
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Membrane folding and shape transformation in biomimetic vesicles†
B. Jelle
Toebes
and
Daniela A.
Wilson
 *
*
Systems Chemistry Department, Institute for Molecules and Materials, Radboud University, Nijmegen, The Netherlands. E-mail: d.wilson@science.ru.nl
First published on 30th December 2020
Abstract
Polymeric self-assembled structures have been a topic of interest in the last few decades, specifically for the use of biomedical applications, such as drug delivery. It is exciting to investigate the formation of various shapes and sizes of such structures, as the morphology is crucial for their function. In this manuscript the important factors to control the morphology during self-assembly and subsequent shape transformation processes are discussed. We describe the main parameters to control and show the practical application of these parameters on biodegradable amphiphilic PEG-PDLLA block-copolymers. Thereby a variety of different morphologies, including micelles, worms, LCVs, discs, rods, stomatocytes, nested vesicles, and spherical vesicles of various sizes are created using only four diblock-copolymers and with careful tuning of two organic solvents. Further advances will lead to formation of more complex structures.
Introduction
Amphiphilic diblock-copolymers self-assemble in aqueous solutions into nano-sized structures, as the hydrophobic parts cluster together to prevent unfavorable solvent interactions, thereby reducing internal energy.1 During self-assembly a variety of different structures can be created, including micellar spheres, worms, and spherical vesicles containing a bilayer membrane, so called polymersomes.2–4 Basically, upon formation of a polymersome, an aqueous lumen is created, surrounded by a protecting membrane bilayer that can be used to shield the inside from harmful conditions. In biology, such compartmentalization is abundantly present in the form of liposomes that consist of many phospholipid molecules.5 Polymersomes, however, are more robust than their phospholipid counterparts and their polymer composition, length and functionality are more easily tuned. This makes them suitable for many different applications, such as drug delivery or nano-reactors.6–8The characteristics of nanoparticles are inextricably linked to their size and shape, comparable to improved oxygen uptake of red blood cells due to their biconcave disc shape, or better cellular uptake of particles with a high aspect ratio.9,10 It is fascinating to explore new possibilities and functions of various nanoparticle morphologies. Therefore, it is necessary to consider the two main processes that affect the morphology of polymersomes, especially when trying to create more intricate designs; the initial self-assembly and the subsequent shape transformation processes. We will describe the important parameters that define these processes from the bottom up.
Important for the morphology during the self-assembly process is how the polymers are packed together (Fig. 1b). If a relatively large hydrophilic corona is created with a small hydrophobic core, the amphiphilic polymers will form small micellar structures. Reducing the corona dimensions by decreasing the repulsive forces between the hydrophilic chains and/or increasing the stretching of the hydrophobic core part will lead to the formation of worms. Further reduction of the corona size and increased stretching of the core chains will eventually result in bilayered membranes that form spherical polymersomes (Fig. 1b).11 Detailed investigation from the group of Eisenberg has resulted in strong insight in the self-assembly and have found several important parameters that need to be discussed. The four main factors that dictate the morphology of polymersomes during self-assembly are; the total length of the amphiphilic polymer, the ratio of the hydrophilic block relative to the hydrophobic block, the repulsion among the hydrophilic corona chains, and the stretching of the hydrophobic core chains.12,13 The total length of the amphiphilic polymer is important for the size of the self-assembly and can easily be tuned during their synthesis.14 The ratio of the respective polymer blocks is also easily controlled during synthesis and affects both size and morphology of the self-assembly, as the ratio of hydrophilic block will determine the corona size and the hydrophobic block will determine the stretching of the core. Detailed manipulation of the last two factors, the repulsion forces of the corona chains and the amount of core stretching, are determined by the interactions with their environments, such as solvents and additives.14 An important parameter to consider here is the Flory–Huggins interaction parameter (χ), which is a measure of the polymer–solvent interaction and is related to their respective solubility parameters (δ) and the dielectric constants (ε).12,15 The miscibility of a polymer with a specific solvent can be estimated from the solubility parameter, as the closer the solubility parameters are to each other, the better they mix and the higher the stretching of the core chains. On the other side, the polarity of the solvent will affect the repulsion between the corona chains, as the hydrophilic part of the polymer is polar. More polar solvents have more interaction with the corona chains and will increase the corona dimensions, whereas solvents with lower polarity have weaker interactions and therefore result in a smaller corona.
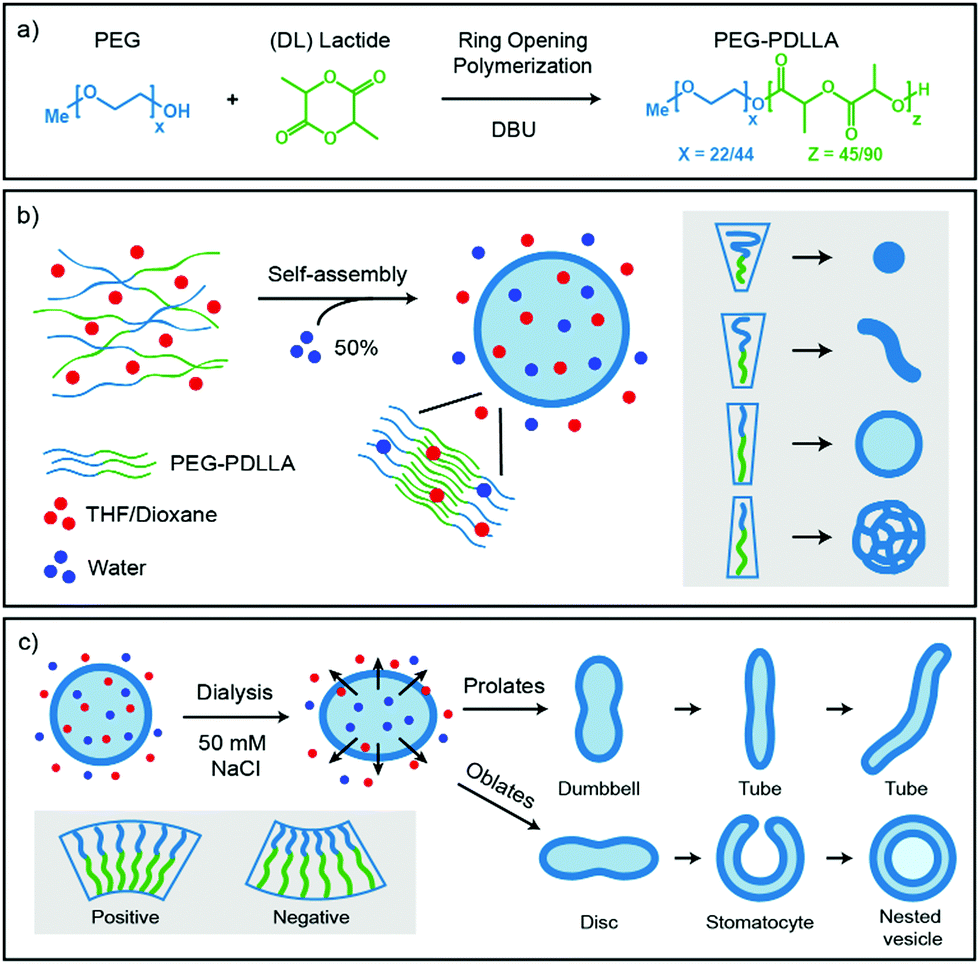 | ||
| Fig. 1 Schematic representation of the synthesis and self-assembly of the amphiphiles (a) synthesis via ring opening polymerisation of PEG-PDLLA diblock-copolymers, (b) self-assembly of PEG-PDLLA polymersomes when water is added, the packing of the polymer in the membrane induces the formation of micelles, worms, spherical polymersomes, or large compound vesicles (LCVs), (c) shape transformation of spherical polymersomes by inducing osmotic pressure. A larger osmotic pressure will result in more deflated structures. A positive membrane curvature results into prolates (rod-like structures) and a negative membrane curvature will result in oblates (disc-like structures). | ||
Once spherical polymersomes have been obtained, their morphology can be changed by inducing an osmotic pressure, thereby creating various more complex shapes, such as rods, discs, stomatocytes, but also stomatocytes within stomatocytes or even polyhedron shaped particles.16–19 During polymersome formation, the membrane is flexible as the solvent composition is similar on the inside compared to the outside of the vesicle. If an osmotic shock is applied by adding solutes during dialysis, such as salts or PEG, the spherical polymersomes are forced to deflate to lower the osmotic pressure at the cost of bending energy.20 During dialysis the organic solvent is removed, but the hydrophobic character of the bilayer hampers influx of water in the polymersomes, which leads to shape transformation. Higher water contents make the hydrophobic blocks more rigid and less permeable to water, trapping glassy polymersomes into their obtained shape in a local minimum of the energy landscape.
So far, the shape transformation of polymersomes has mostly been done with poly(ethylene glycol)-b-polystyrene (PEG-b-PS), as the polystyrene generates glassy vesicles that can be tuned with relative ease.21 The PEG layer provides a stealth character to the vesicle, which avoids the immune system and allows a long blood circulation time, making these polymersomes particularly suitable as drug carrier nanomotors.22,23 Unfortunately, polystyrene is not biodegradable, rendering it useless for medical applications. A promising alternative is found in polylactide, as its characteristics are similar to polystyrene.24,25 However, as the formation and shape transformation of polymersomes is largely dependent on the building blocks, it is important to determine the practical parameters to control the final morphology of polymersomes.
Various theoretical models have predicted shapes of vesicles comprised of amphiphilic molecules by calculating minimal bending energies as a function of the reduced volume under various conditions.26–29 The spontaneous curvature model determines favorable shape conformations depending on the spontaneous curvature of the vesicles. A positive spontaneous curvature promotes the formation of prolates (rod-like structures), whereas a negative spontaneous curvature results in oblates (disc-like structures). Further volume reduction of prolate structures leads to dumbbell shapes that further turn into tubes/rods, while oblates will deflate into biconcave discs, stomatocytes (bowl-shaped vesicles) and can eventually close again to form so called nested vesicles (Fig. 1c).26 This volume reduction is dependent on the osmotic pressure, but also on the exchange rate of the solvents.
The out-of-equilibrium situations that forces spherical polymersomes to deflate into different morphological structures can be explained in terms of bending energy (eqn (1)).
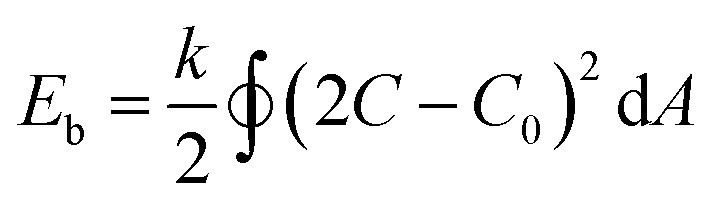 | (1) |
In this equation k is the bending rigidity constant, C is the mean surface curvature and C0 stands for the spontaneous surface curvature. C0 corresponds to the intrinsic curvature of the polymer and is depending on its length and composition. The difference between the intrinsic curvature and the actual mean curvature of the vesicle determines the cost of the bending energy. When an external stimulus is applied to a flexible membrane, the surface curvature can be altered, thereby influencing the bending energy. A positive bending energy will lead to the formation of prolate (rod-like) morphologies, whereas a negative bending energy leads to the formation of oblate (disc-like) morphologies. The hydrophilic and hydrophobic parts of the polymersome largely influence the surface charge, temperature sensitivity, rigidity and permeability and thereby affect the surface curvature response.30 For example, introducing an osmotic shock to spherical poly(ethylene glycol)-b-poly(D,L-lactide) (PEG-b-PDLLA) polymersomes induces shape transformation into elongated nanotubes (prolates), whereas poly(ethylene glycol)-b-polystyrene (PEG-b-PS) forms stomatocyte morphologies (oblates) under similar conditions.17,31,32 Important to note is that not only the membrane curvature, but also the solvent exchange rate is decisive during shape transformation. A fast solvent exchange rate could deflate and trap vesicles via a different route than a slow solvent exchange rate, meaning that factors that influence the solvent exchange rate, such as membrane thickness, play a crucial role.28
The goal of this study was to control the self-assembly process of poly(ethylene glycol)-b-poly(D,L-lactide) (PEG-b-PDLLA) polymersomes and the subsequent shape transformation process to create biodegradable polymersomes with a wide variety of shapes and sizes. Four different PEG-PDLLA block-copolymers were synthesized and self-assembled using mPEG22-PDLLA45 or mPEG44-PDLLA90 with different amounts of PEG. Two block-copolymers were synthesized using mPEG22-PDLLA45 with 6.5 wt% PEG and 13 wt% PEG, respectively. Two block-copolymers were synthesized using the twice as long mPEG44-PDLLA90 with 13 wt% PEG and 26 wt% PEG, respectively. To control the corona dimensions and the core stretching of the amphiphilic polymers the Flory–Huggins parameters of commonly used solvents were investigated. First the solubility parameters of solvents were compared with that of polylactide (δ = 19.3–21.0 [MPa]1/2). The polylactide–solvent interaction is closest to dioxane (δ = 20.5 [MPa]1/2) and then to THF (δ = 18.5 [MPa]1/2), which indicates that the stretching of the core should be highest in dioxane and to a lesser extent in THF. Furthermore, the polarity of dioxane (ε = 2.25) is lower than THF (ε = 7.58), so dioxane should have less interaction with the corona chains and this would lead to smaller corona dimensions. The self-assembly of these polymers was performed with different ratios of THF and dioxane and subsequently dialyzed against Milli-Q or salt solutions to induce an osmotic shock. The samples were characterized by cryo-Transmission Electron Microscopy (cryo-TEM) and DLS.
Experimental
Materials
All compounds were used as received. For synthesis of the four PEG-PDLLA copolymers, methoxy-PEG22-OH (1 kDa) was purchased from Creative PEG Works and methoxy-PEG44-OH (2 kDa) was purchased from Rapp Polymere. D,L-Lactide and the tetrahydrofuran (THF) were purchased from Acros Organics. The ultra-pure Milli-Q water was obtained via Labconco Water Pro PS purification system (18.2 ME). Dialysis membranes of MWCO 12–14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Dalton Spectra/Por were used to remove the organic solvent. Sodium chloride was purchased from Merck. Dioxane was purchased from Biosolve Chimie. All other chemicals were supplied by Sigma-Aldrich.
000 Dalton Spectra/Por were used to remove the organic solvent. Sodium chloride was purchased from Merck. Dioxane was purchased from Biosolve Chimie. All other chemicals were supplied by Sigma-Aldrich.
Methods
:4, THF:Dioxane v/v). The vial was closed with a rubber septum and the mixture was stirred for 30 minutes at 800 rpm. Subsequently, 1 ml Milli-Q water (50 wt%) was added via a syringe pump at 1 ml per hour until a cloudy suspension was obtained. The suspension was transferred to a pre-hydrated membrane (Spectra/Por, molecular weight cut-off: 12–14 kDa) and dialyzed against 1 L of Milli-Q or 50 mM NaCl for 24 hours in a fridge at 4 °C, with a solution change after 1 hour. Samples were stored in the fridge at 4 °C.
Results and discussion
Several PEG-PDLLA diblock-copolymers with different lengths and hydrophilic segments were synthesized using Ring Opening Polymerization (ROP); mPEG22-PDLLA45 (1), mPEG44-PDLLA45 (2), mPEG22-PDLLA90 (3) and mPEG44-PDLLA90 (4). The product compositions were calculated from their respective NMR and GPC spectra (Table S1 and Fig. S1, ESI†). For the formation of polymersomes, the polymers were dissolved in organic solvent, after which an equivalent of water was added slowly at 1 ml h−1, inducing self-assembly. Afterwards, the samples were dialyzed against Milli-Q or salt (50 mM NaCl) to remove the organic solvent and to potentially add an osmotic shock. For analysis with cryo-TEM at least 50 structures were observed for each sample.The self-assembly of the four separate diblock-copolymers was tested in two mixtures of THF and dioxane to see the effect of the organic solvent on the core stretching and corona dimensions (Table 1). First the results obtained with a ratio of 4:1 THF to dioxane are discussed. Both polymer 2 and 4 (PEG44-PDLLA45 and PEG44-PDLLA90) showed the formation of micelles of 25 and 85 nm, respectively (Fig. S4a and S6a, ESI†), whereas polymer 1 (mPEG22-PDLLA45) formed spherical polymersomes of around 450 nm (Fig. S3a, ESI†). Polymer 3 (mPEG22-PDLLA90) formed a polydisperse mixtures of LCVs (Large Compound Vesicles) and aggregates (Fig. S5a, ESI†). Changing the organic solvent composition affected the self-assembly behaviour of the polymers as expected. An increase in size was observed for all samples when a higher dioxane content was used (1:4 THF to dioxane) (Table 1). For polymer 4 a morphology change was observed as well and changed from micelles to worm-like structures (Fig. S6b, ESI†). Higher dioxane contents reduce the corona dimension and increase the core stretching and therefore a morphological change from micelles towards vesicles is seen (Fig. 2a–d).
| Polymer | THF:Diox |
Morphology | Size (nm) | PDI |
|---|---|---|---|---|
| a PS = Polymersomes. | ||||
| (1) PEG22-PDLLA45 | 1:4 |
Spherical PSa | 453 | 0.11 |
| 4:1 |
Spherical PS | 344 | 0.09 | |
| (2) PEG44-PDLLA45 | 1:4 |
Micelles | 22 | 0.49 |
| 4:1 |
Micelles | 18 | 0.37 | |
| (3) PEG22-PDLLA90 | 1:4 |
Aggregates + LCVs | 3290 | 0.73 |
| 4:1 |
Aggregates + LCVs | 457 | 0.43 | |
| (4) PEG44-PDLLA90 | 1:4 |
Micelles, Worms | 97 | 0.14 |
| 4:1 |
Micelles | 65 | 0.1 | |
| (1 + 2) PEG22-PDLLA45 PEG44-PDLLA45 | 1:4 |
Micelles | 34 | 0.17 |
| 4:1 |
Micelles | 24 | 0.24 | |
| (1 + 3) PEG22-PDLLA45 PEG22-PDLLA90 | 1:4 |
Spherical PS | ∼4149 | 0.4 |
| 4:1 |
Irregular PS | ∼1524 | 1.0 | |
| (1 + 4) PEG22-PDLLA45 PEG44-PDLLA90 | 1:4 |
Spherical PS | 380 | 0.13 |
| 4:1 |
Spherical PS | 85 | 0.04 | |
| (3 + 4) PEG22-PDLLA90 PEG44-PDLLA90 | 1:4 |
Spherical PS | 495 | 0.07 |
| 4:1 |
Spherical PS | 304 | 0.11 | |
 | ||
| Fig. 2 Cryo-TEM images of various shapes and sizes of PEG-PDLLA polymersomes. (a) Micelles from polymer 2 (PEG44-PDLLA45), (b) worms from polymer 4 (PEG44-PDLLA90), (c) spherical polymersomes from polymer 1 (PEG22-PDLLA45), (d) large compound vesicles (LCVs) from polymer 3 (PEG22-PDLLA90), (e) tubular polymersomes from polymer 1, (f) stomatocytes from a mixture of polymers 3 and 4. Scale bars are 500 nm. | ||
Addition of salt during the dialysis can have various effects on the morphology of the self-assembled structure. The salt ions have a stronger interaction with the polar corona and therefore will mainly affect the corona dimension and thereby the packing of the polymers. An increase in size and morphology is expected with increasing amounts of salt, due to reduced corona dimensions induced by the salt. This was indeed the case for polymer 4, as small polymersomes and worm structures were observed instead of the micellar and worm structures obtained against dialysis with water (Fig. S6c and d, ESI†). A smaller effect was seen for polymer 2, as a small increase in size of the micellar structures were observed when dialyzed against 50 mM NaCl compared to dialysis against water (Fig S4c and d, ESI†). On the other hand, addition of salt also increases the osmotic pressure in the solution and this will affect the vesicles during dialysis. The increased osmotic pressure will lead to an outflow of water from the vesicles, decreasing their size and forcing the shape transformation. For polymer 1 tubular structures (prolates) were observed when dialyzed against salt with 4:1 THF to dioxane, but a mixture of nested vesicles (oblates) and tubes were formed with 1:4 THF to dioxane (Fig. S3c and d, ESI†). This can be explained as the corona dimensions are larger and the core is shorter for polymersomes with higher THF content. The membrane curvature of these vesicles is more positive and will therefore deflate into prolates. Increasing the dioxane content will lower the membrane curvature and opens up the pathway for oblates. Around zero membrane curvature both pathways are possible, with a slight preference towards prolates. Yet, once a vesicle has been deflated via a specific pathway, the shape is locked due to limited flexibility of the membrane. For polymer 3 again a polydisperse sample was created with aggregates and LCVs (Fig. S5c and d, ESI†).
Although a shift in morphology was observed for polymer 1 from prolates to oblates, still a large quantity of prolate structures were present. Therefore, another possibility was to combine different polymer lengths to tune better the corona dimensions and core length, but also solvent exchange rate. Changing the corona dimensions by mixing polymer 1 with polymer 2 resulted in slightly larger micelles compared to polymer 2 alone (Fig. S7, ESI†). The corona dimensions are larger in this mixture compared to polymer 1 alone and will shift therefore towards micellar structures. The opposite is true for polymer 2, as the corona dimensions are smaller for the mixture and therefore result in larger micelles. Mixing polymer 1 with the longer polymer 4 resulted in spherical polymersomes with a size of 380 nm, in between the sizes of the individual polymers (Fig. S8a and b, ESI†). The membrane thickness (r) of a polymersome is dependent on the polymer block lengths, according to r ∼ NαBN−γA, where NB is the length of the hydrophobic block and NA is the length of the hydrophilic length. The values for α are often significant higher (0.4–0.8) compared to γ (0–0.1), as the hydrophilic block has less effect on the membrane thickness.33 In Fig. 3 the average membrane thickness of the polymersomes are shown from the separate polymers 1 and 4, as well as the mixture of both. The membrane thickness of the mixture (19 nm) is in between those of the separate polymer vesicles (16 and 23 nm, respectively), indicating equal mixing of the polymers throughout the polymersomes. Dialyzing the mixture of polymers 1 and 4 against salt resulted in stomatocytes and nested vesicles with 1:4 THF to dioxane (Fig. S8c and d, ESI†). The incorporation of a longer core forming block in the mixture resulted in a slightly lower membrane curvature and the formation of more oblate structures. Furthermore, this thicker membrane also decreased the solvent exchange rate, resulting in less deflated structure. Where polymer 1 showed mainly nested vesicles, with the mixture of polymers 1 and 4 also stomatocytes were observed. When polymers 3 and 4 were mixed and dialyzed against water, spherical polymersomes with small PDI were obtained, even though polymer 3 alone created structures with very large PDI, showing successful mixing (Fig. S9a and b, ESI†). When dialyzed against salt, elongated oblates and prolates were observed with 4:1 THF to dioxane and stomatocyte structures with 1:4 THF to dioxane (Fig. S9c and d, ESI†). Finally, mixing polymers 1 and 3 showed large polymersomes when dialyzed against Milli-Q water, but uncontrolled shape transformation when dialyzed against salt, as different undefined structures were formed with large PDI (Fig. S10, ESI†).
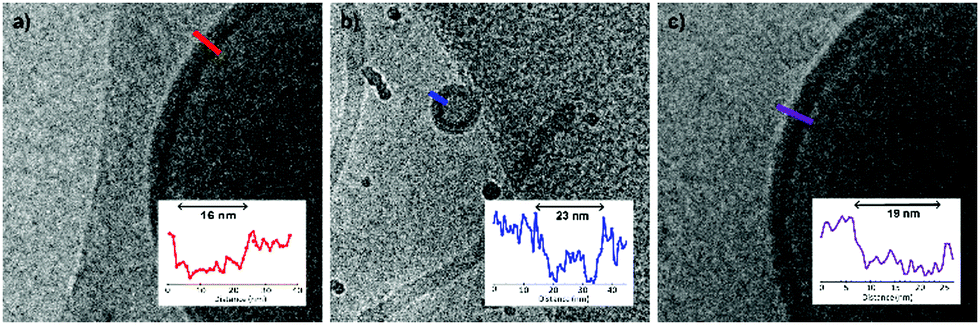 | ||
| Fig. 3 Membrane thickness of different PEG-PDLLA polymersomes based on polymer length. (a) Membrane thickness of polymer 1 (PEG22-PDLLA45) vesicles is 16 nm, (b) membrane thickness of polymer 4 (PEG44-PDLLA90) is 23 nm, (b) membrane thickness of the mixture of polymers 1 and 4 is 19 nm. | ||
From the various morphologies obtained with observations of the samples, a model has been created that shows the effect of important factors during self-assembly and shape transformation (Fig. 4). A larger corona and/or smaller core stretching will lead to a shift from vesicular to micellar structures, whereas an opposite effect is seen for smaller corona dimensions and higher core stretching. In Fig. 4a, the influence of each factor on the packing of PEG-PDLLA polymers during self-assembly is shown. The corona dimensions are enlarged with increasing amounts of NaCl, the wt% PEG, and the THF content, but reduced with a higher dioxane content. The core stretching is increased by the dioxane content and the wt% of PLA but reduced by THF. The molecular weight (Mw) of the total polymer will increase both the corona dimension and the core stretching, increasing the total length of the polymer. However, the polymer will also be more compact and expands sideways, changing the packing towards micellar structures. In Fig. 4b the important factors are listed that affect the shape transformation pathways by changing C0. A larger corona and smaller core stretching will lead towards a positive curvature, whereas smaller corona and core dimensions will lead to a negative curvature. Around zero membrane curvature, the solvent exchange rate plays a determining role, a fast solvent exchange rate will quickly reduce the inner volume of the vesicles, not giving time to reach lower energy state negative curvature vesicles. A slow solvent exchange rate ensures proper folding and provides negative curvature. Important for the solvent exchange rate is the thickness of the membrane, and therefore linked to the Mw of the polymer. A thicker membrane will slow down the exchange rate to the solvents. Another factor that is important here is the osmotic pressure and can be altered by increasing solute concentration, or by changing the ratio of organic solvent to water (Table 2).
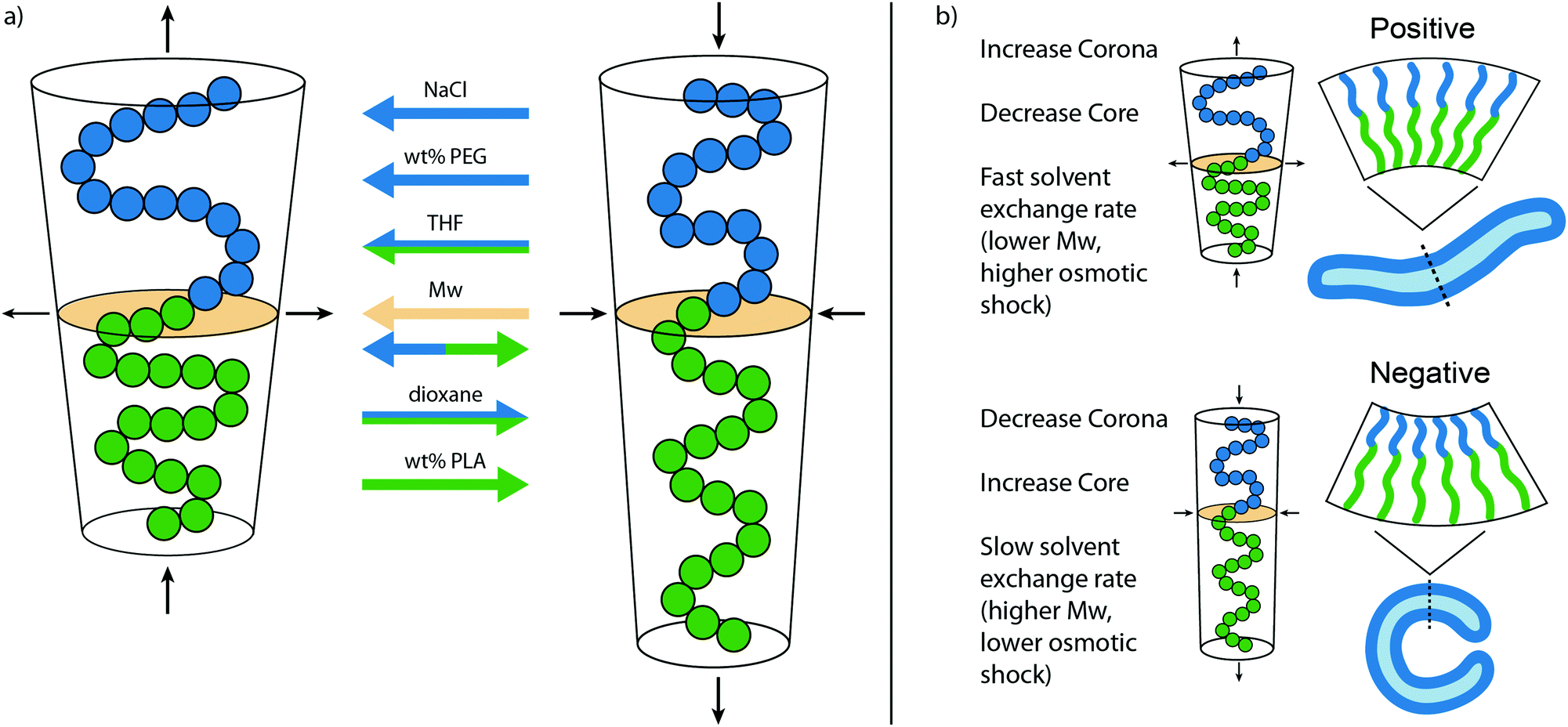 | ||
| Fig. 4 Mechanism for the different morphologies observed with PEG-PDLLA vesicles. (a) Overview of the main factors that affect the packing of PEG-PDLLA during self-assembly. An increase in NaCl, wt% PEG, THF content will result in a larger corona dimension and thereby lead to a shift from vesicular to micellar structures. The opposite effect is seen with a higher dioxane content. The core stretching is influenced by the THF and dioxane content and the wt% of PLA, where an increase in core stretching will cause vesicle structures. The Mw of the total polymer will increase both the corona dimension and the core stretching, increasing the total length of the polymer, but also expands sideways, affecting the leading to micellar structures. (b) Important factors that influence the shape transformation pathways. An increase in corona dimensions will promote a positive curvature, whereas an increase in core stretching will promote a negative curvature. When the membrane curvature reaches zero, the solvent exchange rate is determining for the final morphology. | ||
| Polymer | THF:Diox |
Morphology | Size (nm) | PDI |
|---|---|---|---|---|
| a PS = Polymersomes. | ||||
| (1) PEG22-PDLLA45 | 1:4 |
Nested vesicles + tubes | 432 | 0.08 |
| 4:1 |
Tubes | 248 | 0.1 | |
| (2) PEG44-PDLLA45 | 1:4 |
Micelles | 25 | 0.09 |
| 4:1 |
Micelles | 24 | 0.19 | |
| (3) PEG22-PDLLA90 | 1:4 |
Aggregates + LCVs | 3079 | 0.96 |
| 4:1 |
Aggregates + LCVs | 355 | 0.34 | |
| (4) PEG44-PDLLA90 | 1:4 |
Worms + spherical PSa | 110 | 0.15 |
| 4:1 |
Micelles + worms | 67 | 0.12 | |
| (1 + 2) PEG22-PDLLA45 PEG44-PDLLA45 | 1:4 |
Micelles | 36 | 0.33 |
| 4:1 |
Micelles | 35 | 0.32 | |
| (1 + 3) PEG22-PDLLA45 PEG22-PDLLA90 | 1:4 |
Nested vesicles | 600 | 0.34 |
| 4:1 |
Irregular deflated PS | 278 | 0.41 | |
| (1 + 4) PEG22-PDLLA45 PEG44-PDLLA90 | 1:4 |
Nested vesicles + stomatocytes | 343 | 0.08 |
| 4:1 |
Spherical PS | 145 | 0.12 | |
| (3 + 4) PEG22-PDLLA90 PEG44-PDLLA90 | 1:4 |
Stomatocytes | 250 | 0.11 |
| 4:1 |
Discs | 222 | 0.13 | |
Conclusions
To conclude, we have shown control over the self-assembly and shape transformation of PEG-PDLLA polymersomes. It is possible to create different morphologies during self-assembly by tuning the polymer length, composition and both the hydrophilic corona and hydrophobic core chains. We have been able to show different morphologies (micelles, worms, and polymersomes) with the same diblock-copolymer depending on the self-assembly conditions. Furthermore, we have shown that by mixing different diblock-copolymers, better control over the self-assembly and shape transformation can be obtained. By changing C0 and the solvent exchange rate, the shape transformation of spherical polymersomes could be controlled to both prolate and oblate structures. Connecting and incorporating experimental parameters with more detailed molecular modelling studies can lead to the formation of polymersomes with more intricate designs and shapes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the NWO Chemische Wetenschappen VIDI Grant 723.015.001.References
- Y. Barenholz, D. Gibbes, B. Litman, J. Goll, T. Thompson and F. Carlson, Biochemistry, 1977, 16, 2806–2810 CrossRef CAS.
- M. Krishna, J. Phys. Chem. A, 1999, 103, 3589–3595 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS.
- R. Roodbeen and J. C. Van Hest, BioEssays, 2009, 31, 1299–1308 CrossRef CAS.
- K. T. Kim, J. Zhu, S. A. Meeuwissen, J. J. L. M. Cornelissen, D. J. Pochan, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2010, 132, 12522–12524 CrossRef CAS.
- D. A. Wilson, R. J. Nolte and J. C. van Hest, J. Am. Chem. Soc., 2012, 134, 9894–9897 CrossRef CAS.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS.
- P. B. Canham, J. Theor. Biol., 1970, 26, 61–81 CrossRef CAS.
- A. Albanese, P. S. Tang and W. C. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS.
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS.
- Y. Yu, L. Zhang and A. Eisenberg, Macromolecules, 1998, 31, 1144–1154 CrossRef CAS.
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- A. Choucair, C. Lavigueur and A. Eisenberg, Langmuir, 2004, 20, 3894–3900 CrossRef CAS.
- J.-F. Le Meins, O. Sandre and S. Lecommandoux, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 14 CrossRef.
- Y. Men, W. Li, G. J. Janssen, R. S. M. Rikken and D. A. Wilson, Nano Lett., 2018, 18, 2081–2085 CrossRef CAS.
- S. A. Meeuwissen, K. T. Kim, Y. Chen, D. J. Pochan and J. C. van Hest, Angew. Chem., 2011, 123, 7208–7211 CrossRef.
- R. S. Rikken, H. H. Kerkenaar, R. J. Nolte, J. C. Maan, J. C. van Hest, P. C. Christianen and D. A. Wilson, Chem. Commun., 2014, 50, 5394–5396 RSC.
- C. K. Wong, A. D. Martin, M. Floetenmeyer, R. G. Parton, M. H. Stenzel and P. Thordarson, Chem. Sci., 2019, 10, 2725–2731 RSC.
- R. S. Rikken, H. Engelkamp, R. Nolte, J. Maan, J. Van Hest, D. Wilson and P. Christianen, Nat. Commun., 2016, 7, 12606 CrossRef CAS.
- S. F. Van Dongen, M. Nallani, S. Schoffelen, J. J. Cornelissen, R. J. Nolte and J. C. van Hest, Macromol. Rapid Commun., 2008, 29, 321–325 CrossRef CAS.
- F. Peng, Y. Tu, J. C. M. van Hest and D. A. Wilson, Angew. Chem., Int. Ed., 2015, 54, 11662–11665 CrossRef CAS.
- Y. Li, M. Kröger and W. K. Liu, Biomaterials, 2014, 35, 8467–8478 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS.
- U. Seifert, Adv. Phys., 1997, 46, 13–137 CrossRef CAS.
- W. Góźdź, Langmuir, 2004, 20, 7385–7391 CrossRef.
- H. Yuan, C. Huang and S. Zhang, Soft Matter, 2010, 6, 4571–4579 RSC.
- A. Sakashita, N. Urakami, P. Ziherl and M. Imai, Soft Matter, 2012, 8, 8569–8581 RSC.
- G.-Y. Liu, C.-J. Chen and J. Ji, Soft Matter, 2012, 8, 8811–8821 RSC.
- L. K. E. A. Abdelmohsen, D. S. Williams, J. Pille, S. G. Ozel, R. S. M. Rikken, D. A. Wilson and J. C. M. van Hest, J. Am. Chem. Soc., 2016, 138, 9353–9356 CrossRef CAS.
- B. J. Toebes, L. K. Abdelmohsen and D. A. Wilson, Polym. Chem., 2018, 9, 3190–3194 RSC.
- L. Zhang, R. J. Barlow and A. Eisenberg, Macromolecules, 1995, 28, 6055–6066 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01932a |
| This journal is © The Royal Society of Chemistry 2021 |