
Morphology evolution with polymer chain propagation and its impacts on device performance and stability of non-fullerene solar cells†
Long
Zhang
a,
Xuelong
Huang
 b,
Chunhui
Duan
*ac,
Zhongxiang
Peng
d,
Long
Ye
*ad,
Nigel
Kirby
e,
Fei
Huang
*a and
Yong
Cao
a
b,
Chunhui
Duan
*ac,
Zhongxiang
Peng
d,
Long
Ye
*ad,
Nigel
Kirby
e,
Fei
Huang
*a and
Yong
Cao
a
aInstitute of Polymer Optoelectronic Materials & Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: duanchunhui@scut.edu.cn; msfhuang@scut.edu.cn
bCollege of Pharmacy, Gannan Medical University, Ganzhou 341000, P. R. China
cBeijing National Laboratory for Molecular Sciences, Beijing 100190, P. R. China
dSchool of Materials Science and Engineering, Tianjin University, Tianjin 300350, P. R. China. E-mail: yelong@tju.edu.cn
eAustralian Synchrotron, Clayton, Victoria 3168, Australia
First published on 8th December 2020
Abstract
Active layer morphology is a key factor that dictates the device performance and stability of polymer solar cells (PSCs). Herein, we investigate the morphology evolution of the active layer and the impacts on device performance and stability upon varying the molecular weights of polymer donor (PBDB-T4Cl5S) from 17 to 98 kg mol−1. When blended with a leading non-fullerene acceptor (Y6), both the short-circuit current density and fill factor of the PSCs rise gradually with increasing the polymer molecular weights. Detailed morphology analysis showed that the reduced phase separation scale and improved phase purity accounted for these enhancements. Interestingly, the best device stability was achieved by the polymer with a medium molecular weight of 30 kg mol−1, which is most likely caused by the nearly-optimal miscibility between the polymer donor and Y6. These results demonstrate that the optimization of molecular weights of polymer donors is crucial to achieve high-efficiency and high-stability simultaneously for non-fullerene PSCs.
Introduction
Polymer solar cells (PSCs) have attracted massive interests due to their unique merits such as flexibility, light-weight, and solution processability for achieving high-throughput and large-scale manufacturing.1 Remarkable breakthroughs have been made in recent years owing to the development of non-fullerene acceptors,2–7 which have enabled the power conversion efficiencies (PCEs) of PSCs surpassing 17% in both single junction and tandem devices.8–11 However, the PCEs of PSCs still lag much behind those of copper indium gallium diselenide (CIGS) and perovskite solar cells. Moreover, the poor stability under thermal stress and light illumination is also a critical limitation to the commercialization of PSCs.12,13 The PCEs of PSCs depend mainly on the fundamental optoelectronic properties including energy levels, light absorption capability, and charge mobilities of the photoactive materials.1,14–16 Besides, the packing modes, molecular orientation, crystallinity, and phase separation morphology of the donor:acceptor blends are critical to the device performance of PSCs as well.17,18 Generally, an efficient non-fullerene PSC requires proper phase separation and relatively high phase purity in the donor:acceptor blend to facilitate exciton dissociation and charge transport.19,20 The device stability of PSCs was revealed to be related to the miscibility and phase compositions between the donors and acceptors.21–23 Therefore, it is of significant importance to reveal the factors that affect the active layer morphology and how these factors influence the device performance and stability of PSCs.As a basic attribute of polymers, the molecular weight can significantly influence optical,24,25 electrical,26–28 and mechanical properties29–31 of conjugated polymers. Meanwhile, the impacts of polymer molecular weights on bulk-heterojunction (BHJ) morphology and device performance of PSCs have been extensively investigated.32–38 For example, Janssen et al. reported that the molecular weight of polymer donors could controls the phase separation scale and the formation of fibrillar networks in polymer:fullerene blends. The fiber width declines with increasing polymer molecular weight due to the solubility-governed nucleation-and-growth of free polymers.34 Ye et al. found that highly immiscible polymer:non-fullerene acceptor blend needs to be kinetically quenched to lock in the high-performance BHJ morphology where the compositions of mixed domains get close to the percolation threshold for electron transport. And the highest molecular weight is capable of an early liquid–solid transition to lock in an optimal morphology in non-fullerene PSCs.37 These studies were mainly devoted to the impact of polymer molecular weight on active layer morphology and device performance, while the influence on morphological stability and device stability remains poorly understood,39–41 let alone the influential factors behind.
In this work, we focus on the morphology evolution with the polymer chain propagation and investigate its impacts on device performance and stability of non-fullerene PSCs. A set of chlorinated and alkylthio substituted polymer donors (PBDB-T4Cl5S) with molecular weights ranging from 17 to 98 kg mol−1 was synthesized, which are denoted as P-17k, P-30k, P-53k, and P-98k, respectively. It is worth pointing out that the polymer with the same structure was reported during the course of our study.42 When blended with a leading non-fullerene acceptor Y6, the short-circuit current density (Jsc) and fill factor (FF) of the PSCs increased monotonically with increasing molecular weights of the polymers. Correspondingly, the PCEs of the PSCs increased from 11.2% for P-17k to 15.3% for P-30k, 16.3% for P-53k, and 16.2% for P-98k. Morphology studies showed that the phase separation scale reduced and phase purity increased monotonically in polymer:Y6 blends with lengthening the polymer chains. In contrast, such a monotonical trend was not observed for device stability, and the most stable device upon storage and thermal stress was achieved with P-30k. These intriguing results were further explained from the thermodynamic intermolecular interactions between the polymer donors and Y6. Therefore, this work not only revealed the morphology evolution with polymer chain propagation but also suggested the importance of molecular weight optimization in achieving high-efficiency and high-stability simultaneously in non-fullerene PSCs.
Results and discussion
Polymer synthesis and characterizations
The chemical structure and synthetic route of PBDB-T4Cl5S are shown in Fig. 1a. The details of synthesis and characterizations are presented in the ESI.† The polymers with various molecular weights were synthesized through palladium-catalyzed Stille cross-coupling reactions from BDTT-4Cl5S and BDD monomers by tuning the reaction conditions. The number-average molecular weight (Mn) and molar-mass dispersity (ĐM = Mw/Mn) of the polymers were measured by gel permeation chromatography using ortho-dichlorobenzene (o-DCB) as the eluent at 140 °C. The GPC traces are shown in Fig. S7,† and the numerical data are listed in Table 1. The Mn/ĐMs are 17 kg mol−1/2.6, 30 kg mol−1/1.9, 53 kg mol−1/1.6, and 98 kg mol−1/1.4 for P-17k, P-30k, P-53k, and P-98k, respectively. The solubility of the polymers decreases distinctly with increasing molecular weights. P-17k and P-30k are readily soluble in chloroform (CF) and chlorinated aromatics including chlorobenzene (CB) and o-DCB at room temperature, P-53k is hardly soluble in CF at room temperature but readily soluble in CB at room temperature, while P-98k can only be dissolved in hot CB (or o-DCB) and the solution gelated upon cooling (Fig. 1b). The thermal properties of the polymers were evaluated by thermogravimetric analysis (TGA). The detailed parameters are listed in Table 1, and the corresponding thermograms are shown in Fig. S8.† The polymers exhibit good thermal stability with decomposition temperatures (Td, 5% weight loss temperature) over 350 °C, and the thermal stability improves monotonically with polymer chain propagation, with the lowest Td of 350 °C for P-17k and the highest Td of 381 °C for P-98k.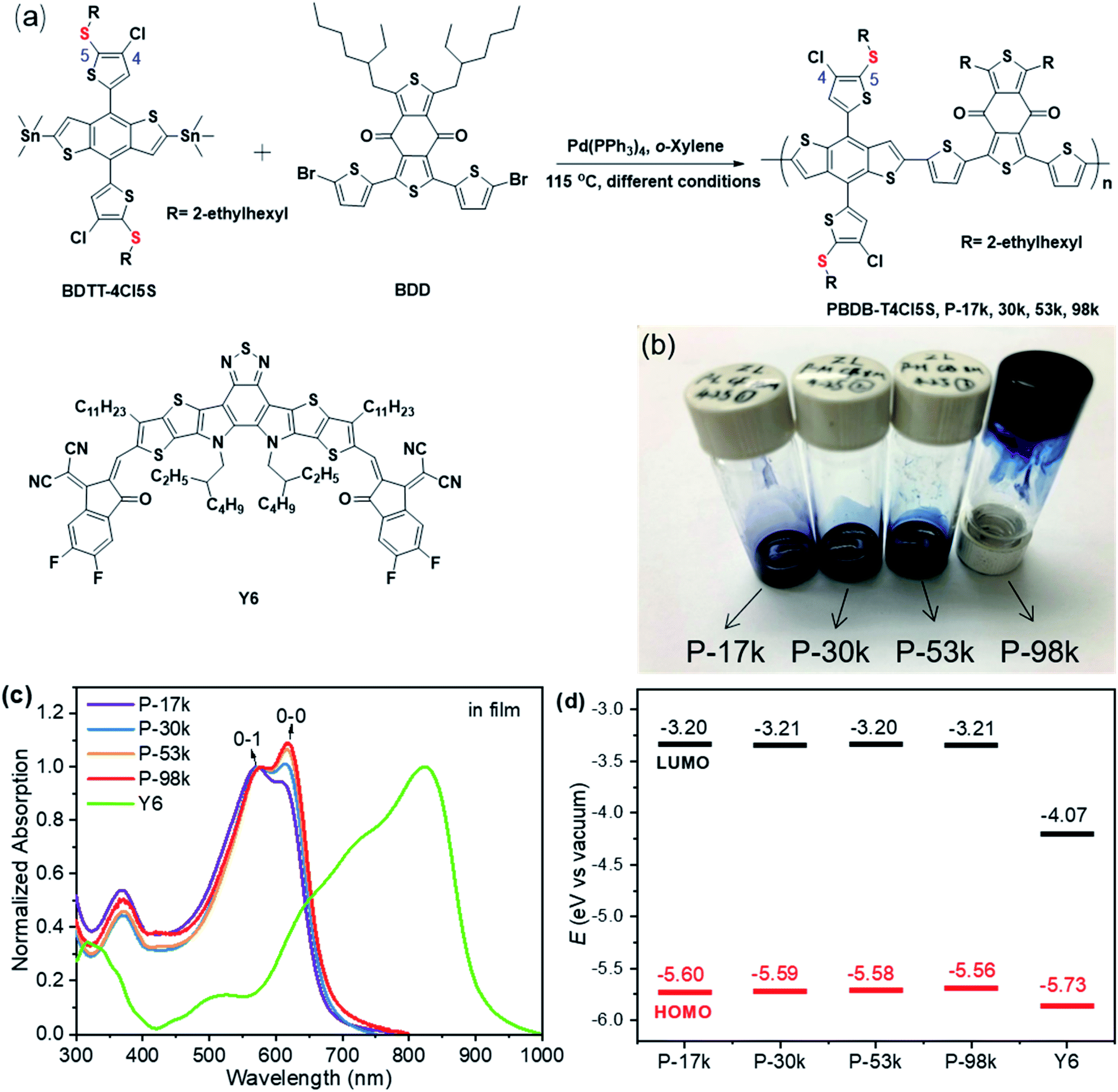 | ||
| Fig. 1 (a) The synthetic route of the polymers and the chemical structure of Y6; (b) the picture for comparing polymer solubility: P-17k and P-30k in CF, P-53k and P-98k in CB; (c) the absorption spectra in films and (d) energy levels of the polymers and Y6. | ||
| Polymers |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n [kg mol−1]
n [kg mol−1] |
Đ M | T d [°C] | Absorption | LUMOb [eV] | HOMOb [eV] | μ h [cm2 V−1 s−1] | |
|---|---|---|---|---|---|---|---|---|
| Solutiona 0-0/0-1 | Filma 0-0/0-1 | |||||||
| a Absorbance ratios of 0-0 peaks and 0-1 peaks. b Determined by SWV, EHOMO = −(Eox + 5.13 − EFc/Fc+) eV, ELUMO = −(Ered + 5.13 − EFc/Fc+) eV, EFc/Fc+ = 0.63 V. c Measured by SCLC method. | ||||||||
| P-17k | 17 | 2.6 | 350 | 1.00 | 0.95 | −3.20 | −5.60 | (1.6 ± 0.6) × 10−4 |
| P-30k | 30 | 1.9 | 369 | 1.10 | 1.01 | −3.21 | −5.59 | (2.4 ± 0.4) × 10−3 |
| P-53k | 53 | 1.6 | 373 | 1.15 | 1.07 | −3.20 | −5.58 | (3.0 ± 0.5) × 10−3 |
| P-98k | 98 | 1.4 | 381 | 1.20 | 1.09 | −3.21 | −5.56 | (5.5 ± 0.2) × 10−3 |
The UV-vis absorption spectra of the polymers are shown in Fig. 1c and S9a.† The absorption onsets of the polymers are all around 664 nm in solutions while those in solid states are slightly red-shifted with the propagation of polymer chains, indicating the improvement of interchain π–π stacking of the polymers in solid states. By normalizing the 0-1 absorption peaks and comparing them with 0-0 absorption peaks in both solutions and solid states, the 0-0 absorption peaks rise apparently with the increase of molecular weights. The relative aggregation degrees of the polymers were further investigated by comparing the ratios between 0-0 and 0-1 peaks. The plots of these ratios with molecular weights are presented in Fig. S9b.† The enhanced aggregation degrees in both solutions and solid states with the polymer chain propagation are observed, further evidencing the improved interchain π–π stacking of the polymers. To further investigate the aggregation behaviors in solutions, temperature-dependent absorption spectra of the polymers were recorded. As shown in Fig. S10,† the 0-0 absorption peaks of the higher molecular weight polymers (P-53k and P-98k) decrease less significantly than the lower counterparts (P-17k and P-30k), confirming the fact that the propagation of polymer chain leads to higher aggregation degree. The higher aggregation degree of long polymer chains probably results from the coiling, cluster-like chain conformation, or entanglement effects while the relatively less aggregation of the lower-weight polymer is due to weak interactions between the shorter polymer chains.30,31
The energy levels of the polymers and Y6 were measured by square wave voltammetry (SWV). The lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) energy levels were calculated by using the energy level of −5.13 eV versus vacuum for ferrocene (Fc/Fc+). The relevant results are presented in Fig. 1d, Table 1, and Fig. S11.† The LUMO levels of the polymers are constant to be −3.20 eV, whereas the HOMO levels up-shifted slightly from −5.60 to −5.56 eV upon increasing the polymer molecular weights, which might be due to the incremental conjugated length with the chain propagation.43 The LUMO–LUMO and HOMO–HOMO offsets between the polymers and Y6 are approximate 0.85 and 0.15 eV, respectively, suggesting that the energetic driving forces for hole and electron transfer among the four polymer:Y6 blends are similar.
The microstructures of the pure polymer films were investigated by two-dimensional (2D) grazing-incidence wide-angle X-ray scattering (GIWAXS). The 2D-GIWAXS patterns and one-dimensional (1D) line profiles are shown in Fig. 2a, b, and c, respectively. The (100) lamellar peaks can be found in both out-of-plane (OOP) and in-plane (IP) directions among all the polymers with an IP (100) lamellar distance of 20–22 Å. The (h00) peaks are also distinct except for P-17k, suggesting higher ordering of the higher molecular weight polymers. The OOP (010) peaks get more pronounced upon increasing the molecular weight from 17 to 98 kg mol−1. Meanwhile, the q values corresponding to the (010) peaks increase slightly from 1.79 Å−1 for P-17k to 1.81 Å−1 for P-98k, and the relevant π–π stacking distances were calculated to be 3.51, 3.49, 3.49, 3.47 Å for P-17k, P-30k, P-53k, and P-98k, respectively. The denser π–π stacking of the higher molecular weight polymers confirmed the enhancement of packing ordering in solid states. Besides, the corrected pole figures44† of the (010) stacking peaks of neat polymer films (Fig. 2d) suggested that the face-on/edge-on ratio increases from ∼0.2 to ∼2.9 with the polymer molecular weight and the highest fraction of face-on orientation is obtained in the highest molecular weight polymer P-98k. The hole mobilities of the polymers in vertical direction were evaluated in hole-only devices by fitting the acquired data to space-charge-limited-current (SCLC) model.45 As shown in Table 1 and Fig. S14a,† the hole mobility of the polymers exhibited the trend of P-17k < P-30k < P-53k < P-98k. The incremental hole mobility with polymer molecular weights is consistent with the higher population of face-on orientation in solid states.
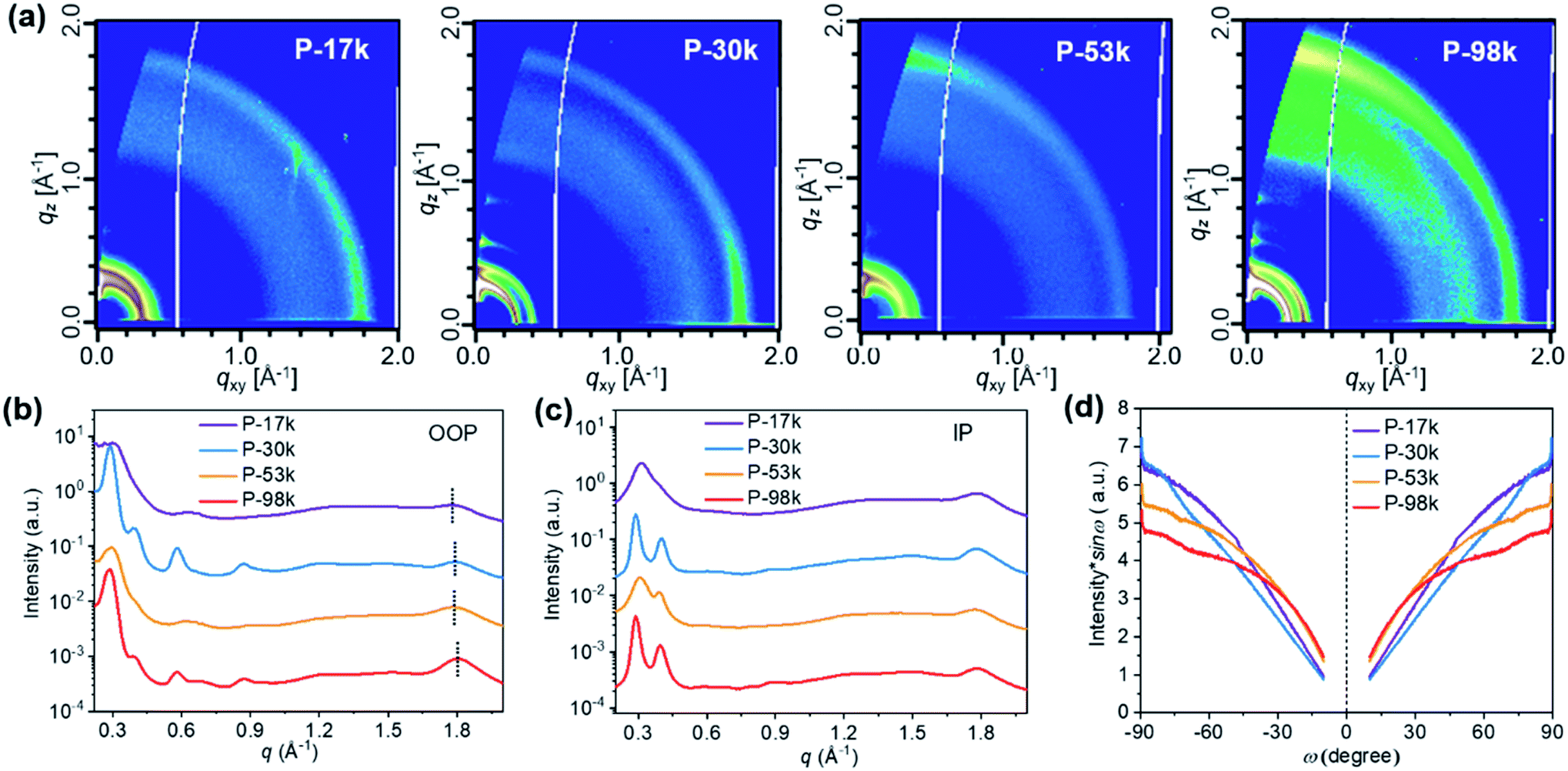 | ||
| Fig. 2 (a) 2D GIWAXS patterns, (b) 1D OOP, and (c) IP profiles of pure polymer films. (d) Pole figure of (010) peak for the pure polymer films. Here ω is the polar angle of (010) peak. | ||
Photovoltaics properties and electrical characteristics
The photovoltaic properties of the polymers were evaluated in PSCs with a device architecture of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS)/polymer:Y6/PNDIT-F3N-Br/Ag, where PNDIT-F3N-Br is poly[(9,9-bis(3′-((N,N-dimethyl)-N-ethylammonium) propyl)-2,7-fluorene)-alt-5,5′-bis(2,2′-thiophene)-2,6-naphthalene-1,4,5,8-tetracaboxylic-N,N′-di(2-ethylhexyl)imide]dibromide.46 The current density–voltage (J–V) characteristics under AM1.5G illumination (100 mW cm−2) and the external quantum efficiency (EQE) spectra of the champion devices are shown in Fig. 3a and b, respectively. The representative device parameters are summarized in Table 2.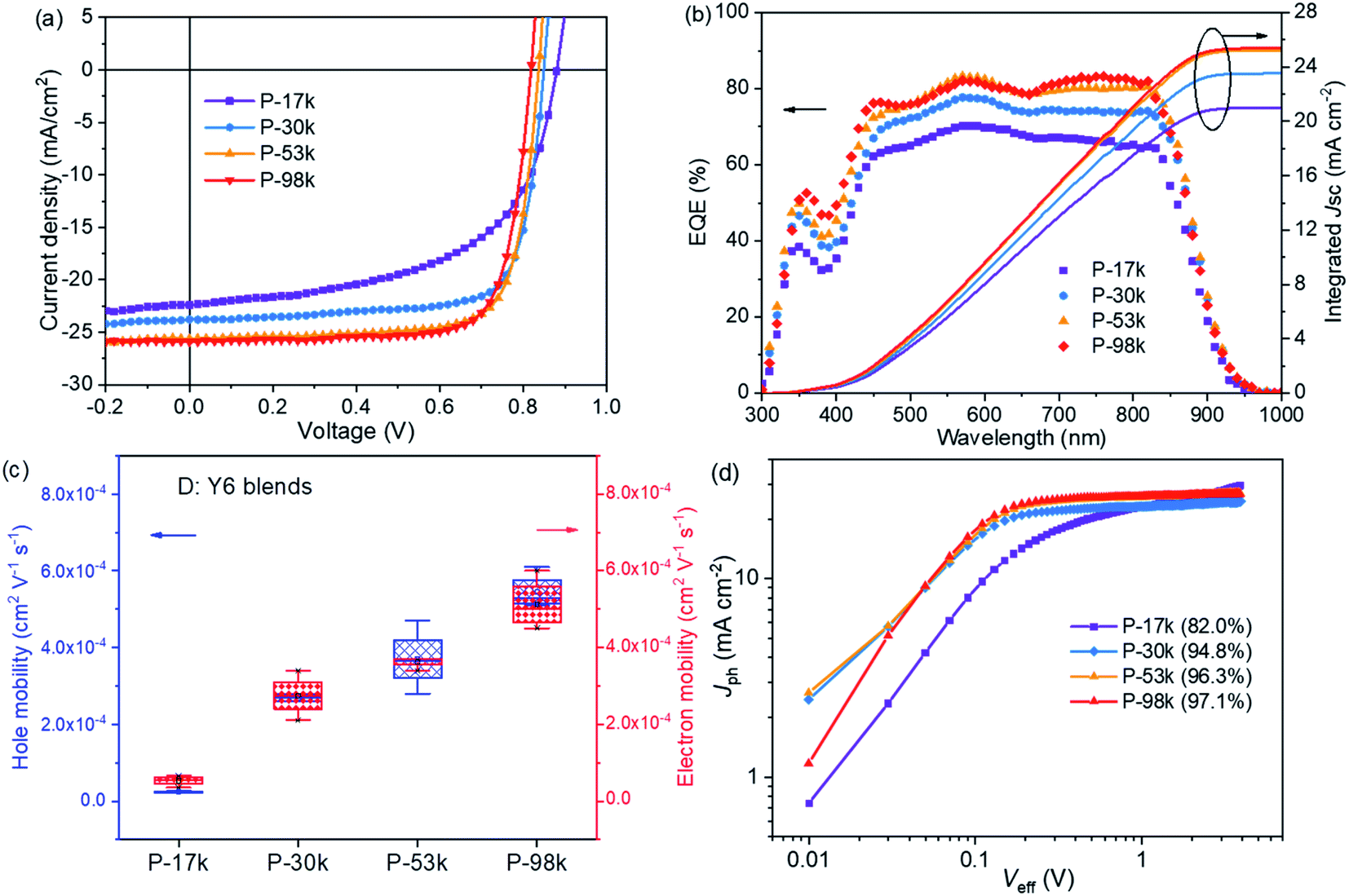 | ||
| Fig. 3 (a) J–V curves and (b) EQE spectra of the polymer:Y6 PSCs; (c) hole and electron mobilities of the polymer:Y6 blends acquired from single-carrier devices; (d) Jphversus Veff of the polymer:Y6 solar cells. | ||
| Polymer | V oc [V] | J sc [mA cm−2] | J sc (EQE)b [mA cm−2] | FF | PCE [%] |
|---|---|---|---|---|---|
| a The data in brackets are the average values and standard deviation of at least 16 independent devices for each blend. b The Jsc (EQE) is obtained from the integration of EQEs to AM1.5G spectra. | |||||
| P-17k | 0.88 (0.88 ± 0.00) | 22.4 (22.5 ± 0.3) | 21.5 | 0.57 (0.55 ± 0.01) | 11.2 (10.9 ± 0.2) |
| P-30k | 0.86 (0.85 ± 0.00) | 23.8 (23.8 ± 0.5) | 23.5 | 0.75 (0.73 ± 0.03) | 15.3 (14.7 ± 0.4) |
| P-53k | 0.84 (0.84 ± 0.01) | 25.6 (25.3 ± 0.3) | 25.2 | 0.76 (0.75 ± 0.01) | 16.3 (15.9 ± 0.3) |
| P-98k | 0.82 (0.82 ± 0.00) | 25.9 (25.8 ± 0.4) | 25.4 | 0.76 (0.76 ± 0.01) | 16.2 (16.0 ± 0.2) |
The Vocs of the PSCs decrease slightly with increasing molecular weights, which can be ascribed to the subtle difference of the energy levels of the polymers. The Jscs increase monotonically with polymer molecular weights, which are 22.4, 23.8, 25.6, and 25.9 mA cm−2 for P-17k, P-30k, P-53k, and P-98k, respectively. This trend was verified by the EQE spectra of the PSCs, where the EQE maxima enhanced clearly for the higher molecular weight polymers, i.e. 70.3% for P-17k, 77.7% for P-30k, 83.3% for P-53k, and 83.2% for P-98k, respectively. Meanwhile, the FF increased significantly from 0.57 for P-17k to 0.75 for P-30k, 0.76 for P-53k, and 0.76 for P-98k. The higher Jsc and FF suggest better charge transport and less charge recombination in the PSCs,47,48 which should be related to the BHJ morphology of the polymer:Y6 blends and will be discussed further in the following section. The highest PCE of 16.3% was offered by P-53k, which is much higher than that of P-17k. The PSCs with IT-4F49 as the electron acceptor were also investigated. The increasing trend of Jsc, FF, and overall PCE was also observed from P-17k to P-53k, and the highest PCE of 13.9% was realized with P-53k (Fig. S13 and Table S4†).
The hole and electron mobilities of the polymer:Y6 blends were measured by fitting the data obtained from single-carrier devices to the space-charge-limited-current (SCLC) model.45 As shown in Fig. 3c and S14,† the four blends exhibited the same trend of P-17k < P-30k < P-53k < P-98k for both hole and electron mobility, thus leading to higher FF for the PSCs. Particularly, there is one order of magnitude enhancement of both hole and electron mobilities from P-17k (∼10−5 cm2 V−1 s−1) to P-30k blend (∼10−4 cm2 V−1 s−1), which should account for the surge of FF from P-17k (0.57) to P-30k (0.75). The J–V curves under the illumination of different light intensities (Plight) were tested and both Jsc and Voc as a function of Plight (Fig. S15a and b†) were plotted to investigate the charge recombination of the PSCs.50 Generally, the relationship between Jsc and Plight can be expressed as Jsc ∝ (Plight)α, where α is the exponential factor. The fitted α value for P-17k is 0.86, which increased to almost unity for the other polymers. These results indicate less bimolecular recombination in PSCs with the higher molecular weight polymers. The relationship of Voc and Plight can be described by Voc ∝ (nkBT/q)ln(Plight), where kB, T, and q is the Boltzmann constant, absolute temperature, and elementary charge, respectively. Trap-assisted recombination is pronounced when the slope of Vocversus ln(Plight) is 2kBT/q and bimolecular recombination is dominating when the slope is kBT/q. As shown in Fig. S15b,† the slopes showed a decreasing trend with increasing polymer molecular weights, suggesting the suppression of trap-assisted recombination in the PSCs with higher molecular weight polymers.
The charge generation and extraction of the PSCs were evaluated by plotting the photocurrent density (Jph) versus the effective voltage (Veff) (Fig. 3d). Jph is given by Jph = JL − JD, where JL and JD are the current density measured under illumination and in the dark, respectively. Veff is defined as Veff = V0 − Vappl, where V0 and Vappl are the voltage at Jph = 0 and applied bias voltage, respectively. For the PSCs based on P-30k, P-53k, and P-98k, the Jph saturated quickly at a low Veff around 0.2 V, whereas the P-17k-based device presented a distinct field-dependent extraction characteristic. Meanwhile, we calculated the charge generation and collection probability P(E,T) using the equation of P(E,T) = Jph/Jsat, where Jsat is the saturated photocurrent density. Under short-circuit conditions (Veff = V0), the P(E,T) values are 82.0%, 94.8%, 96.3%, and 97.1% for P-17k, P-30k, P-53k, and P-98k, respectively (Table S5†). Based on the results demonstrated above, it is clear that the increase of polymer molecular weights led to improved charge mobility, suppressed bimolecular and trap-assisted recombination, and enhanced charge generation and collection probability, which in return resulted in the monotonical increase of Jsc and FF in PSCs.
Molecular packing and bulk-heterojunction morphology
To understand the microstructural origin of the performance difference of the polymer:Y6 PSCs, we used GIWAXS to probe the molecular packing and orientation of the blend films. The 2D patterns and 1D profiles are displayed in Fig. 4. The stacking intensities and d-spacings of OOP (010) peaks and IP lamellar (100) peaks are quite comparable. The notable difference is the orientational texture, and the face-on/edge-on ratios follow the order: P-30k:Y6 > P-17k:Y6> P-53k:Y6 > P-98k:Y6. Among these, P-30k exhibited the highest portion of face-on oriented stacking. In contrast, P-53k and P-98k showed significantly lower face-on/edge-on ratios. It is widely acknowledged that higher fraction of face-on orientation is favorable to OOP charge transport and thus device performance of PSCs. However, the orientation texture characterized here was not well correlated with the device performance of the blend films. This implies that other morphological factors play predominant roles in determining solar cell performance.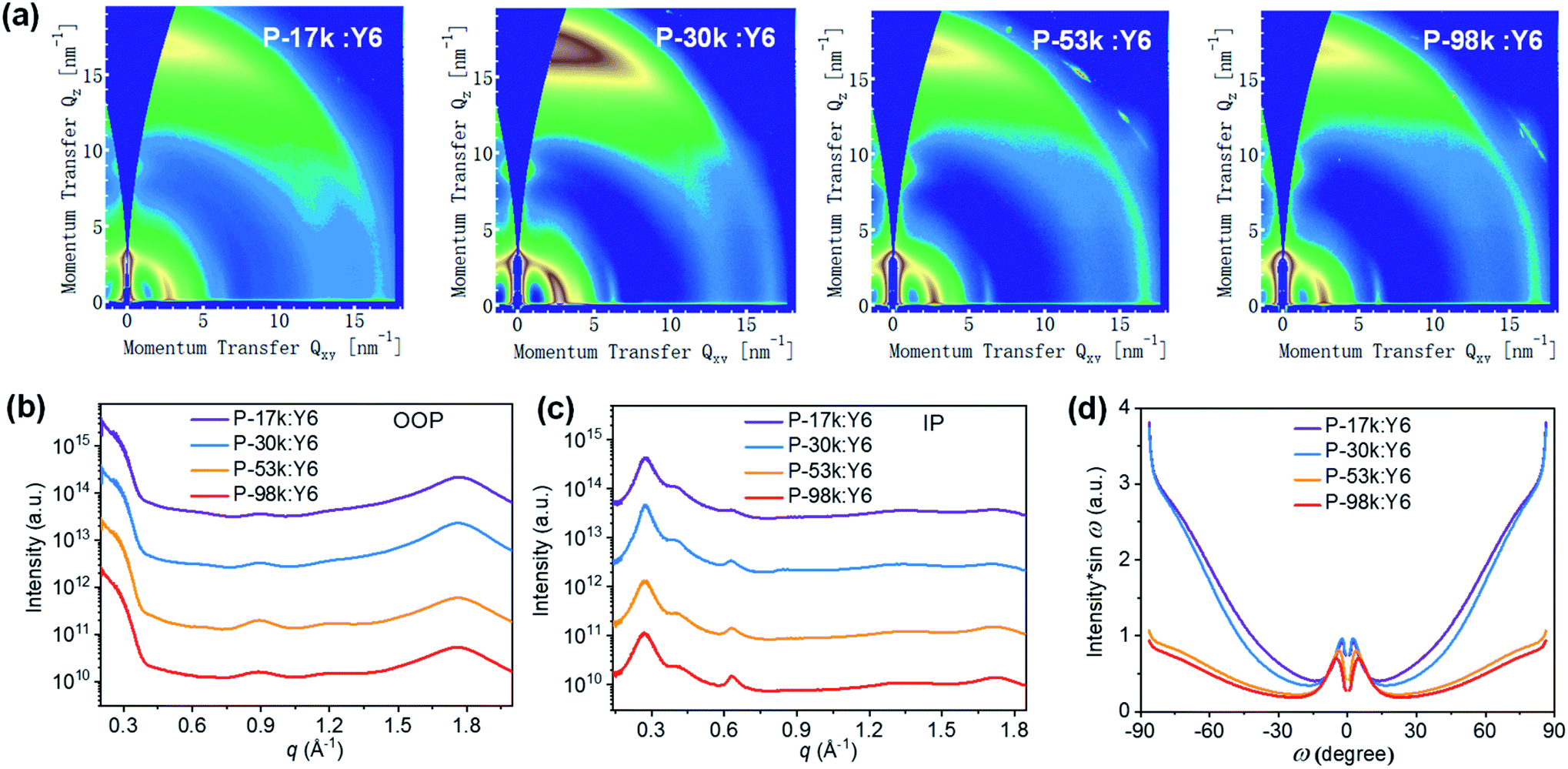 | ||
| Fig. 4 (a) 2D GIWAXS patterns, (b) 1D OOP, and (c) IP profiles of blend films. (d) Pole figure of (100) peak for the blend films. Here ω is the polar angle of (100) peak. | ||
Next, we characterized the mesoscale morphology of the polymer:Y6 blends with resonant soft X-ray scattering (RSoXS). The RSoXS profiles (Fig. 5a) allowed us to compare the long period, generally associated with the center-to-center domain spacing51 of the samples. The scattering peak shifts to higher q value with increasing the polymer molecular weights, corresponding to a decreasing trend of the long period (Fig. 5b). The calculated long periods of the polymer:Y6 blends are 137, 64, 50, 46 nm for P-17k, P-30k, P-53k, and P-98k, respectively. The decrease of the long period of the blends correlates well with the increase of average Jsc of the PSCs. Furthermore, the standard deviations of the acceptor composition (previously referred to as relative domain purities) of the blends can be extracted from the RSoXS profiles of the blends. The relative domain purity was evaluated by normalizing the square roots of the integrated scattering intensities (ISI) values, which are 0.80, 0.90, 0.96, 1.00 for the polymer:Y6 blends based on P-17k, P-30k, P-53k, and P-98k, respectively. A higher domain purity is favorable for facilitating charge transport and suppressing geminate and non-geminate recombination.51 This explained why the Jsc and FF of the PSCs increase monotonically with increasing polymer molecular weights (Fig. 5c).
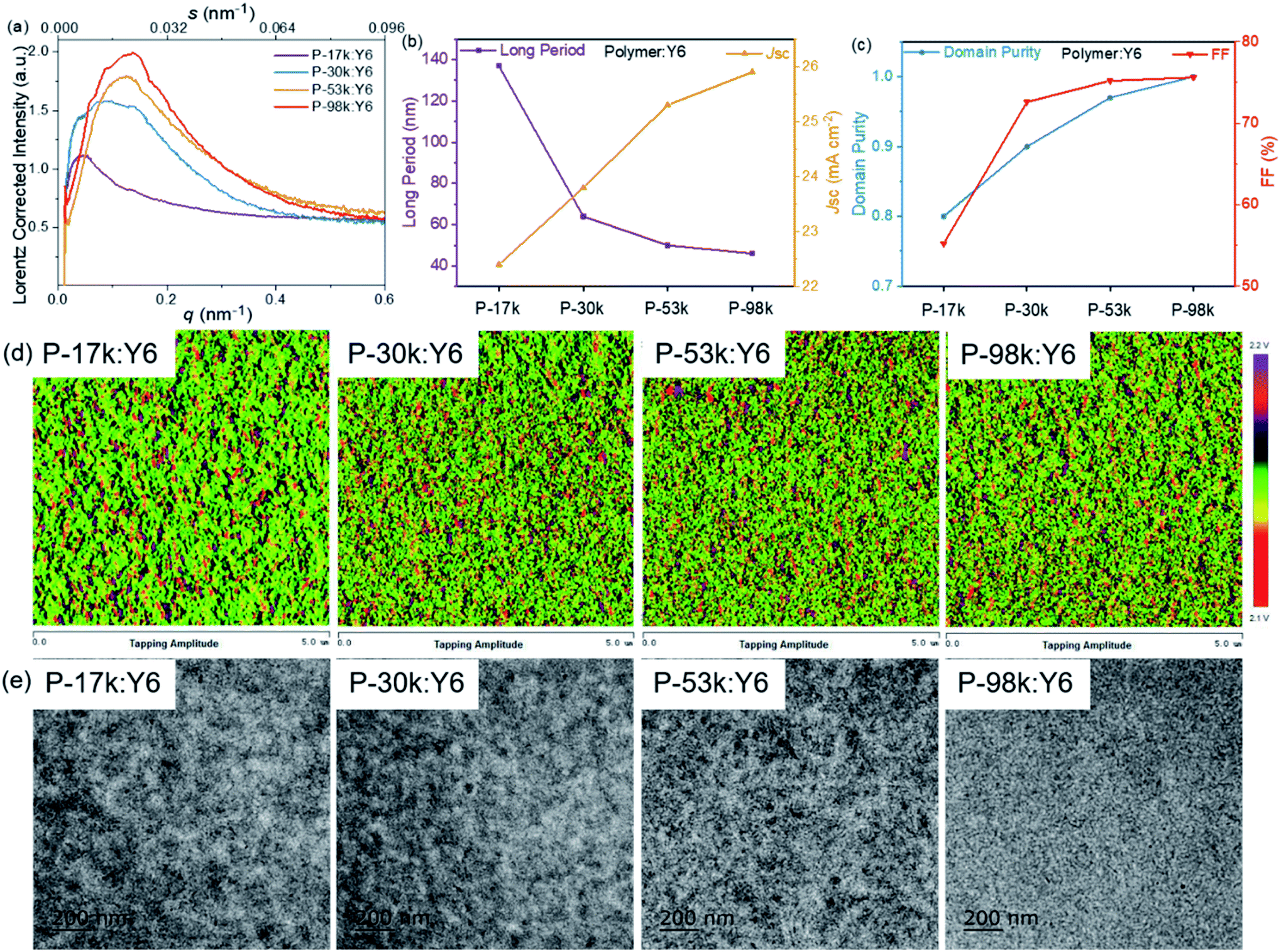 | ||
| Fig. 5 (a) RSoXS profiles of the optimized blend films. (b) Plots of long period and Jsc of the PSCs. (c) Relative domain purity and FF of the PSCs. (d) Tapping mode AFM amplitude images, and (e) TEM images of the optimized polymer:Y6 blends. | ||
The surface and bulk morphology of the polymer:Y6 blends were investigated by atomic force microscopy (AFM) and transmission electron microscopy (TEM), respectively. More intimately mixed morphology with smaller domain sizes can be observed from both AFM and TEM images with increasing polymer molecular weights (Fig. 5d and e), which is in good consistent with the RSoXS results. This morphology evolution with the molecular weights favors charge generation due to the formation of a larger area of donor:acceptor interfaces. Meanwhile, a longer polymer chain can guarantee the connection between ordered regions, which would significantly improve charge transport across the PSCs.28 Overall, these results suggest that it is fruitful to optimize the blend morphology and photovoltaic performance through tuning the molecular weights of polymer donors. The morphology evolution with polymer molecular weights observed in this work agrees well with the solubility controlled formation of fibrillar network in polymer:fullerene blends reported by Janssen et al.34 and kinetically quenched morphology of polymer:non-fullerene acceptor blends reported by Ye et al.37
Devices stability and thermodynamic molecular interactions
Having analyzed the impacts of polymer molecular weights on active layer morphology and device performance, we turned our attention to device stability. The champion devices were stored in dark or heated at 85 °C continuously in nitrogen-filled glovebox. The normalized PCEs as a function of time were plotted in Fig. 6a and b to investigate the long-term and thermal stability of the PSCs. Among the four polymers, P-30k afforded the best long-term device stability with 90% of the initial PCE being maintained after 3000 hours storage in dark, whereas P-17k, P-53k, and P-98k can only offer 65%, 84%, and 84% of the initial PCEs of the PSCs, respectively. The best thermal stability was also achieved by P-30k with 90% of the initial PCE being maintained after continuous heating at 85 °C for 60 hours, whereas the other PSCs only maintained about 70% of their initial PCEs under the same conditions. After extending the thermal annealing duration to 1400 hours, the P-30k:Y6 still exhibited the best device performance.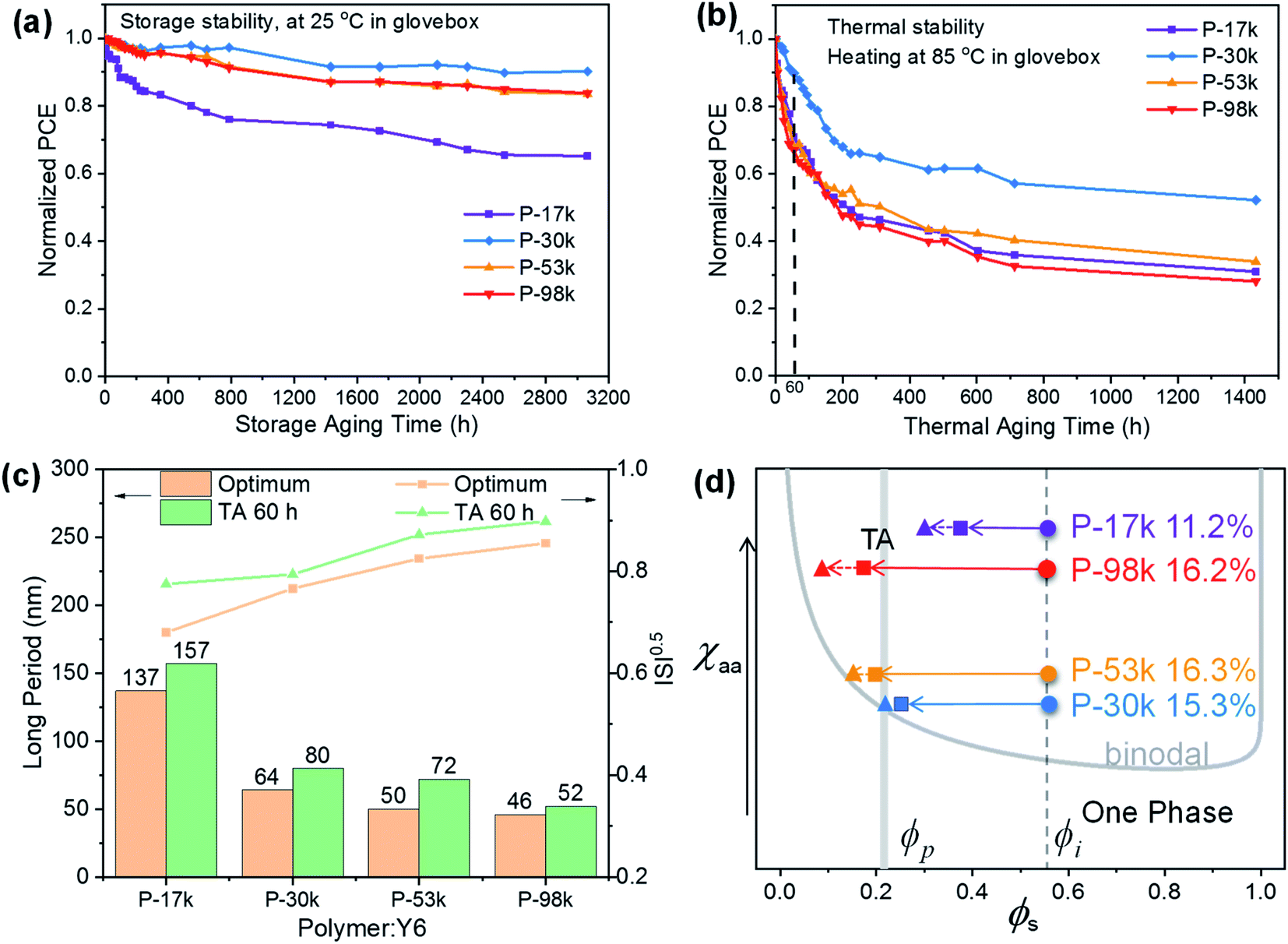 | ||
| Fig. 6 Normalized PCE of the PSCs based on the polymer:Y6 blends as a function of time upon storage in the dark (a) and thermal annealing at 85 °C (b); (c) long period values and relative domain purity of the freshly cast polymer:Y6 blended films (optimum) and the films that continuously heated at 85 °C for 60 hours (TA 60 h). (d) Schematic illustration of the phase evolution of various polymer:Y6 blends on the χ–ϕ plane, modeled after a recent study.52ϕ is the composition of the small molecule. The grey band indicates the percolation threshold (ϕp) for electron transport. ϕi denotes the initial composition of Y6 in the blend. Spheres represent the initial blend films and squares are the quenched films in the PSC devices. Triangles represent the annealed blend films subject to TA 60 hours. | ||
As the interfacial buffer layers and device configuration are the same for all four kinds of PSCs, the superior device stability of P-30k:Y6 blend most likely stems from its better morphological stability than the others. This speculation was verified by comparing the RSoXS profiles of the freshly cast films and the films after consecutive heating at 85 °C for 60 hours. As shown in Fig. S18,† the RSoXS profile of P-30k:Y6 showed the least change among the four blends. We further compared the long periods of the freshly cast films and thermally annealed films (Fig. 6c). The long period of all blends increased after thermal annealing, suggesting that the phase separation scale of the blends got enlarged upon long-time heating. Besides, the ISI value of P-30k:Y6 blend did not vary much after annealing, whereas those of P-17k-, P-53k- and P-98k-based blends increased more evidently (Fig. 6c) upon annealing. These results suggested the occurrence of de-mixing in all blends upon thermal annealing while the P-30k:Y6 blend showed the best morphological stability among the four blends.
The Flory–Huggins interaction parameter (χ) is particularly useful for evaluating the degree of mixing in blends, and a lower χ value suggests better miscibility between the two components.53,54 The χ values were obtained from the empirical relation 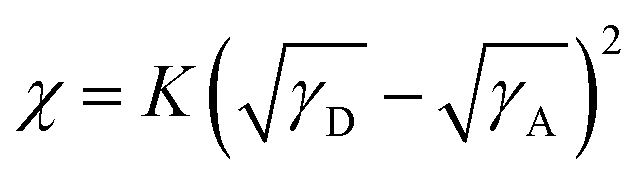 ,55,56 where K is a positive constant, γD and γA were surface tensions obtained from contact angle measurements of two distinct solvents on top of the neat films of the polymers and Y6 (Fig. S19†), respectively.52 The χ values follow the trend of P-30k:Y6 (0.23K) < P-53k:Y6 (0.24K) < P-98k:Y6 (0.44K) < P-17k:Y6 (0.46K) (Table S7†), which agrees well with the RSoXS results. The highest miscibility between P-30k and Y6 is beneficial to reduce the isolated small-scale Y6 domains below the percolation threshold, thus decreasing morphological traps for electron transport.23 Based on the phase purities, χ parameters, and device performance, a self-consistent picture emerges. As schematically illustrated in Fig. 6d, the χ parameter of P-30k:Y6 is most likely close to the ideal value according to the miscibility-function model proposed by Ye and Ade et al.57 The morphology of higher χ systems, namely P-17k:Y6 and P-98k:Y6 should be kinetically frozen to a non-equilibrium state to afford good performance. However, these high χ (hypo-miscibility) systems58 tend to evolve spontaneously into its thermodynamic equilibrium state and thus leads to spontaneous phase separation in solid states.21 Therefore, the P-30k:Y6 blends with nearly optimal-miscibility can be readily stabilized at the percolation condition, which is favorable to achieve a decent performance and superior device stability simultaneously.22,52 Moreover, to further evidence this observation, we also analyze the device stability based on blends of these polymers and another non-fullerene acceptor IT-4F. As shown in Fig. S20,† the relation between device stability and calculated χ parameters (Table S7†) was in good agreement with that of Y6 blends. These results suggested that the control of polymer molecular weight is a simple yet fruitful strategy to achieve high performance and high-stability simultaneously although there is no monotonous dependence between molecular weight and morphology stability.
,55,56 where K is a positive constant, γD and γA were surface tensions obtained from contact angle measurements of two distinct solvents on top of the neat films of the polymers and Y6 (Fig. S19†), respectively.52 The χ values follow the trend of P-30k:Y6 (0.23K) < P-53k:Y6 (0.24K) < P-98k:Y6 (0.44K) < P-17k:Y6 (0.46K) (Table S7†), which agrees well with the RSoXS results. The highest miscibility between P-30k and Y6 is beneficial to reduce the isolated small-scale Y6 domains below the percolation threshold, thus decreasing morphological traps for electron transport.23 Based on the phase purities, χ parameters, and device performance, a self-consistent picture emerges. As schematically illustrated in Fig. 6d, the χ parameter of P-30k:Y6 is most likely close to the ideal value according to the miscibility-function model proposed by Ye and Ade et al.57 The morphology of higher χ systems, namely P-17k:Y6 and P-98k:Y6 should be kinetically frozen to a non-equilibrium state to afford good performance. However, these high χ (hypo-miscibility) systems58 tend to evolve spontaneously into its thermodynamic equilibrium state and thus leads to spontaneous phase separation in solid states.21 Therefore, the P-30k:Y6 blends with nearly optimal-miscibility can be readily stabilized at the percolation condition, which is favorable to achieve a decent performance and superior device stability simultaneously.22,52 Moreover, to further evidence this observation, we also analyze the device stability based on blends of these polymers and another non-fullerene acceptor IT-4F. As shown in Fig. S20,† the relation between device stability and calculated χ parameters (Table S7†) was in good agreement with that of Y6 blends. These results suggested that the control of polymer molecular weight is a simple yet fruitful strategy to achieve high performance and high-stability simultaneously although there is no monotonous dependence between molecular weight and morphology stability.
Conclusions
A set of wide bandgap polymers, PBDB-T4Cl5S, with molecular weights ranging from 17 to 98 kg mol−1 were synthesized to elucidate the impacts of molecular weight on morphology, device performance, and stability of PSCs. These polymers exhibited gradually improved thermal stability, enhanced aggregation in solutions, more ordered packing in solid states, and gradually increased face-on/edge-on ratio with increasing molecular weights. When blended with the leading non-fullerene acceptor Y6, the PSCs realized a PCE of 11.2%, 15.3%, 16.3%, and 16.2% for P-17k, P-30k, P-53k, and P-98k, respectively. The improved device performance was revealed to be caused by the enhanced charge mobility, suppressed bimolecular and trap-assisted recombination, and more efficient charge generation and collection, which can be further ascribed to the reduced phase separation scale and increased phase purity of the polymer:Y6 blends. The most stable PSCs were achieved by the polymer donors with medium molecular weight due to the highest miscibility (lowest χ) between the polymer donor and Y6 acceptor, as suggested by thermodynamic analysis and scattering results. This study highlights that manipulating miscibility between donor and acceptor toward the optimal condition via delicate molecular weight control of polymer donors helps to pave the way for highly-efficient and long-term stable PSCs.Experimental section
Materials
All reagents were purchased from commercial sources (Sigma-Aldrich, J&K, and Derthon, etc) and used as received without further purification unless specifically mentioned.Characterizations
The detailed procedures were described in ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The research was financially supported by the Ministry of Science and Technology of China (2019YFA0705900), the National Natural Science Foundation of China (21875072), and the Fundamental Research Funds for the Central Universities (South China University of Technology, No. D2190310). This work was also supported by Guangdong Innovative and Entrepreneurial Research Team Program (2019ZT08L075) and Beijing National Laboratory for Molecular Sciences (BNLMS201901). L. Y. was supported by the Peiyang Scholar Program of Tianjin University and the Open Fund of the State Key Laboratory of Luminescent Materials and Devices (South China University of Technology, no. 2020-skllmd-11). The authors thank Yiting Guo and Prof. Weiwei Li for the help in GPC measurements, and Prof. Dongqin Bi for the help in elemental analysis. L. Y. acknowledges the merit beamtime (Proposal ID: 15692) approved by Australian Synchrotron. Part of the GIWAXS data were acquired on the SAXS/WAXS beamline at the Australian Synchrotron, part of ANSTO. X-ray data were also acquired at the ALS, which was supported by the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Drs C. Zhu and C. Wang (ALS) was appreciated for assisting with the data acquisition.References
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- C. Duan and L. Ding, Sci. Bull., 2020, 65, 1231–1233 CrossRef CAS.
- Z. Luo, R. Sun, C. Zhong, T. Liu, G. Zhang, Y. Zou, X. Jiao, J. Min and C. Yang, Sci. China: Chem., 2020, 63, 361–369 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094 CrossRef CAS PubMed.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- R. Ma, T. Liu, Z. Luo, Q. Guo, Y. Xiao, Y. Chen, X. Li, S. Luo, X. Lu, M. Zhang, Y. Li and H. Yan, Sci. China: Chem., 2020, 63, 325–330 CrossRef CAS.
- W. R. Mateker and M. D. McGehee, Adv. Mater., 2017, 29, 1603940 CrossRef PubMed.
- H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821–7861 CrossRef CAS PubMed.
- L. Zhang, W. Deng, B. Wu, L. Ye, X. Sun, Z. Wang, K. Gao, H. Wu, C. Duan, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2020, 12, 753–762 CrossRef CAS PubMed.
- C. Duan and L. Ding, Sci. Bull., 2020, 65, 1422–1424 CrossRef CAS.
- C. Duan and L. Ding, Sci. Bull., 2020, 65, 1597–1599 CrossRef CAS.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photon., 2015, 9, 403 CrossRef CAS.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- H. Lee, C. Park, D. H. Sin, J. H. Park and K. Cho, Adv. Mater., 2018, 30, 1800453 CrossRef PubMed.
- F. Zhao, C. Wang and X. Zhan, Adv. Energy Mater., 2018, 8, 1703147 CrossRef.
- N. Li, J. D. Perea, T. Kassar, M. Richter, T. Heumueller, G. J. Matt, Y. Hou, N. S. Güldal, H. Chen, S. Chen, S. Langner, M. Berlinghof, T. Unruh and C. J. Brabec, Nat. Commun., 2017, 8, 14541 CrossRef CAS PubMed.
- M. Ghasemi, H. Hu, Z. Peng, J. J. Rech, I. Angunawela, J. H. Carpenter, S. J. Stuard, A. Wadsworth, I. McCulloch, W. You and H. Ade, Joule, 2019, 3, 1328–1348 CrossRef CAS.
- X. Du, T. Heumueller, W. Gruber, O. Almora, A. Classen, J. Qu, F. He, T. Unruh, N. Li and C. J. Brabec, Adv. Mater., 2020, 32, 1908305 CrossRef CAS PubMed.
- M. S. Vezie, S. Few, I. Meager, G. Pieridou, B. Dorling, R. S. Ashraf, A. R. Goni, H. Bronstein, I. McCulloch, S. C. Hayes, M. Campoy-Quiles and J. Nelson, Nat. Mater., 2016, 15, 746–753 CrossRef CAS PubMed.
- B. B. Fan, L. Ying, Z. F. Wang, B. T. He, X. F. Jiang, F. Huang and Y. Cao, Energy Environ. Sci., 2017, 10, 1243–1251 RSC.
- Z. B. Henson, K. Mullen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS PubMed.
- F. P. V. Koch, J. Rivnay, S. Foster, C. Müller, J. M. Downing, E. Buchaca-Domingo, P. Westacott, L. Yu, M. Yuan, M. Baklar, Z. Fei, C. Luscombe, M. A. McLachlan, M. Heeney, G. Rumbles, C. Silva, A. Salleo, J. Nelson, P. Smith and N. Stingelin, Prog. Polym. Sci., 2013, 38, 1978–1989 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- J. Choi, W. Kim, D. Kim, S. Kim, J. Chae, S. Q. Choi, F. S. Kim, T.-S. Kim and B. J. Kim, Chem. Mater., 2019, 31, 3163–3173 CrossRef CAS.
- N. Balar, J. J. Rech, R. Henry, L. Ye, H. Ade, W. You and B. T. O'Connor, Chem. Mater., 2019, 31, 5124–5132 CrossRef CAS.
- W. T. Li, L. Q. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456–4462 CrossRef CAS PubMed.
- H. Kang, M. A. Uddin, C. Lee, K.-H. Kim, T. L. Nguyen, W. Lee, Y. Li, C. Wang, H. Y. Woo and B. J. Kim, J. Am. Chem. Soc., 2015, 137, 2359–2365 CrossRef CAS PubMed.
- J. J. van Franeker, G. H. L. Heintges, C. Schaefer, G. Portale, W. Li, M. M. Wienk, P. van der Schoot and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 11783–11794 CrossRef CAS PubMed.
- N. Zhou, A. S. Dudnik, T. I. N. G. Li, E. F. Manley, T. J. Aldrich, P. Guo, H.-C. Liao, Z. Chen, L. X. Chen, R. P. H. Chang, A. Facchetti, M. Olvera de la Cruz and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 1240–1251 CrossRef CAS PubMed.
- A. Wadsworth, Z. Hamid, M. Bidwell, R. S. Ashraf, J. I. Khan, D. H. Anjum, C. Cendra, J. Yan, E. Rezasoltani, A. A. Y. Guilbert, M. Azzouzi, N. Gasparini, J. H. Bannock, D. Baran, H. Wu, J. C. de Mello, C. J. Brabec, A. Salleo, J. Nelson, F. Laquai and I. McCulloch, Adv. Energy Mater., 2018, 8, 1801001 CrossRef.
- L. Ye, S. S. Li, X. Y. Liu, S. Q. Zhang, M. Ghasemi, Y. Xiong, J. H. Hou and H. Ade, Joule, 2019, 3, 443–458 CrossRef CAS.
- K. D. Deshmukh, R. Matsidik, S. K. K. Prasad, L. A. Connal, A. C. Y. Liu, E. Gann, L. Thomsen, J. M. Hodgkiss, M. Sommer and C. R. McNeill, Adv. Funct. Mater., 2018, 28, 1707185 CrossRef.
- J. Lee, T. H. Lee, M. M. Byranvand, K. Choi, H. I. Kim, S. A. Park, J. Y. Kim and T. Park, J. Mater. Chem. A, 2018, 6, 5538–5543 RSC.
- C. Zhang, T. Heumueller, W. Gruber, O. Almora, X. Du, L. Ying, J. Chen, T. Unruh, Y. Cao, N. Li and C. J. Brabec, ACS Appl. Mater. Interfaces, 2019, 11, 18555–18563 CrossRef CAS PubMed.
- Z. Li, W. Zhong, L. Ying, F. Liu, N. Li, F. Huang and Y. Cao, Nano Energy, 2019, 64, 103931 CrossRef CAS.
- S. Park, H. Ahn, J.-y. Kim, J. B. Park, J. Kim, S. H. Im and H. J. Son, ACS Energy Lett., 2020, 5, 170–179 CrossRef CAS.
- C. Zhou, Y. Liang, F. Liu, C. Sun, X. Huang, Z. Xie, F. Huang, J. Roncali, T. P. Russell and Y. Cao, Adv. Funct. Mater., 2014, 24, 7538–7547 CrossRef CAS.
- K. A. Page, A. Kusoglu, C. M. Stafford, S. Kim, R. J. Kline and A. Z. Weber, Nano Lett., 2014, 14, 2299–2304 CrossRef CAS PubMed.
- J. C. Blakesley, F. A. Castro, W. Kylberg, G. F. A. Dibb, C. Arantes, R. Valaski, M. Cremona, J. S. Kim and J. S. Kim, Org. Electron., 2014, 15, 1263–1272 CrossRef CAS.
- Z. Wu, C. Sun, S. Dong, X.-F. Jiang, S. Wu, H. Wu, H.-L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2016, 138, 2004–2013 CrossRef CAS PubMed.
- J. A. Bartelt, D. Lam, T. M. Burke, S. M. Sweetnam and M. D. McGehee, Adv. Energy Mater., 2015, 5, 1500577 CrossRef.
- D. Bartesaghi, I. d. C. Pérez, J. Kniepert, S. Roland, M. Turbiez, D. Neher and L. J. A. Koster, Nat. Commun., 2015, 6, 7083 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- A. K. K. Kyaw, D. H. Wang, V. Gupta, W. L. Leong, L. Ke, G. C. Bazan and A. J. Heeger, ACS Nano, 2013, 7, 4569–4577 CrossRef CAS PubMed.
- X. Jiao, L. Ye and H. Ade, Adv. Energy Mater., 2017, 7, 1700084 CrossRef.
- Z. Liang, M. Li, Q. Wang, Y. Qin, S. J. Stuard, Z. Peng, Y. Deng, H. Ade, L. Ye and Y. Geng, Joule, 2020, 4, 1278–1295 CrossRef CAS.
- X. Liu, C. Zhang, C. Duan, M. Li, Z. Hu, J. Wang, F. Liu, N. Li, C. J. Brabec, R. A. J. Janssen, G. C. Bazan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2018, 140, 8934–8943 CrossRef CAS PubMed.
- M. Gao, Z. Liang, Y. Geng and L. Ye, Chem. Commun., 2020, 56, 12463–12478 RSC.
- S. Nilsson, A. Bernasik, A. Budkowski and E. Moons, Macromolecules, 2007, 40, 8291–8301 CrossRef CAS.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 12057–12067 CrossRef CAS PubMed.
- L. Ye, B. A. Collins, X. Jiao, J. Zhao, H. Yan and H. Ade, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- C. R. McNeill, Energy Environ. Sci., 2012, 5, 5653–5667 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta10163j |
| This journal is © The Royal Society of Chemistry 2021 |