
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an electrically responsive hydrogel for programmable in situ immobilization within a microfluidic device†
Rok
Ambrožič
 a and
Igor
Plazl
*ab
a and
Igor
Plazl
*ab
aUniversity of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, 1000 Ljubljana, Slovenia. E-mail: igor.plazl@fkkt.uni-lj.si
bChair of Microprocess Engineering and Technology – COMPETE, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia
First published on 14th June 2021
Abstract
A novel microfluidic channel device with programmable in situ formation of a hydrogel 3D network was designed. A biocompatible hybrid material consisting of iron ion-crosslinked alginate was used as the active porous medium. The sol–gel transition of the alginate was controlled by the oxidation state of Fe ions and regulated by an external electrical signal through an integrated gold plate electrode. The SEM images, FT-IR analysis, and rheological test demonstrated that homogeneous yet programmable hydrogel films were formed. The higher the concentration of the crosslinker (Fe(III)), the smaller the pore and mesh size of the hydrogel. Moreover, the hydrogel thickness and volume were tailored by controlling the deposition time and the strength of electric current density. The as-prepared system was employed as an active medium for immobilization of target molecules, using BSA as a drug-mimicking protein. The reductive potential (activated by switching the current direction) caused dissolution of the hydrogel and consequently the release of BSA and Fe. The diffusion of the entrapped molecules was optimally adjusted by varying the dissolution conditions and the initial formulations. Finally, the altering electrical conditions confirm the programmable nature of the electrically responsive material and highlight its wide-ranging application potential.
1. Introduction
Materials capable of responding to (bio)chemically applied potentials,1,2 electric/magnetic fields,3,4 light,5 temperature changes,6 pH,7,8 and mechanical signals9,10 are currently being developed and optimized for their potential chemical, biomedical, and environmental applications. Of these, electrical stimulus via an induced electric potential is a particularly attractive tool for affecting the shape and properties of biomaterials due to its high transformation selectivity and single-layer oriented mode of operation. The latter, in combination with advanced electrochemical techniques, enables precise 2D and 3D spatial control of the resulting material.11–13 On the other hand, hydrogels are innovative advanced biomaterials that are widely used as programmable active materials due to their unique properties such as high water content, softness, flexibility and biocompatibility.14,15 By combining such properties with electrical sensitivity, electro-responsive hydrogels could be further developed to enable the conversion of electrical energy/stimuli into mechanical energy/transformation. Recent studies from this field explore their use for exogenous stimulus-activated drug delivery,4,16 protein patterning,17 cell cultivation and sorting,18,19 tissue and material engineering,20,21 and as dynamic 3D scaffolds.22,23 In addition, the repeatable/cyclic sol–gel transformation of hydrogels enables the design of smart engineering materials as stimulable catalytic supports with immobilized active sites.24Among others, the design of naturally accessible polysaccharides, such as alginic acid and chitosan, is of particular interest due to their sustainable disposition.5,13,25–27 Sodium or potassium salts of alginic acid form viscous, homogeneous aqueous solutions that are converted to ionotropic hydrogels by crosslinking with divalent or multivalent metal cations, typically Ca2+. Crosslinking of alginate is achieved by chelating metal cations via carboxylate groups of β-D-mannurate (M) and especially α-L-guluronate (G) residues of the alginate. The M2+-crosslinked hydrogel is typically arranged in an “egg-box” 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 helical structure in which domains of four G subunits are bound by an M2+. Associated hydrogels are biocompatible and non-immunogenic, allowing molecules of drugs, biopolymers and whole living cells to be entrapped/immobilized within their structure without affecting their stability and/or functionality. In addition, iron(III) cations also form stable alginate hydrogels by preferentially binding oxygen atoms in negatively charged ligands such as carboxylate, phenolate or hydroxamate groups.25,26,28 These hydrogels have been successfully used for cell cultivation due to their increased cell adhesion compared to Ca2+ crosslinked alginate hydrogels. While Fe2+ cations (Fe(II)) are also capable of crosslinking alginate, the iron(II) ion is known to be a “soft” metal cation, resulting in a weak hydrogel structure. It has been shown that the crosslinking of alginate can be regulated by controlling the oxidation state of iron via the electrochemical potential.25 For example, alginate–Fe(II) forms a homogeneous solution that can be converted to an insoluble Fe(III)-crosslinked hydrogel by electrochemical oxidation. Of course, the process can also be reversed by electrochemical reduction, resulting in controlled dissolution of the hydrogel. Furthermore, the redox transformation can also be photoinduced via UV or visible light.5 Both strategies allow repeatable stimulation of the hydrogel transformation under mild (typically biocompatible) conditions, without any organic solvent, which is beneficial for maintaining the activity of the charged active molecule.
:1 helical structure in which domains of four G subunits are bound by an M2+. Associated hydrogels are biocompatible and non-immunogenic, allowing molecules of drugs, biopolymers and whole living cells to be entrapped/immobilized within their structure without affecting their stability and/or functionality. In addition, iron(III) cations also form stable alginate hydrogels by preferentially binding oxygen atoms in negatively charged ligands such as carboxylate, phenolate or hydroxamate groups.25,26,28 These hydrogels have been successfully used for cell cultivation due to their increased cell adhesion compared to Ca2+ crosslinked alginate hydrogels. While Fe2+ cations (Fe(II)) are also capable of crosslinking alginate, the iron(II) ion is known to be a “soft” metal cation, resulting in a weak hydrogel structure. It has been shown that the crosslinking of alginate can be regulated by controlling the oxidation state of iron via the electrochemical potential.25 For example, alginate–Fe(II) forms a homogeneous solution that can be converted to an insoluble Fe(III)-crosslinked hydrogel by electrochemical oxidation. Of course, the process can also be reversed by electrochemical reduction, resulting in controlled dissolution of the hydrogel. Furthermore, the redox transformation can also be photoinduced via UV or visible light.5 Both strategies allow repeatable stimulation of the hydrogel transformation under mild (typically biocompatible) conditions, without any organic solvent, which is beneficial for maintaining the activity of the charged active molecule.
Recently, stimuli-response hydrogels have been applied for continuous and flow-through applications.29–32 Using the electrical and flow control available in a microfluidic environment, in situ fabrication of 3D hydrogel scaffolds can be programmed in space and time to arrange specific components as an in vitro communicating system.33,34 Furthermore, the flexible in situ on-demand construction of a biocompatible scaffold within microfluidics promises the fabrication of biological components and systems for in vitro analyses and investigations.35,36 By combining various physiochemical functions on a single chip, the miniaturized fluid systems enable process intensification, high throughput, low energy consumption, and reduced waste production. To date, channel-based hydrogels have been assembled as scaffolds for biomolecules or cells,37,38 as membranes,39,40 as separation devices,41,42 for tissue engineering,43 and as innovative sensing devices.44,45
Accordingly, a novel microdevice with programmable sol–gel transformation of active porous media controlled by an external electrical signal was developed. Herein, an Fe(III)–alginate hydrogel was immobilized directly on a gold electrode by an electrode deposition method. By switching the current direction, Fe(III) was reduced to Fe(II) and the hydrogel began to decompose under controlled conditions. In this adaptive process, the solubility of the active material – the hydrogel – changed, which means that its mesh size and thus its capture/release ability can be adjusted as desired. In such a mechanism, a large hydrogel surface area is desirable as it allows a large working space. However, conventional electro–micro devices with electrodes placed on the side walls typically possess small active surface area. To address these drawbacks, a novel flow microfluidic device with a large active electrode surface area was designed by using a plate–plate shaped geometry. The mechanism of the Fe adsorption/desorption process in a flow operation mode triggered by external electrical stimuli was investigated and the related hydrogel materials were characterized in detail. The deposited hydrogel was used as an innovative stimuli-responsive active material for in vitro immobilization and release of target molecules (BSA), mimicking the drug delivery process. Such in situ formation and structural control of the active hydrogel with simultaneous immobilization promises high and long-term activities without leaching of the immobilized structures from the gel during the performance of a specific (bio)process.
2. Experimental
2.1. Materials
Alginic acid sodium salt from brown algae (medium viscosity, ≥2.000 cP, further marked as alginate 96%), sodium sulfate (anhydrous, 99%), sodium acetate (≥98%), iron(II) sulfate heptahydrate (≥98%), iron(III)chloride (≥98%), ascorbic acid, sulfuric acid, hydrochloric acid, o-ohenanthroline hydrochloride monohydrate, and albumin from bovine serum (lyophilized powder, further marked as BSA) were used as received from Merck KGaA (Darmstadt, Germany). All experiments were carried out in ultrapure water (18.2 MΩ cm).2.2. Microfluidic device
Electrochemical measurements were performed in a custom-made transparent microfluidic system with integrated gold (Au) plate electrodes, purchased from commercial sources. The two electrodes were located within poly(methyl methacrylate) (PMMA) layers attached to the top and bottom of a gasket film (polytetrafluoroethylene; PTFE) forming the sidewalls of the microfluidic channel (Fig. 1). The patterned electrodes, on the other hand, formed the ceiling and floor of the microchannel. The channel was 10 mm wide, 750 μm high, and 40 mm long with an active electrode area in the microchannel of 10 mm × 40 mm. Compared to more traditional electro–micro devices with electrodes placed on the side walls, the electrodes placed on the bottom/top of the microfluidic channel allow for a larger electrode area, resulting in more efficient hydrogel formation and more applicable molecular diffusion.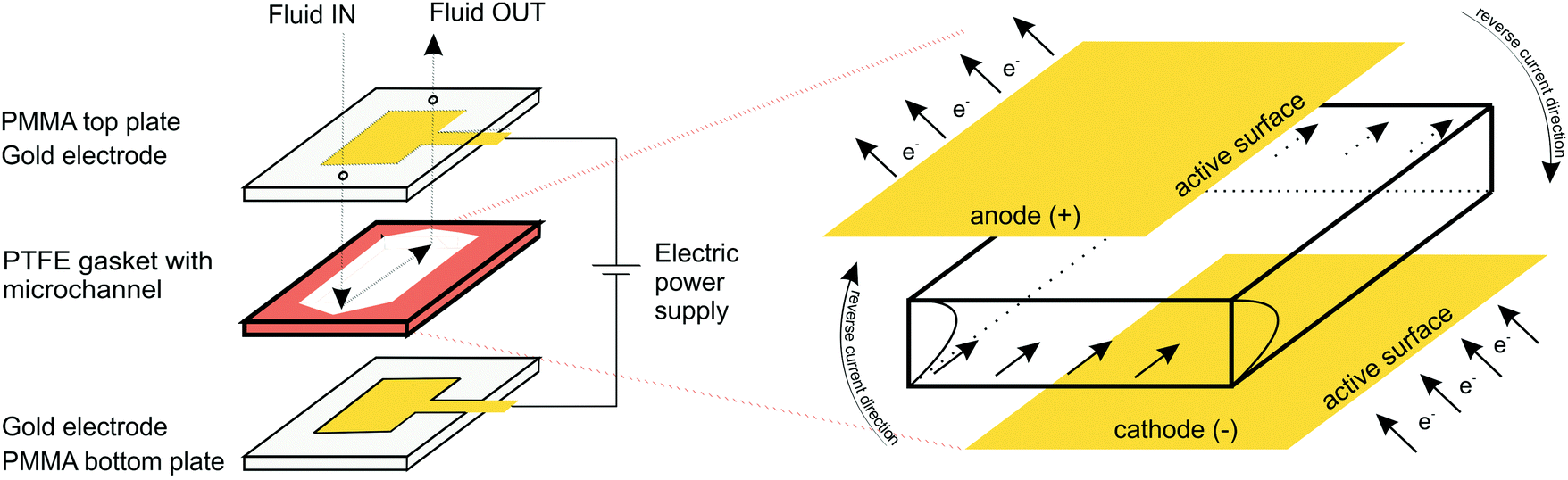 | ||
| Fig. 1 A custom-made transparent microfluidic system with integrated gold (Au) plate electrodes. The two electrodes were located within poly(methyl methacrylate) (PMMA) layers attached to the top and bottom of a sealing film (polytetrafluoroethylene; PTFE) forming the sidewalls of the microfluidic channel. | ||
2.3. Electrodeposition and hydrogel formation
Sodium alginate (0.5, 1.25 and 2.0% w/v) was dissolved in 100 mM Na2SO4 aqueous solution, and stirred overnight. Before each experiment, a different amount of FeSO4 was added to the solution, resulting in different iron ion concentrations (3, 5, 10, 20 and 35 mM). The same procedure, with the addition of BSA (10 mg mL−1), was used for the experiments with the protein-loaded hydrogels.Prior to electrodeposition the microfluidic channel was partially immersed with alginate solution (alginate + FeSO4 + Na2SO4) to ensure that the microfluidic device was completely filled. The flow of solution (100–300 μL min−1) was controlled by a syringe mounted on a syringe pump. An alligator clip was used to connect the plate electrodes to a DC power supply (2400 Sourcemeter, Keithley). The parallel Au plate electrodes served as both cathode (negatively biased electrode) and anode (positively biased electrode). Electrodeposition was performed at a constant current density (2.5, 5, 10 and 15 A m−2) for a specified time interval (3, 4, 5, 6, 8 and 10 min) with typical voltages of 2–3 V. The electrodeposition oxidized the Fe(II) ion to Fe(III), which triggered the formation of ferric–alginate hydrogel on the anode surface. After each electrodeposition step, the microchannel was rinsed several times with alginate-free aqueous Na2SO4 solution and finally with pure water to remove unbound molecules. The total amount of entrapped BSA and crosslinking iron ions was analyzed by dissolving the thin films in the presence of ascorbic acid (10 mM) followed by spectrophotometric analysis of the released BSA and Fe(II) cations.
2.4. Controlled electrochemical dissolution and molecule release
The BSA and iron ions entrapped in the alginate thin films were electrochemically released in Na2SO4 solution (100 mM, pH 6.0) by controlled electrochemical dissolution. For this purpose, the direction of the electric current was inverted, resulting in a reversed electric charge at each Au electrode. The electrochemical dissolution was performed at a constant current density (10 and 15 A m−2) with typical voltages of 2–3 V. The concentrations of electrochemically released Fe(II) cations and BSA were determined spectrophotometrically using standard analytical techniques.2.5. Analytical methods and material characterization
The morphology of Fe–alginate hydrogels was analyzed by field emission scanning electron microscopy (SEM), while scratch analysis of hydrogel (film) thickness was performed by a high-speed optical microscope (Motion Scope). Fourier-transform infrared (FT-IR) spectra of the dried materials were recorded with a Bruker IFS 66/S spectrometer (Billerica, USA) using KBr disks in the range of 4000–400 cm−1.The concentration of trapped and released Fe(II) and/or Fe(III) ions was determined by a standard off-line technique using 1,10-phenantroline as the reagent for colored complex formation. Optical absorbance measurements were performed in a 1 mL poly(methyl methacrylate) (PMMA) cuvette with an optical path length of 1 cm using a UV-2450 spectrophotometer (Shimadzu). First, calibration curves of Fe(II) and Fe(III) were designed to determine the relationship between concentration and UV absorbance required for the quantitative determinations. At least five-point calibration curves within the expected concentration range were constructed for each.
The concentration of entrapped and released protein molecules was determined by on-line UV measurements. The output of the electro-microdevice was connected to a UV flow cell (UV Flow Cells, UV-900) that allowed real-time measurements. A five-point calibration curve of BSA in the expected concentration range was establish prior to measurements.
Rheological tests were performed at 25 °C using an HAAKE RS 150 rheometer equipped with a 50 mm diameter cross-hatch plate (PP50/P2). The thickness of the hydrogel sample corresponded to a distance of 0.5 mm between the plates. Rheological analysis included frequency sweep tests and complex viscosity measurements. Frequency sweep tests were performed within the linear viscoelastic range at constant strain (0.1%) determined by preliminary amplitude sweep tests. Oscillation frequencies varied from 100 to 0.01 Hz. Storage modulus (G′) and loss modulus (G′′) were plotted against pulsation ω (= 2πf) to obtain the mechanical spectrum of viscous and elastic responses over time during the experiment. The obtained experimental data were described using the classical generalized Maxwell models (Maxwell–Wiechert model), which consist of the equilibrium modulus of Ge and the Maxwell element Gi (relaxation moduli).
3. Results and discussion
3.1. Fe(III)–alginate hydrogel formation
In order to verify the role of crosslinking interaction between Fe and alginate in hydrogel formation, different formulations were prepared. Obviously, no gel-like formation was observed when Fe(II) ions were added to the solution to trigger the gelation process. In contrast, hydrogel formation was clearly observed when Fe(III) cations were used. The ionic interaction between positively charged Fe(III) and negatively charged COO− (from the alginate matrix) caused a gelation process.25,28 Since the binding of carboxylate groups by Fe(II) ions is much weaker than their binding by Fe(III), it can be expected that Fe(II) cations have a lower ability to crosslink alginate compared to Fe(III) cations, implying that the oxidation state of iron directly correlates with alginate gelation and its stability. Nevertheless, the critical concentration of Fe(III) that still allowed alginate gelation was 5 mM. No gel-like formation was observed below this iron ion concentration. Obviously, the alginate concentration also plays a major role in gel formation. A formulation with an alginate concentration of 1.25% w/v and an Fe(III) concentration of 10 mM allows the gelation process, while a lower amount of sodium alginate does not trigger precipitation of the homogeneous Fe(III)–alginate complex.A main motivation for our studies was to develop an electrodeposition method that allows controlling sol–gel conversion in microfabricated fluidic devices by an external electrical signal. For this purpose, a flow microfluidic system with bottom/top placed Au electrodes was utilized. In a continuous electrodeposition mode the initial buffer solution (100 mM Na2SO4) was mixed with Na–alginate and an Fe(II) solution and constantly pumped through the microfluidic device. No electrical potential/current was used at this time. Once a stable flow regime was established, a constant electric current was generated by the power supply DC, which triggered the electrodeposition. In this way, the Fe(II) cations were oxidized to Fe(III) cations, which caused the sol–gel transition at the anode surface. The Fe(III)–alginate hydrogels were generated under constant electrodeposition conditions (10 min at a constant current density of 5 A m−2). A formulation with a fixed amount of Na–alginate (1.25% w/v) and different initial Fe(II) ion concentrations (as FeSO4 solution) was used for the gelation process. After the selected deposition time (10 min), the microdevice was opened and the formed hydrogel was analyzed. To determine the Fe concentration within the coordinate complex, a known mass of the hydrogel was dissolved in a small volume of ascorbic acid. The acidic condition causes reduction of Fe(III) to Fe(II) and dissolution of the gel. The Fe(II) concentration was determined by standard spectrophotometry using 1,10-phenanthroline (forms a colored complex). The measured absorption spectra are shown in Fig. 2A and show broad absorption peaks with a maximum at about 510 nm. The signal indicating the formation of the colored Fe–phenanthroline complex is typically observed at this characteristic wavelength.46 The higher the Fe(II) concentration, the higher the UV absorption peak. The pre-made calibration curve (Fig. S2, ESI†) allowed calculation of the exact Fe(II) concentrations. Very good agreement was found between the determined Fe concentration and the bulk Fe concentration (Fig. 2B). Only at higher Fe concentrations (above 25 mM) a slight deviation occurred. Accordingly, the anode signal did not affect the concentration distribution of Fe(II) ions and the sol–gel transition only led to its oxidation but did not affect its total concentration. Assuming complete conversion of Fe(III) to Fe(II) by ascorbic acid addition, the determined Fe(II) concentrations should also signify the Fe(III) concentration trapped in each measured hydrogel.
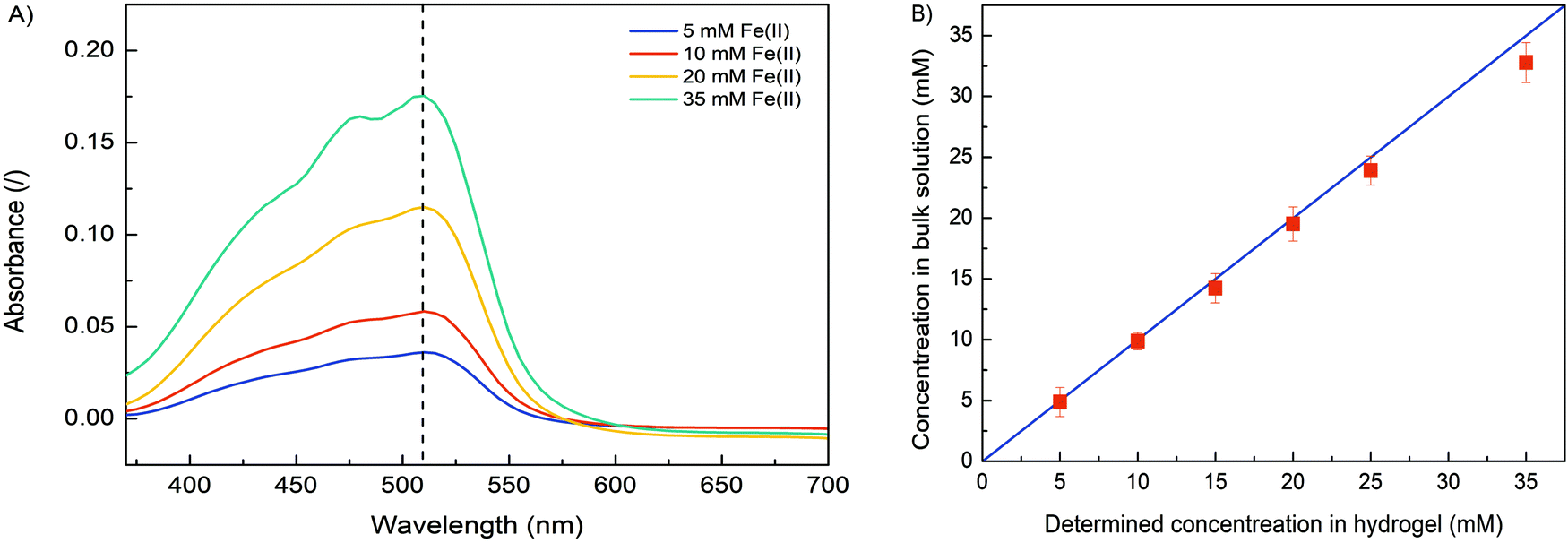 | ||
| Fig. 2 (A) The UV absorption spectra of dissolved Fe(III)–alginate hydrogels at different Fe(II) concentrations (5, 10, 20 and 35 mM). Note that Fe(III) ions were reduced to Fe(II) ions upon addition of ascorbic acid. (B) The agreement between spectrophotometrically determined (red squares) and bulk (blue line) Fe(II) concentrations. | ||
3.2. Evaluation of the electrodeposition process
In order to characterize the influence of deposition conditions (current density, deposition time) on hydrogel formation, a series of experiments were performed. First, electrodeposition was performed at different current densities (2.5, 5, 10 and 15 A m−2) and constant deposition time (4 min) according to the procedure described above. The formulation with a concentration of 10 mM FeSO4 (= Fe(II) concentration), 1.25% w/v Na–alginate and 100 mM Na2SO4 was used. After each experiment, the microdevice was washed with pure buffer solution (100 mM Na2SO4) to wash out all unbound molecules. Afterwards, the microdevices were reopened and the formed hydrogel was analyzed (Fig. 3). To follow the effect of the electric current density on the dimensions of the Fe–alginate hydrogel, the deposited material was analyzed using a high-speed optical microscope. The estimated film thickness increased significantly with increasing current density and ranged from 100 μm to 250 μm. Obviously, Fe(II) oxidation and the resulting deposition process is more effective at higher electric current.47,48 As a result, the volume of the deposited hydrogel also increased. At this point we must state that hydrogel volumes were estimated assuming uniform film thickness. Since no visible irregularities were identified, such an assumption should be reasonable. Besides, alginate and chitosan hydrogels with comparable dimensions have been reported elsewhere.47–49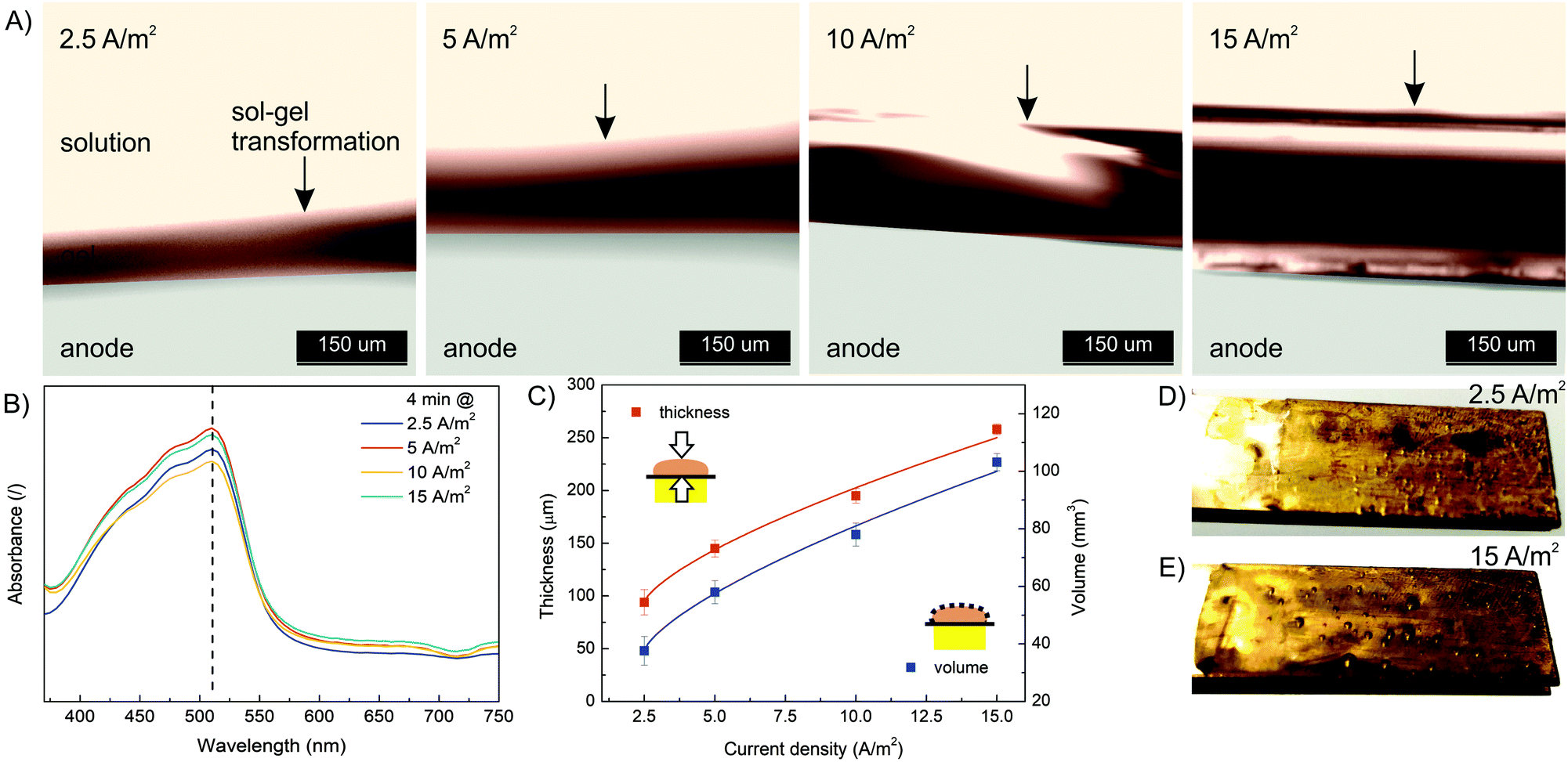 | ||
| Fig. 3 (A) The evolution of hydrogel thickness with a deposition time of 4 min as a function of current density (2.5, 5, 10 and 15 A m−2) analyzed with a high-speed optical microscope. (B) The UV absorption spectra of dissolved Fe(III)–alginate hydrogels formed at different current densities. Note that Fe(III) ions were reduced to Fe(II) ions by addition of ascorbic acid. (C) The thickness and volume of the electrodeposited hydrogel as a function of current density. The optical photo of the electrodeposited hydrogel on the anode surface with a deposition time of 4 min at a current density of 2.5 A m−2 (D) and 15 A m−2 (E). | ||
The results indicate that charge propagation across the film is still possible, leading to continuous growth of the polymer film on the electrode surface as the current density increases. The polymer films with larger thicknesses (or volumes) are obviously beneficial for the entrapment of a larger amount of target molecules and thus mimic the process of drug immobilization. However, the deposition dynamics tend to decrease slightly when thicker polymer films are formed (Fig. 3C). Therefore, the mechanism of charge propagation is thought to be controlled by electrokinetic and ion diffusion, which limits the capture (and release) efficiency of the target molecules. The electrochemical release of specific moieties from the thick polymer films requires optimization of the polymer film thickness by varying the electrodeposition conditions. In addition, the UV absorbance of the dissolved Fe(III)–alginate hydrogels was measured. Nearly identical absorption spectra (Fig. 3B) were observed for all hydrogels tested, regardless of the electrodeposition conditions. Accordingly, the data show that the Fe(III) ions were homogeneously distributed throughout the hydrogel and the induced current density only affected the film thickness but not the Fe(III) ion concentration. A similar observation was made above when an almost identical Fe concentration was found in the bulk solution as in the deposited hydrogel, when the starting Fe(II) concentration varies.
Next, the effect of electrodeposition time was investigated. The same starting formulation as used in the current density effect study was employed (10 mM FeSO4, 1.25% w/v Na–alginate, 100 mM Na2SO4). The electrodeposition time varied from 3 min to 10 min, while the electric current density was constant (10 A m−2). The results presented in Fig. 4 show that the thickness and volume of the anodically deposited alginate hydrogel systematically increased with the deposition time. A hydrogel thickness of 140 μm and 310 μm was approximated for 3 min and 10 min deposition time, respectively. Nevertheless, the results indicate that the anodic deposition was fast (occurring in minutes) and spatially controllable. Compared to the data at other current densities (Fig. 3C), the film volume in this case increased less linearly with deposition time. This could be due to the fact that a larger volume (and thickness) was formed, meaning the electric potential decreased with anode distance, leading to less intense Fe(II) oxidation. In addition, long-term deposition yields less homogeneous film, as some bubbles were trapped in the hydrogel (Fig. 4D and E). Water electrolysis took place on the electrode surface, which generated the oxygen and hydrogen bubbles. The longer the electrolysis time, the more intense the entrapment of the bubbles. Finally, the UV absorption spectra (Fig. 4B) again confirm that the electrodeposition conditions do not affect the ion distribution.
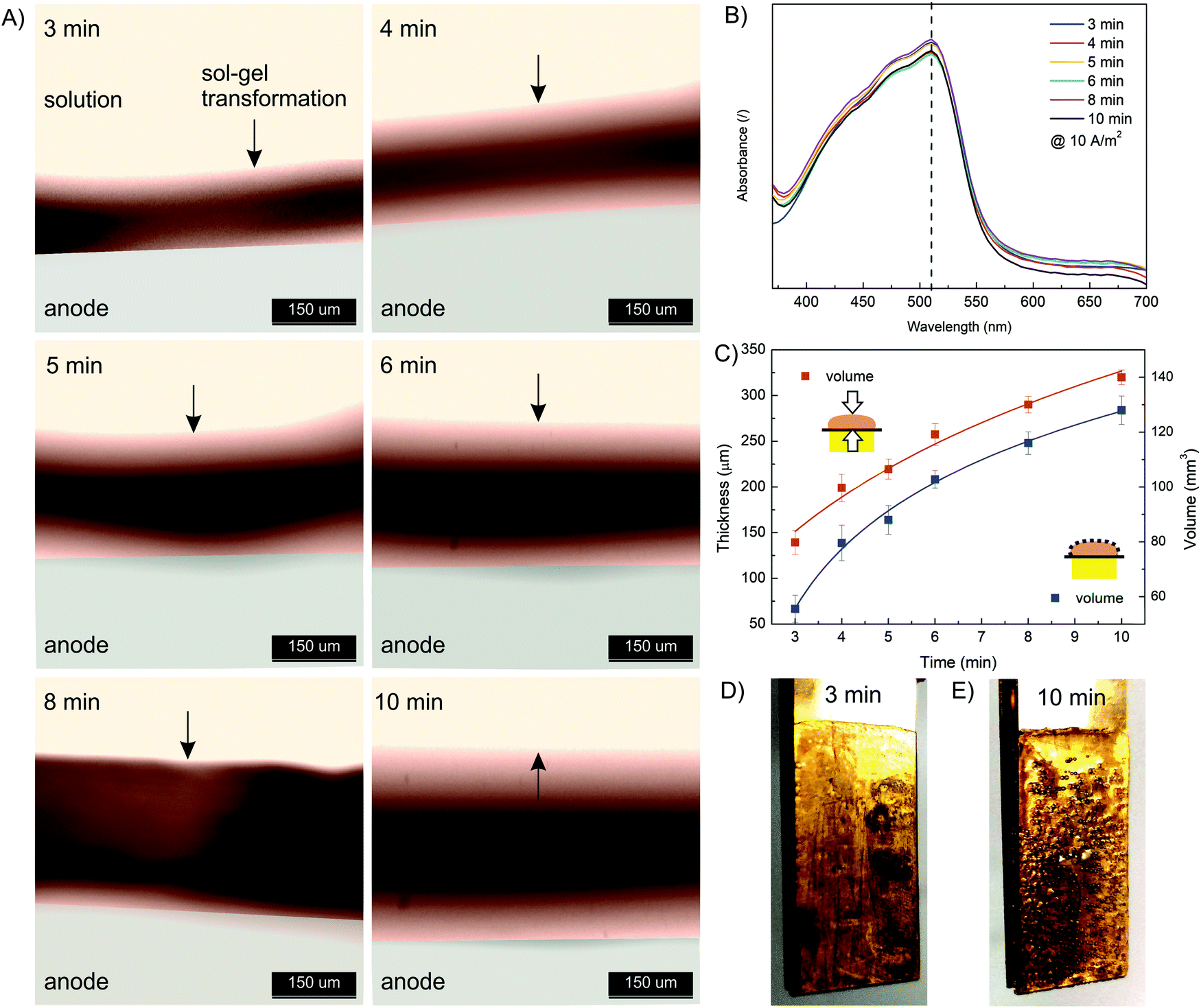 | ||
| Fig. 4 (A) The evolution of hydrogel thickness at a current density of 10 A m−2 as a function of deposition time (3, 4, 5, 6, 8, and 10 min) analyzed using a high-speed optical microscope. (B) The UV absorption spectra of dissolved Fe(III)–alginate hydrogels formed at different deposition times. Note that Fe(III) ions were reduced to Fe(II) ions upon addition of ascorbic acid. (C) The thickness and volume of electrodeposited hydrogel as a function of deposition time. The optical photo of the electrodeposited hydrogel on the anode surface at a current density of 10 A m−2 and a deposition time of 3 min (D) and 10 min (E). | ||
In summary, a tunable external electrical signal allows precise control of the electrodeposition process. Therefore, one can easily adjust the thickness and/or volume of the deposited hydrogel according to specific needs and limitations. Furthermore, the continuous flow mode design provides an additional advantage in integrating such stimuli-responsive materials into various end-use applications.
3.3. Fe(III)–alginate hydrogel characterization
To further confirm chemical interactions in Fe(III)–alginate hydrogels, FT-IR analysis was performed (Fig. S1, ESI†). Prior to analysis, the formed hydrogels and pure sodium alginate were frozen in liquid nitrogen and then lyophilized at −40 °C. Sodium alginate (as received) exhibited two vibrations of the carboxylate group (COO−): an antisymmetric stretching at 1610 cm−1 and a symmetric stretching at 1415 cm−1. Similar observations were made elsewhere.50 In Fe(III)–alginate, both characteristic peaks (at 1610 cm−1 and 1415 cm−1) are slightly shifted. In addition, a new absorption peak appeared at 610 cm−1, which is a characteristic absorption between Fe and the carboxyl group of the alginate. Many studies for the polymer–Fe complex have already shown that hydroxyl groups are bound to metal ions.51,52 Such an interaction causes the formation of a hydrogel complex. As expected, the intensity of this characteristic peak at 610 cm−1 intensified with higher ferric ion concentration. Moreover, the intensity of the broad peak at 3500 cm−1 attributed to the hydroxyl group of sodium alginate decreased slightly and shifted, which is attributed to the newly formed interaction between – OH groups and iron(III) ions. Moreover, a new peak at 1750 cm−1 in the spectrum of Fe(III)–alginate possibly belongs to the absorption vibrations of the carboxyl group of the alginate molecule. The strong polar interaction between Fe(III) and COO− allows the formation of metal–alginate complexes. Naturally, the polar interactions are intensified with a higher oxidation state of Fe ion (Fe3+ > Fe2+), which is due to the higher coordination ability of Fe(III) ions. The broad peak, observed at 1030 cm−1, is attributed to C–O groups. As observed elsewhere,53 its shape and position shifted slightly after the gelation process. Moreover, the band at 2930 cm−1, corresponding to the absorption of the C–H stretching vibration of glucose units in alginate chains, weakened in the spectrum of the Fe(III)–alginate hydrogel. Dong et al.53 suggested that the egg-box structure present in the coordinate complex limits the C–H stretching vibration during the formation of the hydrogel. Nevertheless, the FT-IR result clearly shows that surface gelation of sodium alginate can be formed by the complex of their carboxyl groups with Fe(III) ions in aqueous solution.The morphology of freeze-dried Fe(III)–alginate hydrogels was investigated by SEM microscopy. Except for water removal, no visible changes were observed during freeze-drying. The surface images (Fig. 5) showed that the hydrogels formed an intricate sponge-like structure with abundant crosslinks. The average diameter of the surface pores was in the range of a few nm, while the average diameter of interconnected pores ranged from 200 nm to a μm scale. Similar results have been reported in a related study.28 Moreover, an increase in the crosslinker/polymer ratio (Fe(III) to Na–alginate) resulted in a decrease in the average pore and/or mesh size. This could be due to a more intense gelation process, which was later confirmed via mesh size calculations. Nevertheless, the water-permeable pore structure of the hydrogel matrix was expected to provide many surface-active sites, enabling fast diffusion of small molecules with effective Fe adsorption/desorption activity.
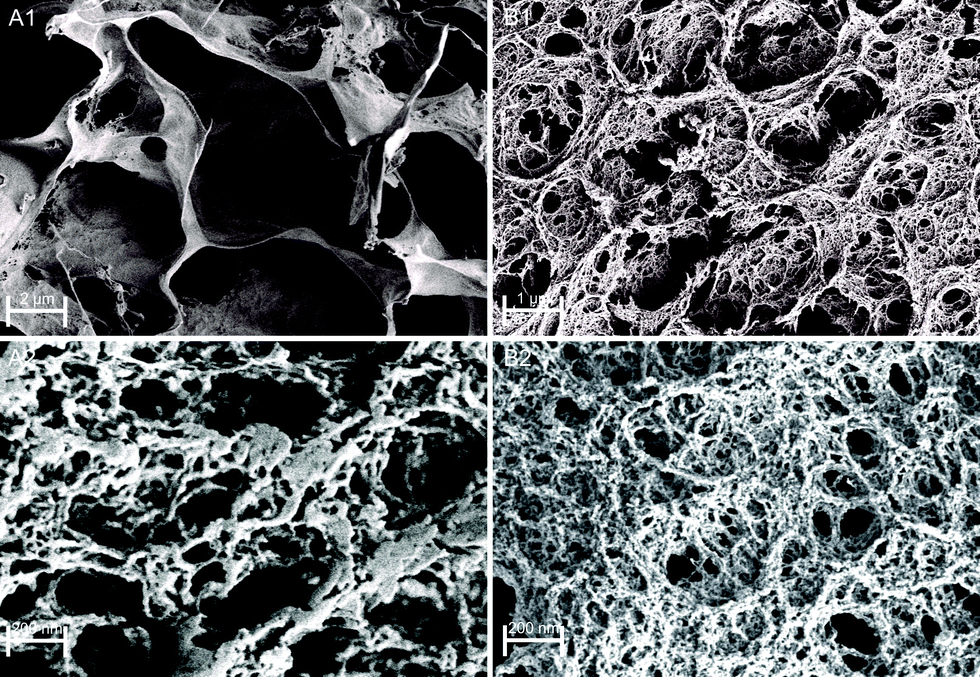 | ||
| Fig. 5 SEM images of freeze-dried Fe(III)–alginate hydrogels as a formulation with 1.25% w/v alginate concentration and crosslinked with 10 mM (A) and 35 mM (B) Fe(III) ions. | ||
Obviously, the mechanical strength of Fe(III)–alginate hydrogels should be sufficient for its integration into flow microdevices. To investigate the shear deformation and flow behavior of Fe(III)-crosslinked alginate hydrogels, rheological analysis was performed (Fig. 6). The viscoelastic properties under shear deformation were to provide information about the behavior of the hydrogel under flow conditions (typical for microchannel operation). The hydrogel samples were prepared by applying electrodeposition at a constant current density of 10 A m−2 for 30 minutes. Such electrodeposition conditions were selected to stimulate the formation of a homogeneous hydrogel with suitable dimensions. The mechanical spectra of the frequency sweep test illustrated the influence of the concentrations of crosslinker (Fe(III)) and polymer (Na–alginate) on the evaluation of the storage (G′) and loss (G′′) moduli. As expected, the G′ and G′′ values of the crosslinked Fe(III)–alginate samples increased with the addition of crosslinker. The higher the addition of a crosslinker, the higher the values of storage and loss modulus (Fig. 6A). The ionic interactions between positively charged Fe(III) and negatively charged COO− of Na–alginate lead to the formation of a coordination complex. Obviously, its intensity and amount increased with a higher number of iron ions. Similarly, the G′ and G′′ moduli also increased with higher Na–alginate content (Fig. 6B). Both phenomena, which are well documented,54,55 should generate additional intra- and interconnections between the individual polymer chains and the crosslinker, yielding higher crosslinking densities.
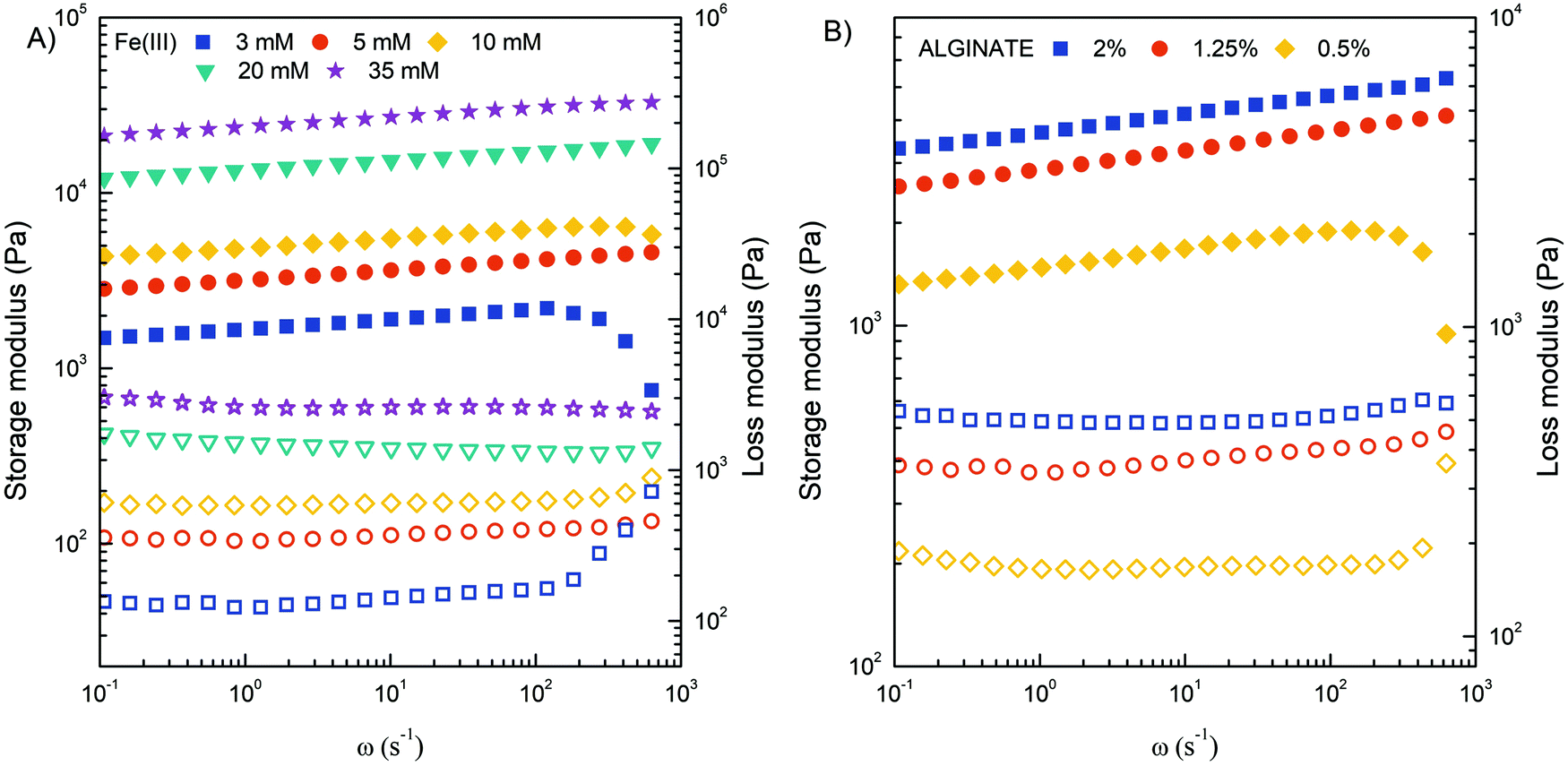 | ||
| Fig. 6 Storage modulus G′ (filled symbols) and loss modulus G′′ (open symbols) of crosslinked Fe(III)–alginate hydrogels at different concentrations of Fe(III) ions (A) and Na–alginate (B). | ||
In addition, the frequency sweep test also provides information about viscoelastic behavior. Since frequency is the reciprocal of time, at low frequencies the samples have more time to relax and respond, which means that the material flow section dominates. As frequency increases, the sample behaves more and more elastically.54 Therefore, the high frequency range corresponds to short term behavior (simulated by rapid motion), while the low frequency range mimics long term behavior. The greater the dependence of G′ on frequency, the weaker the strength of the gel. The results showed that the concentration of 5 mM Fe(III) and 1.25% w/v Na alginate are limited (critical) values for stable hydrogel network formation. Below this value, storage and loss modulus varied more significantly with frequency increase. The results are in excellent agreement with macroscopic observations where a poor gel-like structure was observed at critical concentrations. It is assumed that the extent of ionic interactions was probably too low to properly activate the sol–gel transition. All other hydrogels tested showed adequate mechanical strength with a slight dependence of G′ on applied frequency.
Furthermore, it can be observed that the values of G′ are larger than G′′, confirming the more viscoelastic solid nature of the prepared hydrogels. Accordingly, the ratio of G′′ to G′ for each crosslinked hydrogel was in the range of 0.10–0.30. This ratio also increased dramatically below the critical Fe(III) and Na alginate concentration at 3 mM and 0.5% w/v, respectively. At higher value of angular frequency, the storage and loss modulus profile nearly intersect, indicating a predominant flow-dominated behavior and liquid-like characteristics. In addition, the results of the amplitude sweep test (Fig. S3, ESI†), which is commonly used to determine the linear viscoelastic region, can also provide valuable information about the material yield stress (the stress required to initiate the flow of the material). By monitoring how the loss factor (tanδ) changes with applied stress, one can determine the transition point that is directly related to the yield stress. Such calculations perfectly complement the frequency-dependent results and confirm that the higher the concentrations of Fe(III) and/or alginate, the more elastic the hydrogels behaved. Indeed, the value of yield stress changed from 200 to 1000 Pa when the Fe(III) concentration increased from 5 to 35 mM, and from 200 to 400 Pa when the alginate concentration increased from 0.5 to 2%. Finally, the complex viscosity (η*) of all investigated hydrogel samples decreased with an increase of the angular frequency (Fig. S4, ESI†), confirming that all hydrogels were pseudoplastic fluids with shear-thinning behavior. On the other hand, the value of complex viscosity increased with increasing Fe(III) and alginate concentration throughout the range of measurements, which can be attributed to an enhancement of intermolecular interactions within the gel matrix.
3.4. Electro dissolution process
Due to its stimuli-responsive nature, dissolution of the Fe–alginate hydrogel can also be controlled by an external electrical signal. In this case, the electrical current direction was reversed, i.e., an anode (carrying a deposited hydrogel) became the cathode and vice versa. As a result, entrapped Fe(III) ions were reduced to Fe(II) ions, which have much weaker alginate crosslinking ability, leading to gradual dissolution of the hydrogel. Nevertheless, standard anodic electrodeposition of the Fe–alginate hydrogel was performed in the first step (10 min at 10 A m−2). The formulation containing 10 mM FeSO4, 1.25% w/v Na–alginate and 100 mM Na2SO4 was used as the initial solution. After the indicated electrodeposition time (10 min), the formed hydrogel was washed with 20 mL of pure buffer solution (100 mM Na2SO4) to rinse all untreated molecules out of the microdevice. Such a procedure was then applied for all electro dissolution experiments to ensure repeatable initial conditions. Fe ions entrapped inside the hydrogel were electrically released in Na2SO4 solution (100 mM, pH 6.0) and their concentration was determined spectrophotometrically as a function of dissolution time.First, the effect of electric current density on the dynamics of hydrogel dissolution was investigated. No Fe(II) was detected in the effluent of the microdevice when no electric current was applied. This indicates that a stable and adherent hydrogel with excellent adhesion to the gold electrode was formed. Since no degradation of the gel occurred when fluid flow (300 μL min−1) was applied, such a method could easily be used for other flow-based applications. However, the results shown in Fig. 7 clearly confirm the dissolution process of the hydrogel once an externally induced electric current was applied. Namely, the Fe(II) concentration detected in the microdevice effluent increased gradually, reached its maximum, and then decreased rapidly. A slow increase in concentration in the initial region may be due to higher diffusion limitation. At the beginning of the electro dissolution process, the thickness of the hydrogel was larger, so the reduced Fe(II) ions had to overcome a longer distance to the bulk solution. However, once the hydrogel became more deformed, ion release was facilitated. Nevertheless, the higher the current density, the faster and more intense the dissolution of the hydrogel and thus the Fe(III) reduction. The higher electric current density caused higher electric potential between both electrodes. Accordingly, the hydrogel degraded faster, releasing larger amounts of Fe ions in a shorter amount of time. For example, the maximum Fe concentration was found after 51 min at a current density of 15 A m−2 and after 89 min at a current density of 10 A m−2.
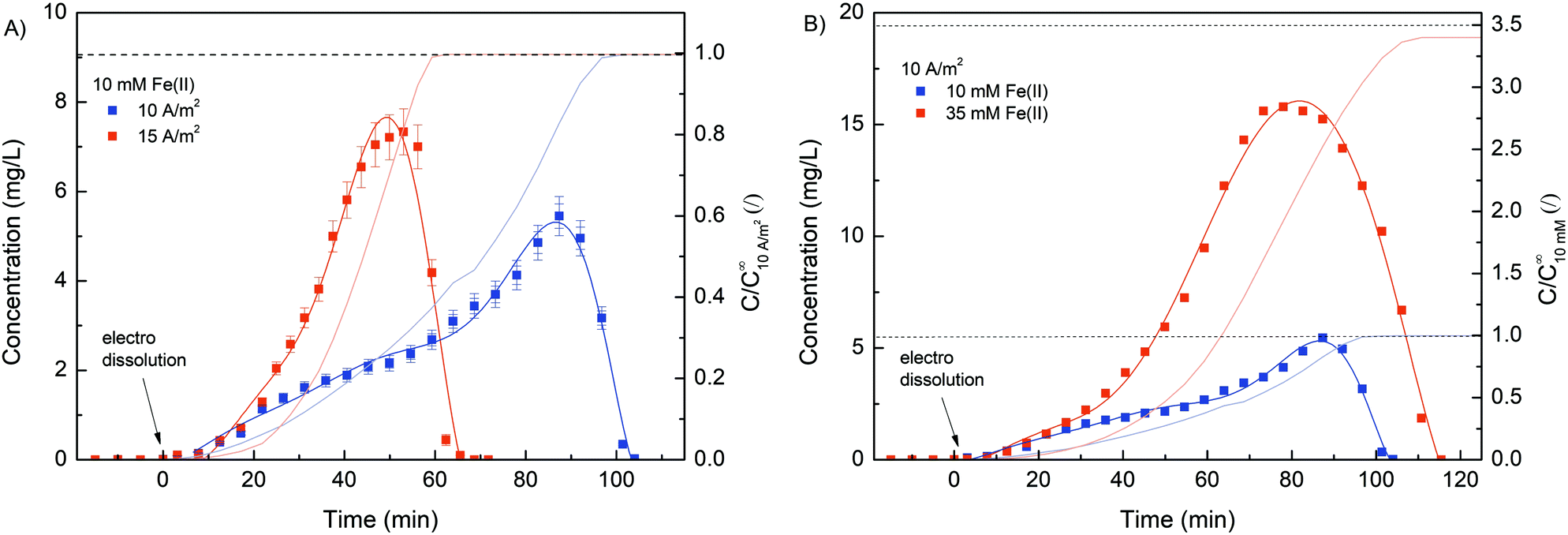 | ||
| Fig. 7 (A) Evaluation of the Fe(II) concentration profile during the electro dissolution process at different electric current densities (10 and 15 A m−2). Note that a constant Fe(II) concentration of 10 mM was applied for the electrodeposition process. (B) Evaluation of the Fe(II) concentration profile during the electro dissolution process at various electrodeposition Fe(II) concentrations (10 and 35 mM). Note that a constant electric current density of 10 A m−2 was applied for the electro dissolution process. | ||
Furthermore, the total amount of Fe ions released was determined by integrating the concentration curve, while considering the flow rate. The calculated data, summarised in Table 1, show excellent reproducibility between individual Fe masses. Since the same deposition procedure was used with the same starting materials, a comparable amount of coordinated iron was expected in the hydrogel. In addition, the Fe concentration in the hydrogel was calculated. Here, the volume of the hydrogel was estimated using the thickness analysis from the optical microscope data. Considering the deposition conditions (10 min at 10 A m−2), the hydrogel volume was estimated to be 128 mm3. Finally, the molar concentration was calculated considering the molar mass of Fe. The obtained concentrations are in good agreement with the bulk Fe concentrations, which is already evident from Fig. 1B.
| Current density [A m−2] | Fe(II) conc. for deposition [mM] | Integrated value [mg L−1 min] | Calculated mass of Fe(II) [mg] | Volume of hydrogela [mm3] | Calculated conc. of Fe(II) [mM] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Based on thickness analysis from optical microscope data. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 10 | 236 | 0.073 | 128 | 9.91 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | 10 | 240 | 0.075 | 128 | 9.98 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 35 | 815 | 0.245 | 128 | 34.27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Interestingly, a more asymmetric concentration distribution was observed (Fig. 7B) when a higher concentration of Fe(II) (35 mM) was used for the electrodeposition process. The rheological analysis suggests that the mechanical strength of the hydrogel can be effectively increased by using a higher concentration of crosslinker. Therefore, the dissolution of the hydrogel was better controlled when a higher Fe(II) concentration was applied. The calculated Fe concentration in the case of 35 mM Fe(II) was slightly lower than the concentration in the bulk solution. Similar was the case when ascorbic acid was used for hydrogel dissolution (Fig. 1B). We assumed that slight repulsive interactions occurred between the positively charged anode and Fe(II) ions due to the electrical potential. Naturally, this effect becomes more dominant at larger Fe concentrations.
To investigate the potential use of deposited hydrogel as an innovative stimuli-responsive material for drug capture and release, a series of experiments were designed with the addition of BSA. For this purpose, BSA (10 mg mL−1) was added to the initial solution (10 mM FeSO4, 1.25% w/v Na–alginate and 100 mM Na2SO4, pH 6) before each electro deposition process and pumped through a custom-made microchannel. Subsequently, electro deposition of Na–alginate was activated by an external electrical signal (10 A m−2 for 10 min), resulting in Fe(II) oxidation and consequent trapping of BSA molecules. Next, the deposited hydrogel was rinsed by pumping approximately 20 mL of 100 mM Na2SO4 through the microchannel. Finally, dissolution of the hydrogel was stimulated by reverse electric current (cathodic signal), which caused the release of entrapped BSA. The effluent samples were continuously collected, chromatographically separated, and analyzed by flow UV detector. Prior to analysis, a BSA calibration curve was prepared (Fig. S5, ESI†). Initially, the deposited hydrogel was characterized. To determine the total amount of BSA entrapped in the alginate film, the hydrogel was chemically reacted with ascorbic acid (10 mM, 30 min), resulting in complete Fe(III) reduction and film dissolution. The total amount of BSA encapsulated in the composite film was 2.1 ± 0.2 mg, which is significantly higher compared to other Fe–alginate systems (20–403 μg).25 The continuous set-up with large electrode surface employed here proved to be an excellent plateau for significantly improving the amount of encapsulated protein.
After the characterization of the deposition segment, the electrical dissolution was performed with two different current densities (10 and 15 A m−2). Again, integration of the release curve yields comparable BSA masses (∼2 mg) regardless of the current density used. Obviously, the amount of entrapped BSA was the same in both cases since the same deposition procedure was applied. Considering the BSA concentration in the initial solution (10 mg mL−1), the deposition time (10 min) and the flow rate (300 μL min−1), the total amount of pumped BSA was estimated to be about 30 mg. Since the mass of BSA entrapped was approximately 2 mg, only 7% of the total flow-through BSA was captured inside the hydrogel network. However, when considering hydrogel volume (128 mm3) and calculating an associated BSA concentration (16 mg mL−1) employing a calibration curve, a positive concentration shift compared to starting concentration (10 mg mL−1) was observed. The isoelectric point of BSA in aqueous solution is typically approximated at 4.8, which means that at working conditions (pH = 6.0), the net surface area of BSA was negatively charged. Therefore, the attractive electrostatic interactions between BSA and the positively charged anode should be formed, and the BSA concentration at the anode surface was increased accordingly.
Furthermore, the results (Fig. 8A) complement the findings from the release analysis of iron ions. The higher the electric current density was, the faster and more intense the gel dissolution and thus the BSA release. However, the concentration profile was rather inconsistent. In contrast to the Fe concentration time profiles, where Fe was released slowly at first, followed by a more rapid release, the BSA was released somewhat more intensely in the early phase of dissolution. Since the direction of electric charge was reversed, gel dissolution and thus BSA release was triggered by the cathode signal. Consequently, the interactions between the BSA and cathode were repulsive (different from during deposition), which could facilitate the rinsing of the protein in the early stage. Moreover, when the Fe(II) concentration at the BSA concentration peak was compared (after 21 and 30 min for 15 and 10 A m−2 current density, respectively), an almost identical value was found, which was about 1.5 mg L−1. This indicates that even a relatively low Fe(III) conversion (∼15%) makes the hydrogel sufficiently permeable to release most of the protein molecules. Although the average pore size, as determined by SEM analysis, was somehow higher (∼50 nm) than the average hydration radius of BSA (∼5 nm), the hydrogels still effectively entrapped the BSA molecule. This could be due to the morphology of the hydrogel network. It is possible that a closed pore structure with dead-end pores was present within the porous hydrogel. Similar observations have been reported for related systems consisting of alginate hydrogel and target biomolecule (BSA,25 Lyzocim26). In addition, these assumptions are also consistent with the observed intense BSA release during the initial dissolution stage, as only a small increase in mesh size would be required for BSA liberation.
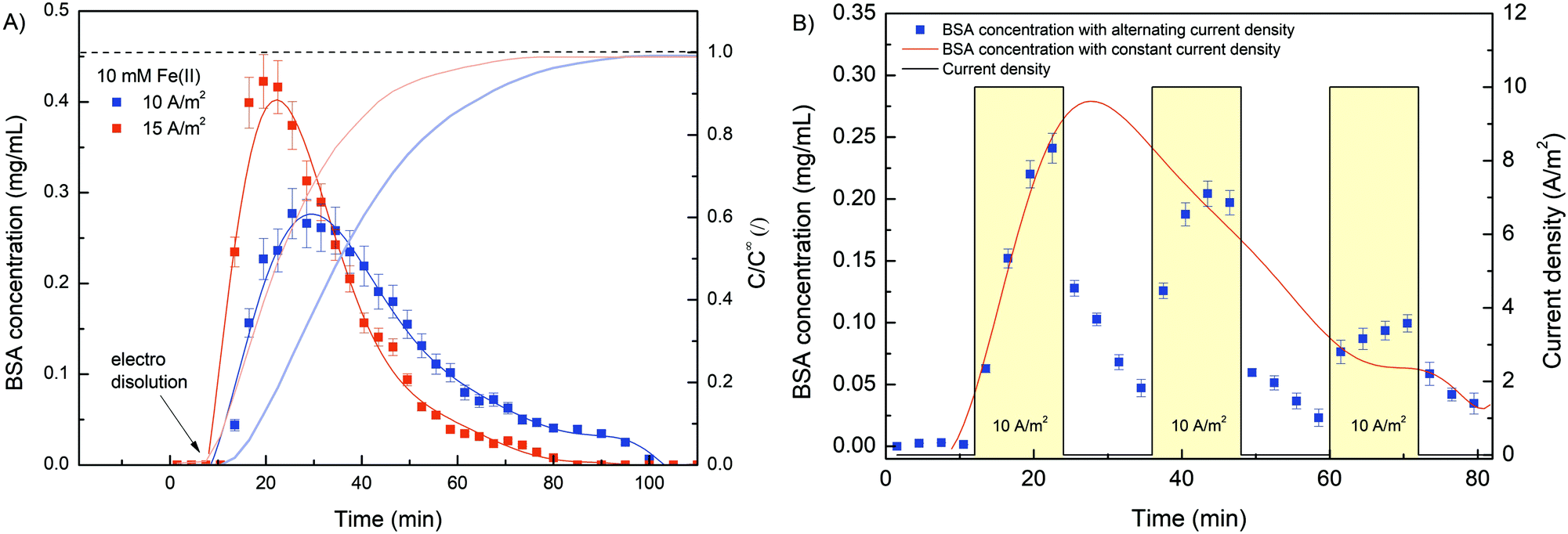 | ||
| Fig. 8 (A) Evaluation of the BSA concentration profile during the electro dissolution process at different electric current densities (10 and 15 A m−2). (B) Evaluation of the BSA concentration profile during the electro dissolution process at an alternating and constant electric current density of 10 A m−2. Note that in all cases, a constant Fe(II) concentration of 10 mM was applied for the electrodeposition process. | ||
The sensitivity and reproducibility of the stimuli responsive system also contribute strongly to its usability. In this sense, a cyclic electro dissolution process was simulated (Fig. 8B). Namely, three cycles (each 24 min long) were performed during which the electric current density alternated between 10 A m−2 and 0 A m−2, and the BSA dissolution was evaluated. During the first electrically induced BSA release its concentration profile followed almost exactly the same concentration profile obtained for constant current density release (red curve in Fig. 8B). Since an exact dissolution condition was applied in both cases, such an outcome was expected, yet it was further evidence of the reproducibility of the system. However, when the electric current was set to 0, the BSA concentration gradually decreased. We assumed that previously released BSA molecules were slowly washed out during this phase. During this process, the BSA concentration approached 0. However, as soon as the electric potential was restored, the dissolution of the hydrogel continued and the BSA concentration increased accordingly. Similarly, although to a lesser extent, was the case during the last cycle. During the 2nd and 3rd cycles, the BSA concentration reached higher values in the dissolution phase compared to the normal dissolution (with constant current density). Naturally, BSA dissolution was minimal without an applied electric current, which means that a higher amount of BSA was still trapped at a given time. As soon as gel dissolution was stimulated, the BSA concentration increased. In summary, the results show the great potential of the Fe–alginate hydrogel as a stimuli responsive material, while the generality of the system allows its implementation in various chemical and biological environments.
3.5. Protocols and algorithm for system implementation
The results of the experiments performed above allow the design of a specific algorithm routine for the implementation of the Fe–alginate hydrogel system as a programmable support media with immobilized active volume. The simple mathematical approach permits the determination of appropriate benchmarks and thus further process design optimization. By doing so, a design of experiment (DoE) and a determination of the process operating range can be performed, which is particularly important in the development phase and can serve as a basis for the quality-by-design (QbD) concept typically required in the pharmaceutical and biotechnology industries during drug development. First, the mesh size of each measured hydrogel network can be estimated based on the rheological frequency sweep test. This involves fitting a generalized Maxwell model to the results of the frequency sweep test, which allows the approximation of the shear modulus (the sum of Maxwell's elastic elements). Such a calculation was performed for our hydrogels and the results are shown in Table 2. Subsequently, the shear modulus can be directly correlated with the crosslink density of the material according to Flory's theory: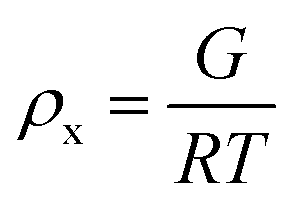 | (1) |
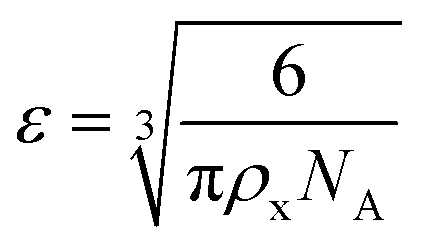 | (2) |
| C Fe [mM] | G [kPa] | ρ x [mol m−3] | ε [nm] |
|---|---|---|---|
| 1 | 0.56 | 0.226 | 24.1 |
| 3 | 2.20 | 0.888 | 15.3 |
| 5 | 10.61 | 4.282 | 9.1 |
| 10 | 23.54 | 9.501 | 6.9 |
| 20 | 48.99 | 19.773 | 5.4 |
| 35 | 78.37 | 31.631 | 4.6 |
Moreover, the deposition conditions (time and current density) effectively control the hydrogel thickness. Since hydrogels with homogeneous thickness were formed over the entire available electrode the active volume of the hydrogel can be easily estimated. Obviously, a higher active volume provides a larger amount of entrapped target molecules (BSA in our case). Therefore, tuning the process conditions allows the formation of the desired hydrogel and, at the same time, a regulated target molecule immobilization. Finally, electro-dissolution can be used to expand the hydrogel network and thus control the molecule release. In this way, the diffusion boundaries are loosened, which significantly improves mass transfer. Accordingly, a hydrogel whose mesh size is controlled by external stimuli can be applied for various end-use applications. For example, it can be utilized as a catalytic support for enzymatic reactions (the mesh size is designed to allow free diffusion of small molecules to the enzyme surface but prevent enzyme leakage), for controlled drug release (once immobilized in the hydrogel network, the drug component can be released in a controlled manner by an external electrical signal), for cell separation (based on the size exclusion effect), and so on. The above proposed protocol for system implementation is schematically shown in Fig. 9. Such an algorithm has great potential for process automation, which could be particularly useful in pharmaceutical and/or related biotechnological environments.
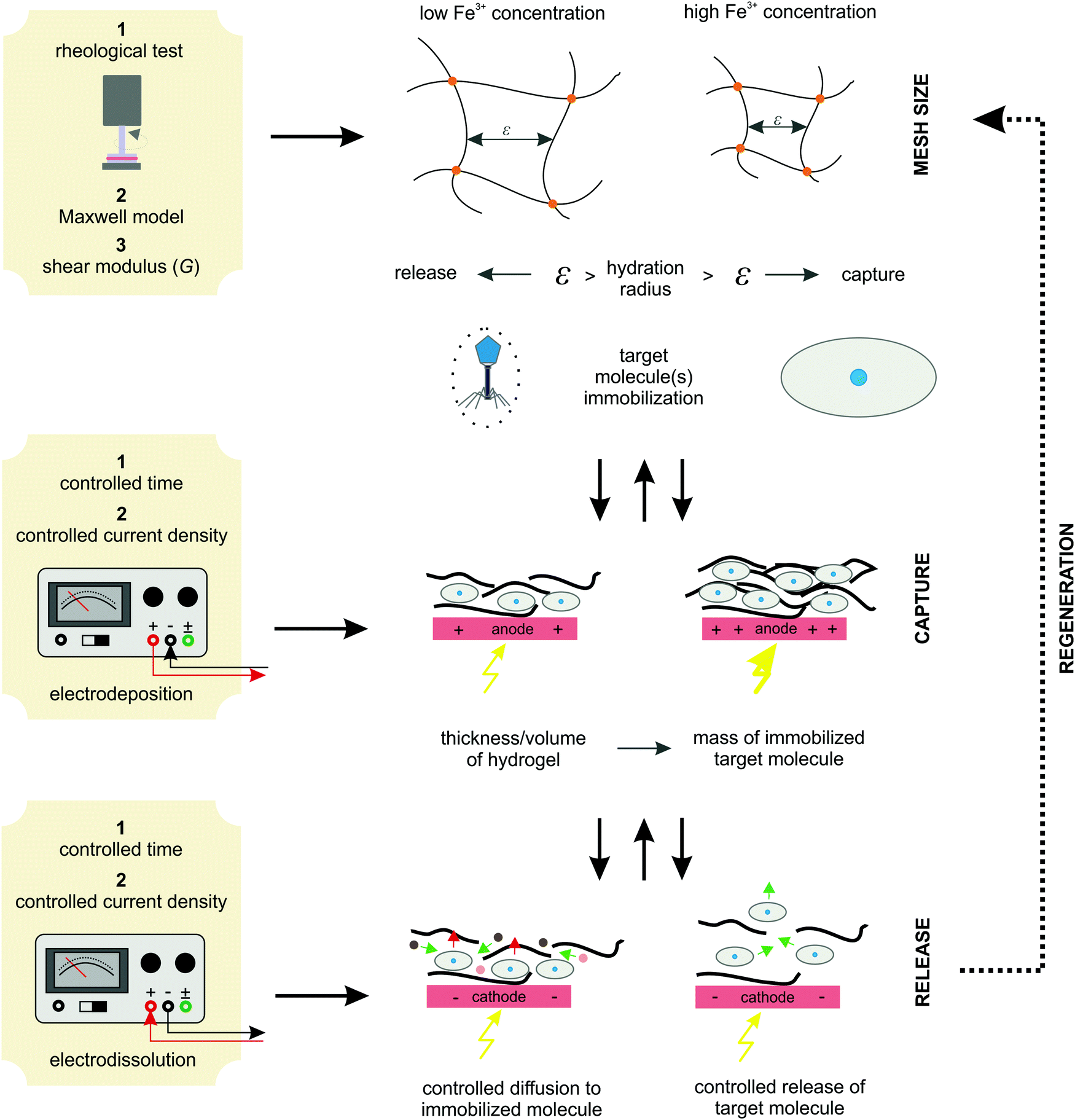 | ||
| Fig. 9 The proposed algorithm protocol for the implementation of an Fe-alginate hydrogel as an innovative stimuli-responsive active medium for biotech environments. | ||
4. Conclusions
The electrochemical deposition and dissolution of an Fe-crosslinked alginate hydrogel was investigated as a general approach to a controlled drug release process exemplified by BSA release. The sol–gel transition was triggered by an external electrical signal causing a change in the oxidation state of Fe ions. While Fe(III) cations lead to the formation of an alginate hydrogel, Fe(II) ions have a weak interaction with alginate and their electrochemical formation leads to the dissolution of the alginate hydrogel. The electrical signal stimulating the sol–gel transition was induced by a plate-shaped golden electrode placed inside the fabricated microchannel. The system allows the flow set-up and thus a continuous and/or cyclic operation mode. We have shown that the thickness of the in situ deposited hydrogel can be effectively controlled on the μm scale by deposition conditions, such as time and current density. For example, when the deposition time was changed from 3 min to 10 min, the thickness of the deposited film increased from 140 μm to 310 μm. Similarly, the increase in current density caused the increase in hydrogel thickness. The rheological study allows an estimation of the hydrogel mesh size, which ranges from 5 nm to 25 nm for an Fe concentration of 35 mM and 1 mM, respectively. Such a network could potentially be used as a porous medium for immobilization of large biological molecules, such as enzymes, cells and/or viruses. Since the mesh network can be easily adapted to the requirements of the application, one can design a nanostructure that stores active molecule(s), while allowing free diffusion of other small molecules through the hydrogel.The electrochemically induced alginate hydrogel formation was accompanied by the entrapment of BSA in the hydrogel thin film on the electrode surface. The electrochemical reduction of the crosslinking Fe(III) ions led to the dissolution of the alginate hydrogel and consequent release of BSA. The higher the current density, the more intense the hydrogel dissolution and thus the BSA and Fe release. Accordingly, by controlling Fe(III) reduction (via an electrical signal), hydrogel dissolution can be targeted and programmed. The latter was demonstrated by changing the electrical conditions, and the programmable hydrogel dissolution was clearly confirmed by an altering external electrical signal. Since hydrogel dissolution allows controlled release such a system could be directly applied on various advance systems.
Author contributions
Rok Ambrožič: conceptualization, methodology, investigation, data curation, writing – original draft preparation. Igor Plazl: conceptualization, data curation, supervision, validation, writing-reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Slovenian Research Agency (Research Core Funding No. P2-0191 and projects L2-7633, N2-0067, and BI-DA/20-22-003). The support through the H2020 project COMPETE, Slovenia (Grant No. 811040) and CELSA 2020 is also acknowledged. The authors also thank Dr Marjan Marinšek for SEM images.References
- W. Lu, X. X. Le, J. W. Zhang, Y. J. Huang and T. Chen, Chem. Soc. Rev., 2017, 46, 1284–1294 RSC.
- M. L. Wei, Y. F. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- J. Kolosnjaj-Tabi, L. Gibot, I. Fourquaux, M. Golzio and M. P. Rols, Adv. Drug Delivery Rev., 2019, 138, 56–67 CrossRef CAS PubMed.
- H. Palza, P. A. Zapata and C. Angulo-Pineda, Materials, 2019, 12 DOI:10.3390/ma12020277.
- R. P. Narayanan, G. Melman, N. J. Letourneau, N. L. Mendelson and A. Melman, Biomacromolecules, 2012, 13, 2465–2471 CrossRef CAS PubMed.
- A. Rucigaj, R. Ambrozic and M. Krajnc, Macromol. Mater. Eng., 2020, 305 DOI:10.1002/mame.202000463.
- Q. Tang, D. L. Zhao, H. Y. Yang, L. J. Wang and X. Y. Zhang, J. Mater. Chem. B, 2019, 7, 30–42 RSC.
- J. Qu, X. Zhao, P. X. Ma and B. L. Guo, Acta Biomater., 2017, 58, 168–180 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. L. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- Z. X. Deng, Y. Guo, X. Zhao, P. X. Ma and B. L. Guo, Chem. Mater., 2018, 30, 1729–1742 CrossRef CAS.
- Q. Lu, S. M. Bai, Z. Z. Ding, H. Guo, Z. Z. Shao, H. S. Zhu and D. L. Kaplan, Adv. Mater. Interfaces, 2016, 3 DOI:10.1002/admi.201500687.
- M. Y. Choi, Y. Shin, H. S. Lee, S. Y. Kim and J. H. Na, Sci. Rep., 2020, 10 DOI:10.1038/s41598-020-59318-3.
- Z. H. Geng, X. Wang, X. C. Guo, Z. Zhang, Y. J. Chen and Y. F. Wang, J. Mater. Chem. B, 2016, 4, 3331–3338 RSC.
- F. Ullah, M. B. H. Othman, F. Javed, Z. Ahmad and H. M. Akil, Mater. Sci. Eng., C, 2015, 57, 414–433 CrossRef CAS PubMed.
- S. Mondal, S. Das and A. K. Nandi, Soft Matter, 2020, 16, 1404–1454 RSC.
- R. Bernasconi, E. Mauri, A. Rossetti, S. Rimondo, R. Suriano, M. Levi, A. Sacchetti, S. Pane, L. Magagnin and F. Rossi, Mater. Des., 2021, 197 DOI:10.1016/j.matdes.2020.109212.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef CAS PubMed.
- M. A. Mohamed, A. Fallahi, A. M. A. El-Sokkary, S. Salehi, M. A. Akl, A. Jafari, A. Tamayol, H. Fenniri, A. Khademhosseini, S. T. Andreadis and C. Cheng, Prog. Polym. Sci., 2019, 98 DOI:10.1016/j.progpolymsci.2019.101147.
- M. Tanaka, M. Nakahata, P. Linke and S. Kaufmann, Polym. J., 2020, 52, 861–870 CrossRef CAS.
- A. Kirillova, R. Maxson, G. Stoychev, C. T. Gomillion and L. Ionov, Adv. Mater., 2017, 29 DOI:10.1002/adma.201703443.
- B. Xue, V. Kozlovskaya and E. Kharlampieva, J. Mater. Chem. B, 2017, 5, 9–35 RSC.
- P. Baei, S. Jalili-Firoozinezhad, S. Rajabi-Zeleti, M. Tafazzoli-Shadpour, H. Baharvand and N. Aghdami, Mater. Sci. Eng., C, 2016, 63, 131–141 CrossRef CAS PubMed.
- J. X. Yan, Y. T. Miao, H. P. Tan, T. L. Zhou, Z. H. Ling, Y. Chen, X. D. Xing and X. H. Hu, Mater. Sci. Eng., C, 2016, 63, 274–284 CrossRef CAS PubMed.
- H. J. Wen, J. Y. Li, G. F. Payne, Q. Feng, M. H. Liang, J. X. Chen, H. Dong and X. D. Cao, Biofabrication, 2020, 12 DOI:10.1088/1758-5090/ab7e74.
- Z. Y. Jin, G. Guven, V. Bocharova, J. Halamek, I. Tokarev, S. Minko, A. Melman, D. Mandler and E. Katz, ACS Appl. Mater. Interfaces, 2012, 4, 466–475 CrossRef CAS PubMed.
- Z. Y. Jin, A. M. Harvey, S. Mailloux, J. Halamek, V. Bocharova, M. R. Twiss and E. Katz, J. Mater. Chem., 2012, 22, 19523–19528 RSC.
- G. F. Wu, K. Y. Jin, L. Liu and H. X. Zhang, Soft Matter, 2020, 16, 3319–3324 RSC.
- Y. G. Kang, H. C. Vu, T. T. Le and Y. S. Chang, Chem. Eng. J., 2018, 353, 736–745 CrossRef CAS.
- F. A. Vicente, I. Plazl, S. P. M. Ventura and P. Znidarsic-Plazl, Green Chem., 2020, 22, 4391–4410 RSC.
- T. Menegatti and P. Znidarsic-Plazl, Micromachines, 2019, 10 DOI:10.3390/mi10120867.
- N. Milozic, G. Stojkovic, A. Vogel, D. Bouwes and P. Znidarsic-Plazl, New Biotechnol., 2018, 47, 18–24 CrossRef CAS PubMed.
- J. M. Bolivar and F. Lopez-Gallego, Curr. Opin. Green Sustainable Chem., 2020, 25 DOI:10.1016/j.cogsc.2020.04.010.
- C. B. Goy, R. E. Chaile and R. E. Madrid, React. Funct. Polym., 2019, 145 DOI:10.1016/j.reactfunctpolym.2019.104314.
- M. Yamada, S. Sugaya, Y. Naganuma and M. Seki, Soft Matter, 2012, 8, 3122–3130 RSC.
- N. Milozic, M. Lubej, M. Lakner, P. Znidarsic-Plazl and I. Plazl, Chem. Eng. J., 2017, 313, 374–381 CrossRef CAS.
- R. Wohlgemuth, I. Plazl, P. Znidarsic-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef CAS PubMed.
- J. Nie, Q. Gao, Y. D. Wang, J. H. Zeng, H. M. Zhao, Y. Sun, J. Shen, H. Ramezani, Z. L. Fu, Z. J. Liu, M. X. Xiang, J. Z. Fu, P. Zhao, W. Chen and Y. He, Small, 2018, 14 DOI:10.1002/smll.201802368.
- Y. H. Hsiao, C. Y. Huang, C. Y. Hu, Y. Y. Wu, C. H. Wu, C. H. Hsu and C. C. Chen, Sci. Rep., 2016, 6 DOI:10.1038/srep32454.
- E. Choi, I. Jun, H. K. Chang, K. M. Park, H. Shin, K. D. Park and J. Park, Lab Chip, 2012, 12, 302–308 RSC.
- X. L. Luo, D. L. Berlin, J. Betz, G. F. Payne, W. E. Bentley and G. W. Rubloff, Lab Chip, 2010, 10, 59–65 RSC.
- N. Bhattacharjee, L. F. Horowitz and A. Folch, Appl. Phys. Lett., 2016, 109, 163702 CrossRef CAS PubMed.
- A. Hatch, D. M. Pesko and S. K. Murthy, Anal. Chem., 2012, 84, 4618–4621 CrossRef CAS PubMed.
- Y. B. Q. Qi, T. Zou, W. L. Dissanayaka, H. M. Wong, L. E. Bertassoni and C. F. Zhang, J. Endod., 2021, 47, 52–61 CrossRef.
- J. Aymerich, A. Marquez, L. Teres, X. Munoz-Berbel, C. Jimenez, C. Dominguez, F. Serra-Graells and M. Dei, Biosens. Bioelectron., 2018, 117, 736–742 CrossRef CAS PubMed.
- D. Semenova, A. C. Fernandes, J. M. Bolivar, I. P. R. Grundtvig, B. Vadot, S. Galvanin, T. Mayr, B. Nidetzky, A. Zubov and K. V. Gernaey, New Biotechnol., 2020, 56, 27–37 CrossRef CAS PubMed.
- H. Nawaz, W. G. Tian, J. M. Zhang, R. N. Jia, Z. Y. Chen and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 2114–2121 CrossRef CAS PubMed.
- Y. Cheng, X. L. Luo, J. Betz, S. Buckhout-White, O. Bekdash, G. F. Payne, W. E. Bentley and G. W. Rubloff, Soft Matter, 2010, 6, 3177–3183 RSC.
- K. M. Gray, B. D. Liba, Y. F. Wang, Y. Cheng, G. W. Rubloff, W. E. Bentley, A. Montembault, I. Royaud, L. David and G. F. Payne, Biomacromolecules, 2012, 13, 1181–1189 CrossRef CAS PubMed.
- Y. Cheng, X. L. Luo, J. Betz, G. F. Payne, W. E. Bentley and G. W. Rubloff, Soft Matter, 2011, 7, 5677–5684 RSC.
- G. Lawrie, I. Keen, B. Drew, A. Chandler-Temple, L. Rintoul, P. Fredericks and L. Grondahl, Biomacromolecules, 2007, 8, 2533–2541 CrossRef CAS PubMed.
- J. Liu, H. T. Wu, J. F. Lu, X. Y. Wen, J. Kan and C. H. Jin, Chem. Eng. J., 2015, 262, 803–812 CrossRef CAS.
- G. X. Li, Y. M. Du, Y. Z. Tao, H. B. Deng, X. G. Luo and J. H. Yang, Carbohydr. Polym., 2010, 82, 706–713 CrossRef CAS.
- Y. C. Dong, W. J. Dong, Y. N. Cao, Z. B. Han and Z. Z. Ding, Catal. Today, 2011, 175, 346–355 CrossRef CAS.
- A. R. Patel, P. Dumlu, L. Vermeir, B. Lewille, A. Lesaffer and K. Dewettinck, Food Hydrocolloids, 2015, 46, 84–92 CrossRef CAS.
- S. M. Hashemnejad and S. Kundu, Soft Matter, 2019, 15, 7852–7862 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm00510c |
| This journal is © The Royal Society of Chemistry 2021 |