanostructure of the photoanode was examined by high resolution scanning electron microscope (HRSEM, FEI Verios) and transmission electron microscope (TEM, Tecnai 12G2 TWIN, 120 kV). The crystal structure was examined by PANalytical's Empyrean multipurpose diffractometer. Diffuse reflectance spectrum (DRS) of the photoanode was recorded by Ocean Optics UV-Vis spectrophotometer (DH-2000-BAL) using a reflectance spectroscopy accessory (QR400-7-UV/VIS). Calibration was performed with a Spectralon standard (SRS-99-010). To assess the impact of the presence of nanotube arrays on the process, photoanodes were also examined after the nanotubes were peeled off by an adhesive tape.30 The adhesive tape was applied to the top surface of the photoanode and then removed mechanically. Subsequently, the photoanode was washed with acetone and deionized water and was left to dry overnight.
Photoelectrochemical measurements
The photoelectrochemical experiments were performed with a Gamry (G™300) potentiostat in a custom made three-electrode sealed cell. The experiments were conducted at room temperature in 0.05 M H2SO4 solution, using a TiO2NTA, Pt wire and Ag/AgCl (3 M NaCl) as the working, auxiliary and reference electrodes, respectively. The working electrode was positioned in front of a quartz window, through which it was illuminated by a Newport Oriel Hg(Xe) lamp at a light intensity of 100 mW cm−2. Prior to the experiments, the solution was purged by the desired gas (methane or argon) for at least half an hour in order to remove dissolved oxygen. The effect of pH on the photoanode performance was examined in 0.1 M KOH and 0.1 M phosphate buffer solutions (pH = 13 and 6.4, respectively). All the measured potentials were converted to the reversible hydrogen electrode (RHE) scale using the following equation:| E(vs. RHE) = E(measured by Ag/AgCl) + 0.197 V + 0.059pH | (1) |
The chopped light linear sweep voltammetry (LSV) was carried out at a scan rate of 5 mV s−1. The dark–light time intervals were 10 s. Chronoamperometry (CA) was conducted while Ar or CH4 was purged into the solution at a flow rate of 20 mL min−1. Electrochemical impedance spectroscopy (EIS) measurements were performed at 0.3 V by applying a sinusoidal voltage of 10 mV, and the spectra were recorded in the frequency range from 100 kHz to 10 mHz. The EIS 300 software (Gamry) was used for data collection, and the obtained impedance plots were fitted with the chosen equivalent circuit by Echem Analyst (Gamry) software. Two trapping experiment sets, in the absence and presence of methane, were conducted at 0.6 V. Each set of experiments involved the addition of a specific scavenger at concentration of 0.2 mM to the PEC system. The experiments were performed using CA while switching between dark and light repeatedly at intervals of 100 s. Products characterization was conducted in a batch mode operation by applying different biased potentials for 2 hours at 1 atm. Gas samples were taken from the headspace with a 0.5 mL gas-tight syringe and were injected manually to a gas chromatograph (GC) column equipped with a flame ionization (with methanizer) and thermal conductivity detectors. Detection of dissolved oxygen as the competitive by-product was conducted using a dissolved oxygen probe (Thermo Scientific Orion™-9708) connected to a pH meter.
Analysis of liquid products
The formation of methane oxygenated products in the solution was verified and quantified by 1H-NMR at the end of the batch mode operation by a procedure we reported previously.31 This analysis was performed with a Bruker AVANCE III 500 MHz spectrometer equipped with a broad band 1H decoupling probe (BBO) at room temperature. Water suppression was achieved using the Bruker pulse program zgpr. A relaxation delay of 10 s was employed, and the number of scans was typically 56. Chemical shifts and products concentrations were obtained using DMSO as an internal standard with known concentration.Photo-fuel cell
A simple home-made cell was constructed. The cell was comprised of two glued polystyrene cuvettes, with a Nafion 117 (Ion Power, Inc.) membrane (thickness of ∼175 μm) in the middle, separating between the anodic and the cathodic chambers. A quartz window was positioned in front of the photoanode for the transition of UV light. A cooling jacket was constructed around the cell and it was maintained at room-temperature by a thermostat. Both chambers were filled with 0.05 M H2SO4 and the electrical circuit was connected between the TiO2NTA photoanode and a platinum mesh cathode. CH4/Ar mixtures and oxygen were supplied to the anodic and the cathodic chambers respectively, at a flow rate of 10 mL min−1. The steady state potential between the electrodes was measured using a voltmeter and rheostat. The gaseous phase products were analyzed by GC from gas samples collected by a sampling loop that was connected to the anodic chamber outlet stream. The electrolyte from the anodic chamber was analyzed by 1H-NMR. A structure of a gas phase photo-fuel cell using an anodized Ti fiber felt gas diffusion photoanode was also designed (details in p. S11†).Results and discussion
The nanotubes obtained by anodization, as characterized by SEM and TEM, are with an approximate length of 2.3 ± 0.2 μm, wall thickness of 20 ± 2 nm, and diameter of 70 ± 1 nm (Fig. 1A and B). The Raman spectrum of the photoanode (Fig. S1A†) exhibits four bands located at 198, 396, 517 and 638 cm−1 and a very sharp and intense peak at 148 cm−1 which are associated with the anatase phase.32 The X-ray diffraction (XRD) pattern (Fig. 1C) shows a strong peak at 25.3° that corresponds to the (101) plane of the anatase phase. In addition, more anatase planes can be seen at 38° and 48° respectively.33,34 TiO2 is known by its wide band gap that requires UV light for the photogeneration of electron–hole pairs in the space charge layer.35 The obtained photoanode is characterized by an estimated band gap of 3.2 eV, as measured by diffuse reflectance UV-Vis absorbance (Fig. S1B†) and in accordance with reported values.35 | ||
| Fig. 1 (A) High and low (inset) magnification of SEM images, (B) TEM image, and (C) XRD characterization of the prepared TiO2NTA photoanode. | ||
The PEC behavior of the TiO2NTA photoanode was tested at room temperature in a half-cell configuration (all potentials are referred to RHE). Linear sweep voltammograms (LSVs) of the photoanode, performed under chopped illumination of 100 mW cm−2 and conducted in a solution of 0.05 M H2SO4 with a continuous flow of inert gas (Ar) or CH4, are presented in Fig. 2A. It can be seen that the chopped light LSV curve in the presence of CH4 exhibits a significant current density increase as compared to that obtained in its absence (saturation current densities of 0.54 and 0.27 mA cm−2, respectively). When shifting to dark, the current rapidly decreased close to null values for both cases. However, the spike-like feature observed after illumination was observed only in the absence of CH4 (Fig. 2A and zoom-in/Fig. S2†). This suggests that methane might serve as a hole scavenger and that recombination processes are probably inhibited by its presence.34 CA at a biased potential of 0.3 V showed reversibility as well as fast response of the photoanode when switching the gas flow from Ar to methane (steady state currents obtained within ∼2 min), as displayed in Fig. 2B.
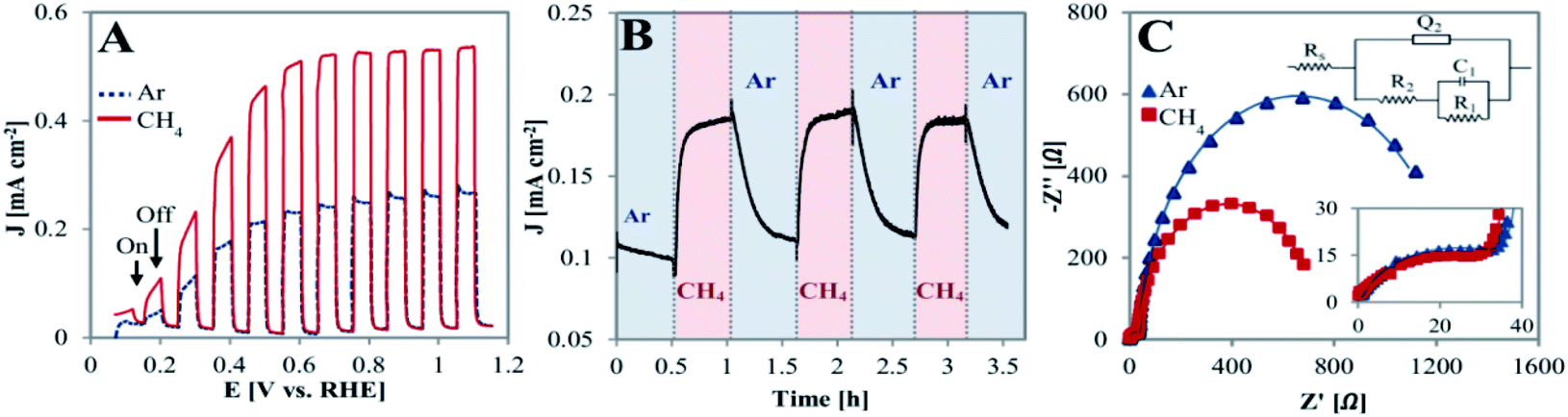 | ||
| Fig. 2 Photoelectrochemical oxidation of methane by a TiO2NTA photoanode at room temperature in 0.05 M H2SO4 under illumination intensity of 100 mW cm−2. (A) Chopped light LSV for TiO2NTA in the absence (Ar, dashed blue line) and presence (solid red line) of methane at scan rate of 5 mV s−1. (B) CA curve for TiO2NTA biased at 0.3 V with alternative shifting the Ar/CH4 flow gas. (C) EIS Nyquist plot obtained for TiO2NTA biased at 0.3 V under illumination in the absence and presence of methane (insets: equivalent circuit and zoom-in the high frequency region. Fitted model data are presented by solid lines). | ||
The charge transfer nature of the semiconductor/electrolyte interface was evaluated by electrochemical impedance spectroscopy (EIS). Fig. 2C depicts the Nyquist plots for the TiO2NTA photoanode in the absence and presence of methane during illumination at 0.3 V. It can be seen that the Nyquist plot is characterized by two semicircles. Based on this observation, the equivalent circuit adopted for the data consists of the electrolyte resistance in series with two RC components (inset in Fig. 2C, fitting parameters in Table S1†). These two components are needed to take into account for the resistive–capacitive processes occurring at the interface of photoanode: charge transfer at the semiconductor/electrolyte interface and charge transport in the oxide layer (R1C1 and R2Q2 elements, respectively).36–39 While the resistance of the high frequency semicircle in the presence of methane is almost identical to the one in its absence (R2 = ∼43 and 46 Ω, respectively, zoom-inset in Fig. 2C), the presence of methane clearly decreases the charge transfer resistance at the semiconductor/electrolyte interface (R1 = ∼0.7 and 1.2 kΩ, in the presence of CH4 and its absence, respectively). Enhancement of charge transfer by methane may indicate the ability of CH4 in capturing holes and inhibiting charge recombination.
In order to determine the nature of the active species in the PEC oxidation reaction of methane, trapping experiments of these species were performed using 0.2 mM of tert-butyl alcohol (TBA), benzoquinone (BQ) or triethanolamine (TEOA) as ˙OH, ˙O2− and h+ scavengers, respectively.40 The photocurrent responses were deduced from CA curves, as presented in Fig. S3,† and comparison of their values in the absence and presence of methane (Fig. 3A) indicate the different behavior of the trapping agents: while BQ decreases the photocurrents both in the absence and presence of CH4, these are increased in the presence of TBA or TEOA. Methane in the absence of scavenger increases current by liberating more electrons to the external circuit. The opposite effect of BQ is explained by the fast removal of ˙O2− radicals in both environments, which in turn accelerates O2 reduction, and eventually reduces the number of electrons that flow in the external circuit.41 The increase of current in both environments by TBA or TEOA seems to indicate that each of them may compete with CH4 as a scavenger of the PEC oxidation reaction products. However, the higher current response observed in the presence of methane when adding TEOA (current increment of 26.3% as compared to 9.6% for TBA) suggests a more crucial role of h+ than ˙OH radicals in governing the PEC methane oxidation reaction.
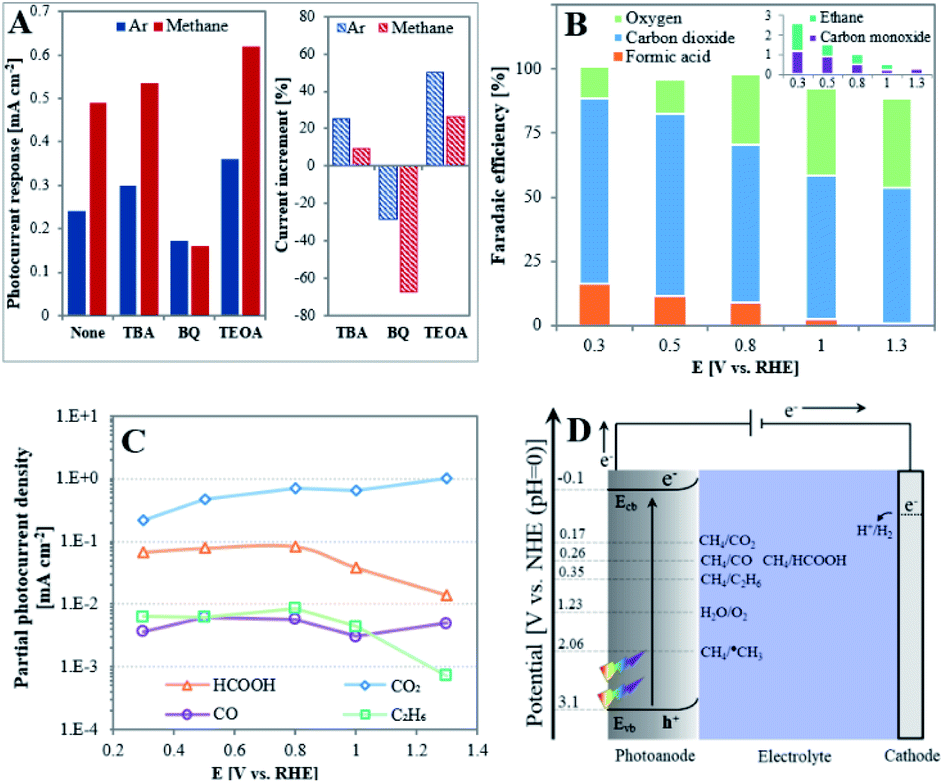 | ||
| Fig. 3 (A) Photocurrent responses in the absence (blue) and presence of methane (red) with 0.2 mM trapping agents obtained using TiO2NTA in 0.05 M H2SO4 at room temperature and under illumination intensity of 100 mW cm−2 (left image) and current increment after adding the trapping agent (right image); (B) FEs of the main products of methane PEC oxidation at different applied potentials (inset: results obtained for minor products). (C) Logarithmic scale of the partial photocurrent density of methane PEC oxidation products as function of the applied potential. (D) Potential scheme for the photoelectrochemical processes. | ||
The evaluation of the products obtained in 0.05 M H2SO4 by methane oxidation on an illuminated TiO2NTA photoanode was investigated at different applied biased potentials in a batch PEC cell maintained at room temperature. Fig. S4† presents the chronoamperograms obtained at different applied potentials. It can be seen that a significant higher PEC response is observed at higher bias potential due to its effect on decreasing hole–electron recombination. Stable current densities were obtained with no visible substantial decay over the examined time period. Gas phase samples analyzed by GC indicated the formation of CO2, O2, CO and C2H6 (Fig. S5†). Formic acid (with traces of methyl hydroperoxide, CH3OOH) was detected in the aqueous phase by 1H-NMR (Fig. S8†). Fig. 3B depicts the faradaic efficiencies (FEs) of the products as function of the applied potential. It can be seen from Fig. 3B that the FE of carbon-based products increases and FE of oxygen obtained by water oxidation decreases at low biased potentials. Under the present experimental conditions, a remarkable 91% FE of carbon-based products is obtained at 0.3 V. This value is quite high when considering the low solubility of methane in the reaction aqueous medium.42 Consequently, at 1.3 V the FE of carbon-based products decreases to 54%. Fig. 3B also shows that carbon dioxide is the main product in the whole potential range. This differs from the behavior of TiO2 photoanodes obtained by ALD which have been reported to generate CO at low applied potential, possibly due to the presence of Ti3+ sites, while CO2 formation was obtained at high potential.13 This seems to indicate that the unique surface of NTA plays a key role in dictating the CH4 oxidation mechanism. In this respect, the thin wall of the nanotubes which is in the magnitude of the space charge layer can provide a path to channel the photoexcited electrons in the vertical direction.24,25,43 This unique feature enhances the mobility of the charge carriers even at low applied bias,44 which in turn alters the reaction mechanism toward the complete oxidation of methane.
Fig. 3C correlates between the partial photocurrent density of each product (Jproduct,i) and the applied potential. In average, JCO2 is higher than JHCOOH by an approximate order of magnitude and by two orders of magnitude than JCO and JC2H6. This means that the formation of CO2 by TiO2NTA is kinetically favored over the rest of the products in the examined potential window while JCO seems to be independent on the applied potential. However, although JHCOOH and JC2H6 are almost constant at E ≤ 0.8 V, their values significantly decrease at higher potentials. This sharp decrease, occurring once saturation currents are reached, are consistent with the tendency of hydrocarbons and their oxygenated derivatives to undergo degradation at the harsh oxidative conditions existing at high potentials.
An energy scheme for the PEC processes is depicted in Fig. 3D. It has been established that upon illumination, holes generation in metal oxide semiconductors electrodes, such as TiO2, initiates oxidation processes on the surface/electrolyte interface.11,45 The external bias potential in this half-cell configuration facilitates the photoinduced charge separation at the anode to produce carbon-based products and transfer of electrons to the cathode accompanied by hydrogen formation. Holes can react with methane to form a methyl radical in the following reaction:1,10
| CH4 + h+ → ˙CH3 + H+, E0 = +2.06 V vs. RHE46 | (2) |
The redox couple potential of CH4/˙CH3 is the energy barrier that needs to be overcome in order to drive the conversion of methane.10 Under illumination of UV light, the high band gap energy of TiO2 anatase results in a valence band (VB) potential of 3.1 V (ref. 47) which is sufficiently higher than E0(CH4/˙CH3) to drive reaction 2. Table S2† summarizes the redox potentials of the reactions leading to oxygenated products (potentials in the 0.17–0.35 V range). It can be seen that they can all occur since they are within the boundaries of the TiO2 VB potential range and that the pathways leading to CO2, HCOOH and CO as products are thermodynamically favored over C2H6 which results from homocoupling of methyl radicals. Furthermore, the present experimental setup using an aqueous electrolyte has also important impact on the reaction pathway. The formation of methyl radical in the presence of water has been considered to be the route for oxygenated products2,14 which also explains the formation of C2H6 as a minor product (Fig. 3B).
To show that the unique nanostructure surface of the TiO2NTA plays an important role in dictating the PEC behavior toward CH4 oxidation, experiments were conducted with TiO2NTA photoanodes after the nanotubes have been peeled off. An oxide barrier layer is left and it is characterized by a pattern of pits (Fig. 4A) with an average diameter (100 ± 20 nm) close to that of the peeled nanotubes (70 ± 1 nm). The voltammograms obtained under illumination for such a photoanode (Fig. 4B) show only a moderate increase of saturation photocurrent density (Jlight − Jdark) in the presence of methane in comparison to that obtained in its absence (0.31 and 0.24 mA cm−2, respectively), indicating that the photocurrent response in both cases results mainly from water oxidation. Moreover, the TiO2 layer in this case behaves differently from TiO2NTA as deduced from EIS (Fig. 4C and Table S1†). The same equivalent circuit, as suggested for TiO2NTA (inset of Fig. 2C), was adapted to the EIS data obtained for TiO2 devoid of NTA. The fit reveals that the charge transfer resistance of the compact oxide layer (R2) is ∼twofold higher in the presence of the NTA than in their absence, probably as the result of the additional oxide layer supplied by the surface area of the tubes bottom. However, the charge transfer resistance at the semiconductor/electrolyte interface (R1) is higher in the absence of NTA than it is in the presence of the nanotubes (∼1.9 and 1.2 kΩ, respectively) in Ar saturated solution and further increases to ∼2.1 kΩ in the presence of methane (instead of a decrease to 0.7 kΩ for TiO2NTA).
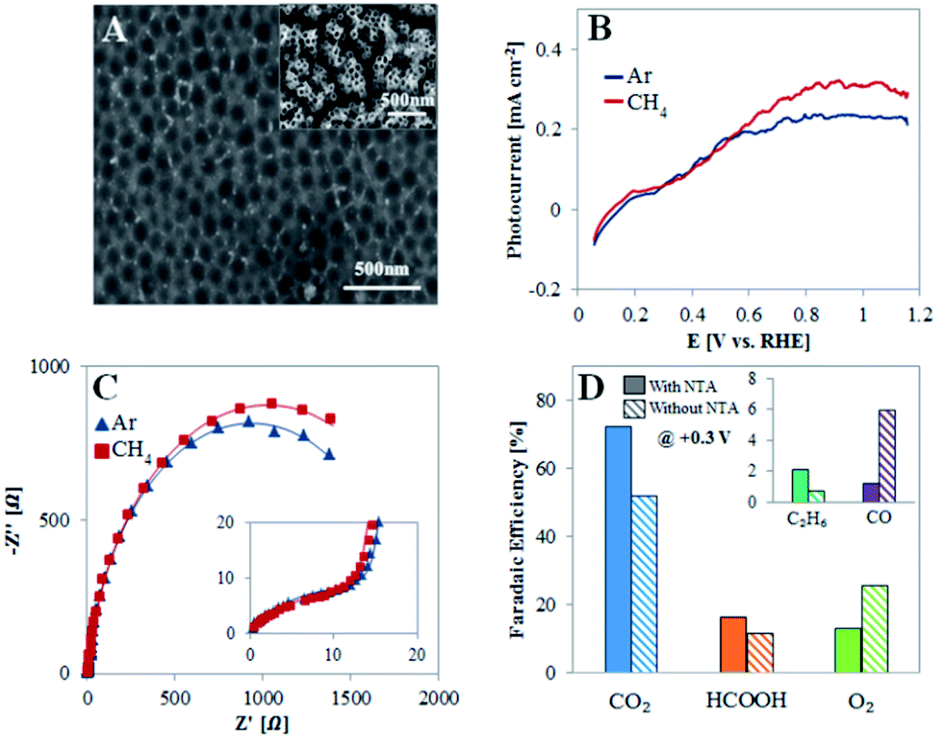 | ||
| Fig. 4 Photoelectrochemical analysis of the TiO2NTA after peeling off the nanotube arrays. (A) HRSEM image of the TiO2NTA photoanode after peeling off the nanotubes (inset: TiO2NTA before the peeling procedure). (B) LSV voltammograms obtained at a scan rate of 5 mV s−1 during illumination while purging Ar (blue) or CH4 (red) to the solution at a flow rate of 20 mL min−1. (C) EIS Nyquist plots obtained for the photoanode biased at 0.3 V under illumination in the absence and presence of methane (inset: zoom-in the high frequency region. Fitted model data are presented by solid lines). (D) Faradaic efficiency of the products of methane PEC oxidation obtained at an applied potential of 0.3 V before (solid filled columns) and after (columns with stripes) removing the NTA (inset: FE for C2H6 and CO). | ||
Products analysis was performed at 0.3 V for a TiO2NTA photoanode after the NTA have been removed (Fig. 4D). The analysis revealed a selectivity decrease toward carbon-based products (FE of 91 and 70% before and after the peeling, respectively). This result is in accordance with the different behavior observed by LSV and EIS which indicated a lower PEC selective activity toward CH4 of TiO2 devoid of NTA. Moreover, the FE of CO after the peeling was much higher than that obtained with the nanotubes (6.0 and 1.2%, respectively). This corroborates with the suggested role of nanostructured TiO2 in dictating the reaction path towards CO2 rather than to CO production which is promoted by the compact oxide layer obtained after the NTA removal. Such different behavior can stem not only from charge transfer considerations but also from improved adsorption and methane diffusion pathways as exerted by NTA as a porous nanostructured surface.35,48,49
The high FE for total oxidation of CH4 to CO2 at TiO2NTA allowed us to consider this reaction to be achieved galvanically in a room temperature photo-fuel cell (PFC) performing in acidic conditions. Such conditions were found to provide higher current responses for PEC methane oxidation as compared to those obtained in neutral and alkaline ones (Fig. S9†). The schematics of the cell structure and energy diagram for the PFC are presented in Fig. 5A. Theoretically, this cell can produce a maximum voltage of about ∼1.3 V due to the potential difference between the conduction band (CB) edge of TiO2 (−0.1 V vs. RHE) and the reduction potential of O2 (1.23 V vs. RHE).47 Illuminating the photoanode with light that has higher energy than the band gap energy of the semiconductor initiates the photogeneration of electrons and holes. The latter oxidize the fuel to produce methane oxygenates and protons which migrate through the membrane to the cathode. The photogenerated electrons migrate through the external electrical wiring to participate in the reduction reaction of oxygen and protons to form water at the cathode.12,50
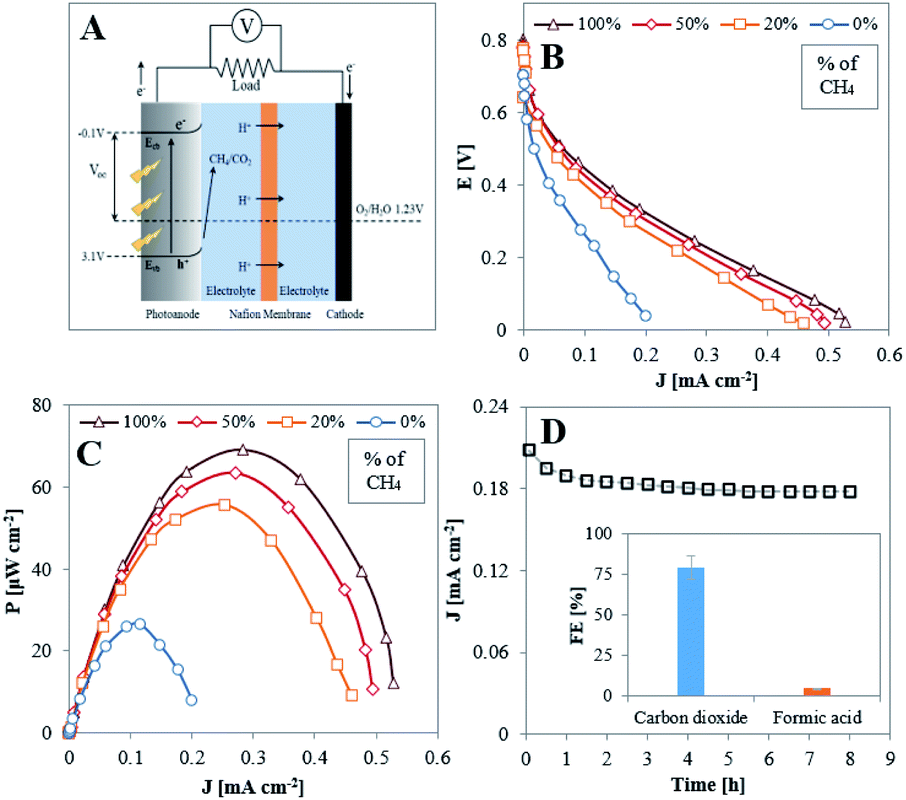 | ||
| Fig. 5 Methane-based PFC performance operating at room temperature under illumination of 100 mW cm−2 with 0.05 M H2SO4 as electrolyte and Nafion membrane as separator. (A) Schematic representation of the photo-fuel cell and energy diagram for the conversion of methane oxidation to electricity. (B) Polarization and (C) power density vs. current density curves with different CH4/Ar mixtures. (D) Long-term performance of the photo-fuel cell at maximum power condition (load of 1000 Ω); inset: faradaic efficiencies of CO2 and formic acid generated by the PFC at steady state conditions. | ||
Polarization and power vs. current density curves obtained under illumination, as depicted in Fig. 5B and C, respectively, demonstrate the performance of a methane-based PFC with CH4/Ar mixtures and O2, fed to the anode and cathode, respectively. Initially, in the absence of methane, the measured power density represents the energy induced by illumination.51–54 The cell performance improves significantly when CH4 is present in the anode gas feed, clearly indicating that it is consumed as fuel in the PFC.
The stability of the PFC performance was examined with pure methane fed to the anode and oxygen to the cathode and operating at maximum power conditions (applying 1000 Ω between the electrodes) for 8 hours. As deduced by Fig. 5D, the TiO2NTA photoanode demonstrates high stability over the examined time period. The current density decreased by only 8.7% from its initial value during the first hour, probably due to initial changes occurring at the photoanode–electrolyte interface, and then remained constant at a value of ∼0.18 mA cm−2 for the rest of the stability test. Comparison of SEM surface examination, before (Fig. 1A) and after (Fig. S10†) the stability test reveals that no significant physical degradation of the nanotubes has occurred during the process. Analysis for the carbon-based products conducted in steady state conditions showed that the overall FE for both CO2 in the gas phase and formic acid in the electrolyte attained a value of ∼84% (FEs of 79.3 and 4.3%, respectively, inset of Fig. 5D). The potential of the photoanode at these conditions was determined (by inserting an Ag/AgCl reference electrode during PFC operation) to be 0.22 V vs. RHE. According to the half-cell results (Fig. 3B), the FE for formic acid at 0.3 V is 16.3% and it is expected to exceed this value at lower potentials. The much lower FE for this product obtained during the operation of the PFC (4.3%) can possibly be related to the formation of a higher amount (∼40 fold) of methyl hydroperoxide (CH3OOH) than found in the half-cell experiments (Fig. S8†). It has been reported that this species can be formed due to oxidation of methane by hydrogen peroxide.55–57 It seems, therefore, that more H2O2 is formed in the PFC configuration by side-reactions. Hydrogen peroxide may originate from several sources, such as PEC 2e-oxidation of water on the TiO2NTA surface58 or from O2 diffusing from the cathodic chamber59 and partially reduced to H2O2 at the anode by conduction band electrons.58 Consequently, the low FE of formic acid is attributed to the more oxidative conditions which prevail in the PFC anodic chamber.
Table 1 summarizes the values of the open circuit potential (VOC), short circuit current density (JSC) and the maximum power density (PMAX) of the PFC. The values increase with increasing the concentration of methane in the anodic chamber. This tendency, corroborated by the EIS and trapping measurements, implies that methane acts as a good scavenger of holes. The highest power density output (69 μW cm−2) from the PFC was obtained when supplying 100% CH4 to the photoanode. The obtained value competes well with those reported for PFCs which operate on liquid ethanol51 and methanol53 as fuels (23 and 30 μW cm−2 respectively).
| CH4 concentration [% vol] | V OC [V] | J SC [mA cm−2] | P MAX [μW cm−2] |
|---|---|---|---|
| 0 | 0.70 | 0.21 | 26 |
| 20 | 0.78 | 0.47 | 56 |
| 50 | 0.78 | 0.5 | 64 |
| 100 | 0.80 | 0.53 | 69 |
Being aware of the possible performance improvement using a gas phase PFC,14,60 the cell was operated with an illuminated anodized porous Ti fiber felt photoanode for methane oxidation (SEM images in Fig. S11†), a commercial Pt/C as gas diffusion cathode and Nafion as solid polymer electrolyte (details in p. S11 of the ESI†). This configuration was able to increase only moderately PMAX by 19%. The obtained PMAX (82 μW cm−2, Fig. S12†) is within the range of reported power densities (11–82 μW cm−2) for PFCs with both illuminated photo-anode and -cathode and fed with solutions of dissolved fuels.61 However, it is lower than that reported for systems operating with photoanodes with much smaller optical band gap (such as W-doped BiVO4) and glucose or biomass-related compounds as fuels (up to 400 μW cm−2).54 Although attempting to reduce the band gap of the photoanode in the present configuration by proper doping may be important to improve performance, it seems that the power density of the present configuration will still be limited by the high energetics involved in methane oxidation, as well by its low diffusion coefficient and slow sorption kinetics on the anode in these conditions.62
Conclusions
The effective PEC activity of a TiO2NTA photoanode toward methane oxidation in acidic electrolyte and ambient conditions has been demonstrated. This activity is characterized in a half-cell configuration by a high FE toward carbon-based products, mainly CO2 and formic acid in the gas and liquid electrolyte, respectively. In the absence of the TiO2 nanotubes, the total FE of carbon-based products decreases while a higher tendency for the production of CO is observed. These results clearly indicate that the special surface architecture of the nanostructured TiO2 photoanode has a significant impact on the methane oxidation reaction pathways. This is exploited for the first time operation of a room-temperature PFC in which methane gas is consumed as a fuel and provides electricity. The maximum output power density obtained using a gas phase photo-fuel cell, (82 μW cm−2), is suggested to be limited both by the photoanode optical properties as well by the difficulties associated with methane gas adsorption on the electrode surface and its subsequent oxidation. Prospects for possible future performance improvement can be expected using the unique features of electrochemically-based TiO2NTA photoanode coatings, as we demonstrated for liquid-fuelled PFCs.63Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to the Israeli Ministry of Energy for funding this research. Y. K. would like to thank Pratt Foundation for their financial support with a doctoral scholarship.References
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS.
- L. Yuliati and H. Yoshida, Chem. Soc. Rev., 2008, 37, 1592 RSC.
- V. Havran, M. P. Duduković and C. S. Lo, Ind. Eng. Chem. Res., 2011, 50, 7089–7100 CrossRef CAS.
- S. Xie, S. Lin, Q. Zhang, Z. Tian and Y. Wang, J. Energy Chem., 2018, 27, 1629–1636 CrossRef.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC.
- J. Baltrusaitis, I. Jansen and J. D. Schuttlefield Christus, Catal. Sci. Technol., 2014, 4, 2397 RSC.
- D. A. Wood, C. Nwaoha and B. F. Towler, J. Nat. Gas Sci. Eng., 2012, 9, 196–208 CrossRef CAS.
- K. Aasberg-Petersen, I. Dybkjær, C. V. Ovesen, N. C. Schjødt, J. Sehested and S. G. Thomsen, J. Nat. Gas Sci. Eng., 2011, 3, 423–459 CrossRef CAS.
- N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19–28 RSC.
- H. Song, X. Meng, Z. j. Wang, H. Liu and J. Ye, Joule, 2019, 3, 1606–1636 CrossRef CAS.
- T. Berger, M. Sterrer, O. Diwald, E. Knözinger, D. Panayotov, T. L. Thompson and J. T. Yates, J. Phys. Chem. B, 2005, 109, 6061–6068 CrossRef CAS.
- P. Lianos, J. Hazard. Mater., 2011, 185, 575–590 CrossRef CAS.
- W. Li, D. He, G. Hu, X. Li, G. Banerjee, J. Li, S. H. Lee, Q. Dong, T. Gao, G. W. Brudvig, M. M. Waegele, D. Jiang and D. Wang, ACS Cent. Sci., 2018, 4, 631–637 CrossRef CAS.
- F. Amano, A. Shintani, K. Tsurui, H. Mukohara, T. Ohno and S. Takenaka, ACS Energy Lett., 2019, 4, 502–507 CrossRef CAS.
- L. Yuliati, T. Hattori, H. Itoh and H. Yoshida, J. Catal., 2008, 257, 396–402 CrossRef CAS.
- L. Yuliati, T. Hattori and H. Yoshida, Phys. Chem. Chem. Phys., 2005, 7, 195–201 RSC.
- H. Yoshida, N. Matsushita, Y. Kato and T. Hattori, J. Phys. Chem. B, 2003, 107, 8355–8362 CrossRef CAS.
- Y. Kato, H. Yoshida and T. Hattori, Chem. Commun., 1998, 2389–2390 RSC.
- L. Li, G. D. Li, C. Yan, X. Y. Mu, X. L. Pan, X. X. Zou, K. X. Wang and J. S. Chen, Angew. Chem., Int. Ed., 2011, 50, 8299–8303 CrossRef CAS.
- S. Murcia-López, K. Villa, T. Andreu and J. R. Morante, ACS Catal., 2014, 4, 3013–3019 CrossRef.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
- M. A. Gondal, A. Hameed, Z. H. Yamani and A. Arfaj, Chem. Phys. Lett., 2004, 392, 372–377 CrossRef CAS.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS.
- R. Mohammadpour, A. Irajizad, N. Taghavinia, M. Rahman and M. M. Ahadian, J. Mater. Sci. Technol., 2010, 26, 535–541 CrossRef CAS.
- M. Z. Ge, C. Y. Cao, J. Y. Huang, S. H. Li, S. N. Zhang, S. Deng, Q. S. Li, K. Q. Zhang and Y. K. Lai, Nanotechnol. Rev., 2016, 5, 75–112 CAS.
- H. Dang, X. Dong, Y. Dong, Y. Zhang and S. Hampshire, Int. J. Hydrogen Energy, 2013, 38, 2126–2135 CrossRef CAS.
- J. R. Rumble, CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data, CRC Press, 2019 Search PubMed.
- M. Li, Y. Liu, L. Dong, C. Shen, F. Li, M. Huang, C. Ma, B. Yang, X. An and W. Sand, Sci. Total Environ., 2019, 668, 966–978 CrossRef CAS.
- B. C. H. Steele, Nature, 1999, 400, 619–621 CrossRef CAS.
- T. S. Kang, A. P. Smith, B. E. Taylor and M. F. Durstock, Nano Lett., 2009, 9, 601–606 CrossRef CAS.
- Y. Bochlin, E. Korin and A. Bettelheim, ACS Appl. Energy Mater., 2019, 2, 8434–8440 CrossRef CAS.
- K. A. Smith, A. I. Savva, C. Deng, J. P. Wharry, S. Hwang, D. Su, Y. Wang, J. Gong, T. Xu, D. P. Butt and H. Xiong, J. Mater. Chem. A, 2017, 5, 11815–11824 RSC.
- S. Halevy, E. Korin and A. Bettelheim, RSC Adv., 2016, 6, 87314–87318 RSC.
- J. Yu and B. Wang, Appl. Catal., B, 2010, 94, 295–302 CrossRef CAS.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS.
- J. Lin, R. Zong, M. Zhou and Y. Zhu, Appl. Catal., B, 2009, 89, 425–431 CrossRef CAS.
- D. Regonini, A. Groff, G. D. Sorarù and F. J. Clemens, Electrochim. Acta, 2015, 186, 101–111 CrossRef CAS.
- P. Acevedo-Peña and I. Gonźalez, J. Electrochem. Soc., 2013, 160, H452–H458 CrossRef.
- A. R. C. Bredar, A. L. Chown, A. R. Burton and B. H. Farnum, ACS Appl. Energy Mater., 2020, 3, 66–98 CrossRef CAS.
- Y. Pang, Y. Li, G. Xu, Y. Hu, Z. Kou, Q. Feng, J. Lv, Y. Zhang, J. Wang and Y. Wu, Appl. Catal., B, 2019, 248, 255–263 CrossRef CAS.
- Y. Pang, G. Xu, C. Fan, J. Lv, J. Liu and Y. Wu, RSC Adv., 2016, 6, 61367–61377 RSC.
- T. Stoll, G. Zafeiropoulos and M. N. Tsampas, Int. J. Hydrogen Energy, 2016, 41, 17807–17817 CrossRef CAS.
- I. Paramasivam, H. Jha, N. Liu and P. Schmuki, Small, 2012, 8, 3073–3103 CrossRef CAS.
- X. Zhou, N. Liu and P. Schmuki, ACS Catal., 2017, 7, 3210–3235 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- X. Pan, X. Chen and Z. Yi, Phys. Chem. Chem. Phys., 2016, 18, 31400–31409 RSC.
- W. Jiang, Y. Zhu, G. Zhu, Z. Zhang, X. Chen and W. Yao, J. Mater. Chem. A, 2017, 5, 5661–5679 RSC.
- B. Hokkanen, S. Funk, U. Burghaus, A. Ghicov and P. Schmuki, Surf. Sci., 2007, 601, 4620–4628 CrossRef CAS.
- J. M. Macak, M. Zlamal, J. Krysa and P. Schmuki, Small, 2007, 3, 300–304 CrossRef CAS.
- B. Viswanathan, V. Subramanian and J. S. Lee, Materials and Processes for Solar Fuel Production, Springer, New York, 2014, vol. 174 Search PubMed.
- M. Antoniadou and P. Lianos, Appl. Catal., B, 2010, 99, 307–313 CrossRef CAS.
- M. Antoniadou, D. I. Kondarides, D. Labou, S. Neophytides and P. Lianos, Sol. Energy Mater. Sol. Cells, 2010, 94, 592–597 CrossRef CAS.
- X. Li, G. Wang, L. Jing, W. Ni, H. Yan, C. Chen and Y. M. Yan, Chem. Commun., 2016, 52, 2533–2536 RSC.
- B. Zhang, J. Shi, C. Ding, R. Chong, B. Zhang, Z. Wang, A. Li, Z. Liang, S. Liao and C. Li, ChemSusChem, 2015, 8, 4049–4055 CrossRef CAS.
- S. Al-Shihri, C. J. Richard and D. Chadwick, ChemCatChem, 2017, 9, 1276–1283 CrossRef CAS.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef CAS.
- H. Song, X. Meng, S. Wang, W. Zhou, X. Wang, T. Kako and J. Ye, J. Am. Chem. Soc., 2019, 141, 20507–20515 CrossRef CAS.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- A. Jablonski, P. J. Kulesza and A. Lewera, J. Power Sources, 2011, 196, 4714–4718 CrossRef CAS.
- F. Amano, H. Mukohara, A. Shintani and K. Tsurui, ChemSusChem, 2019, 12, 1925–1930 CrossRef CAS.
- B. Zhang, W. Fan, T. Yao, S. Liao, A. Li, D. Li, M. Liu, J. Shi, S. Liao and C. Li, ChemSusChem, 2017, 10, 99–105 CrossRef CAS.
- M. Joglekar, V. Nguyen, S. Pylypenko, C. Ngo, Q. Li, M. E. O'Reilly, T. S. Gray, W. A. Hubbard, T. B. Gunnoe, A. M. Herring and B. G. Trewyn, J. Am. Chem. Soc., 2016, 138, 116–125 CrossRef CAS.
- S. Halevy, E. Korin and A. Bettelheim, Nanoscale Adv., 2019, 1, 4128–4136 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0se00984a |
| This journal is © The Royal Society of Chemistry 2021 |