DOI:
10.1039/D1TA02165F
(Paper)
J. Mater. Chem. A, 2021,
9, 11691-11704
Oxygen vacancy enriched NiMoO4 nanorods via microwave heating: a promising highly stable electrocatalyst for total water splitting†‡
Received
15th March 2021
, Accepted 13th April 2021
First published on 13th April 2021
Abstract
It is indeed necessary to develop a suitable bi-functional catalyst for total water splitting. Here, in this work, we demonstrate a microwave-assisted formulation of NiMoO4 nanorods within 30 minutes of the reaction time. As synthesized NiMoO4 nanorods grown on nickel foam were treated with NaBH4 to create internal oxygen vacancies [NiMoO4(Vo)] that favoured OER and HER with very merginal applied overpotential. The generation of oxygen vacancies in the lattice generally leads to the formation of an effective electronic structure for proceeding OER activity in a sustainable way. When vacancy-enriched NiMoO4(Vo) nanorods were applied for OER and HER under alkaline conditions, it demands only 220 and 255 mV overpotential at 50 mA cm−2 current density, respectively. Having observed their phenomenal response in both OER and HER, they were analysed for the real device as the two-electrode system (NiMoO4(Vo) nanorods as anode and cathode); they needed 490 mV as overpotential at 50 mA cm−2. Based on the molecular orbital and band-structured theories, it has been understood that the band gap state (BGS) led to the formation of antibonding states with a very low electron population that favoured rich OER and HER kinetics, as observed from the electrochemical results.
Introduction
Constant use of fossil fuels produces a large amount of greenhouse gases, mainly CO2 and as a consequence of this, the level of CO2 in the environment is increasing day by day. This affects the earth's climatic condition and an example of which is the melting of ice in the Arctic area that caused an increase in the water level in the sea.1 Hence, while searching for alternative fuels, it is desired to go for carbon-neutral fuels instead of carbon-containing ones, and surprisingly researchers have found that hydrogen (H2) with high specific energy density (120–140 MJ Kg−1) can be used as an alternative. Hydrogen can be generated by various processes such as steam reforming of fossil fuels, the hydrolysis of metal hydrides, photoelectrochemical water splitting and the electrolysis of water.2 Out of various available methods, electrolysis of water is the most studied one owing to its environment-friendly nature with no carbon emission. Electrolysis of water consists of two half-cell reactions viz oxygen evolution reaction (OER) at the anode and hydrogen evolution reaction (HER) at the cathode. The standard electrode potential of water splitting is 1.23 V vs. RHE, which is derived from thermodynamics without considering the kinetic aspects of the reaction which vary with respect to the mechanistic steps involved in both OER and HER.3
By considering complex mechanisms, multiple electron transfer and various kinetic parameters of OER and HER, these cannot be proceeding in the thermodynamically-derived potentials and always demand excess potentials over the theoretical potential value. This excess potential is termed as ‘overpotential’ of the reaction3 and to minimize the required overpotential, there is an urgent need for designing suitable electrode materials for OER at a very low overpotential at a certain current density. Noble metal oxides (RuO2 and IrO2) and Pt are considered to be state-of-the-art catalysts for OER and HER, respectively.4,5 However, low abundance made their industrialization problematic due to increasing costs. In this regard, due to the high abundance and low cost of transition metal-based catalysts, they are promising as an alternative to noble metals-based catalysts. Recently, transition metal oxides,6–8 hydroxides,9,10 chalcogenides11–13 and phosphide12,14–16 based materials have been reported as efficient catalysts towards water splitting at different pH conditions. However, the formed oxide or hydroxide phase during OER will only act as a real catalytic surface of various corresponding catalysts mentioned above.
Out of various transition metals-based electrocatalysts, nickel-based oxides showed the most bulbous OER activity, particularly in alkaline environments. The combination of the 4d transition metal with 3d transition metal-based oxide or other compounds has exhibited predominant electrocatalytic activity.17–21 The addition of tungsten or molybdenum metal or their oxides in combination with 3d transition metal ensured enhancement in electrocatalytic performance. In this regard, molybdate-based materials have taken much consideration for total water splitting. Out of various molybdate-based materials, nickel molybdate (NiMoO4) catalysts have taken much consideration owing to the electronic structure of nickel for promoting OER and HER effectively. Zhang et al. reported NiMoO4−x/MoO2 as an efficient water-splitting catalyst and it required just 320 mV overpotential to reach 100 mA cm−2 current density.22 On the other hand, Chen et al. reported iron-doped NiMoO4 for efficient water oxidation reaction and reached 10 mA cm−2 of current density and it required 299 mV of overpotential.23 But, the problem associated with all NiMoO4-based materials is that for efficient synthesis, it requires high temperature and pressure conditions. Hence, it is highly desirable to find an alternative and easy way for synthesizing it within a short time scale with a pronounced activity.
Here, in this work, for the very first time, we have developed NiMoO4 nanorods grown over nickel foam by utilizing microwave conditions within 30 min of reaction time. The as-prepared NiMoO4 was treated with 0.1 M NaBH4 solution to create internal oxygen vacancy, which further enhanced the electrocatalytic performance by changing the surface electronic structure. Vacancy-enriched NiMoO4 was applied for both OER and HER in alkaline conditions. For OER, the vacancy-enriched nickel molybdate [NiMoO4(Vo)] demanded only 220 mV of overpotential, whereas it required 255 mV as overpotential for HER to reach 50 mA cm−2 of current density. Further, NiMoO4(Vo) was used both as the anode and cathode for total water splitting and it required just 360 and 490 mV of overpotential to reach 10 and 50 mA cm−2, respectively. Based on the molecular orbital and band structure theories it can be observed that BGS led to the accumulation of fewer electrons in antibonding states and because of which they ensured a high rate for OER and HER activities in alkaline electrolytes with large-scale stability.
Experimental section
Synthesis of NiMoO4 on nickel foam via microwave heating methods
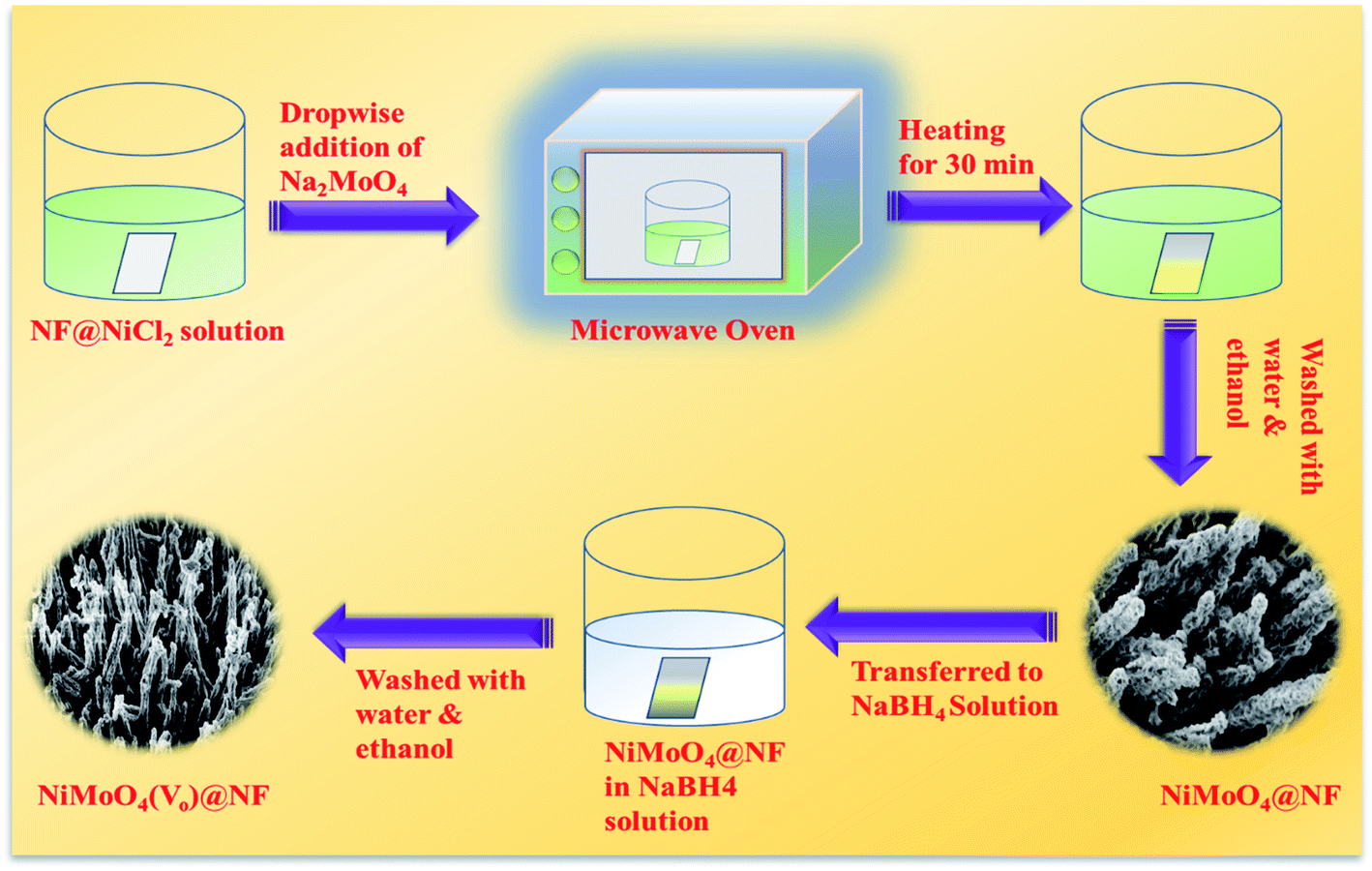
At first, nickel foam (NF) was washed with 3 M HCl solution by ultrasonication and later the ultrasonicated NF was washed with a water–ethanol mixture several times followed by drying at room temperature. Then, the washed NF was directly transferred to a 100 mL beaker containing 20 mL of 0.01 (M) NiCl2 solution. Later on, it was transferred to a microwave oven and it was heated for 30 s at a power of 150 W. To this solution, 2 mL of 0.01 M Na2MoO4 was added dropwise and the heating was continued for 1 min. Likewise, all the Na2MoO4 solution was added in the same manner and heated for 1 min in each interval. After the addition of all the molybdate solution to the reaction mixture, it was additionally heated for 10 s with total heating for 2 min with a gap of 10 seconds for avoiding the spillage of the solution. Afterward, yellow-colored NiMoO4 coated NF was taken out from the reaction mixture and washed with water and ethanol mixture several times followed by drying at 60 °C for 6 h.
Synthesis of NiMoO4(Vo) on nickel foam via wet chemical route
NiMoO4(Vo) was synthesized by a simple wet chemical method. Typically, NiMoO4 coated NF was dipped in 0.1 (M) of sodium borohydride (NaBH4) solution taken in a 50 mL beaker for one hour. After one hour of dipping in NaBH4 solution, the soaked NF coated with NiMoO4 was taken out and washed with water several times followed by drying at 60 °C for 6 hours. The whole synthetic procedure is shown in Scheme 1.
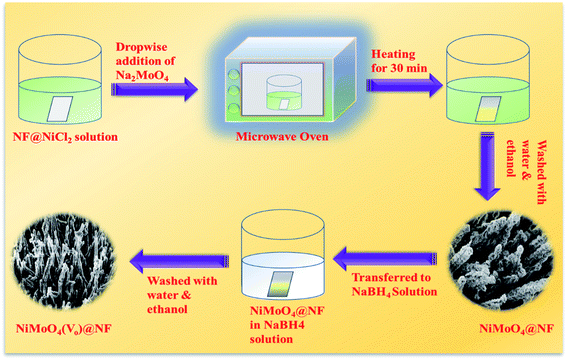 |
| | Scheme 1 A synthetic scheme for synthesizing NiMoO4 and NiMoO4(Vo) via a microwave heating method. | |
Results and discussion
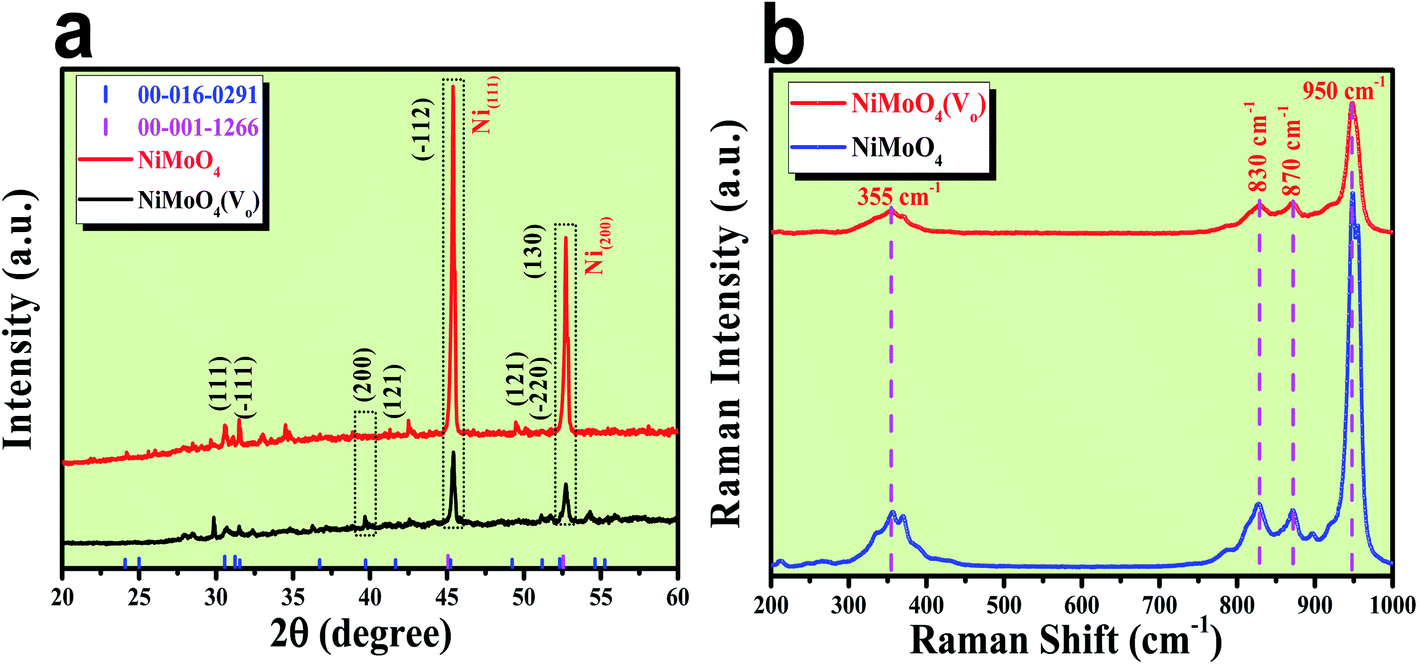
After the successful synthesis of NiMoO4 and NiMoO4(Vo), they were characterized using various advanced characterization tools. First, the formation of both the molybdate materials was preliminarily characterized by XRD analysis and the obtained results are shown in Fig. 1a. Fig. 1a shows the diffraction peaks for various planes like (111), (−111), (200), (121), (−112), (−220) and (130) corresponding to NiMoO4, which were matched with ICDD card no: 00-016-0291. NiMoO4(Vo) shows the same XRD pattern, but the intensity of some of the peaks decreased, which might be due to the creation of a large number of vacancies on the surface of NiMoO4. In addition to all the corresponding peaks of NiMoO4, two large intense peaks appeared on 2θ values of 45.46 and 52.7°, which corresponded to metallic nickel that was used as a coating substrate matching with the ICDD card number of 00-001-1266.21 In order to investigate the surface functional groups, Raman spectroscopic study was conducted on both NiMoO4 and NiMoO4(Vo) and the spectra are shown in Fig. 1b. The peaks at 950, 870 and 830 cm−1 correspond to the symmetric and anti-symmetric stretching of Mo–O bonds, whereas the peak at the frequency value of 355 cm−1 originated from the bending vibration of Mo–O bonds.24 Raman spectra of NiMoO4 and NiMoO4(Vo) are the same but in the case of NiMoO4(Vo), the intensity of all peaks decreased, which might be due to the creation of oxygen vacancies over the surface.
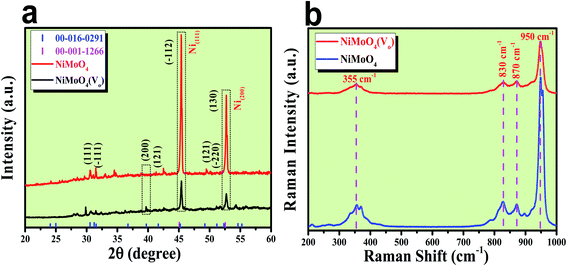 |
| | Fig. 1 (a) XRD patterns of rod shaped NiMoO4 and NiMoO4(Vo), (b) Raman spectra of rod shaped NiMoO4 and NiMoO4(Vo). | |
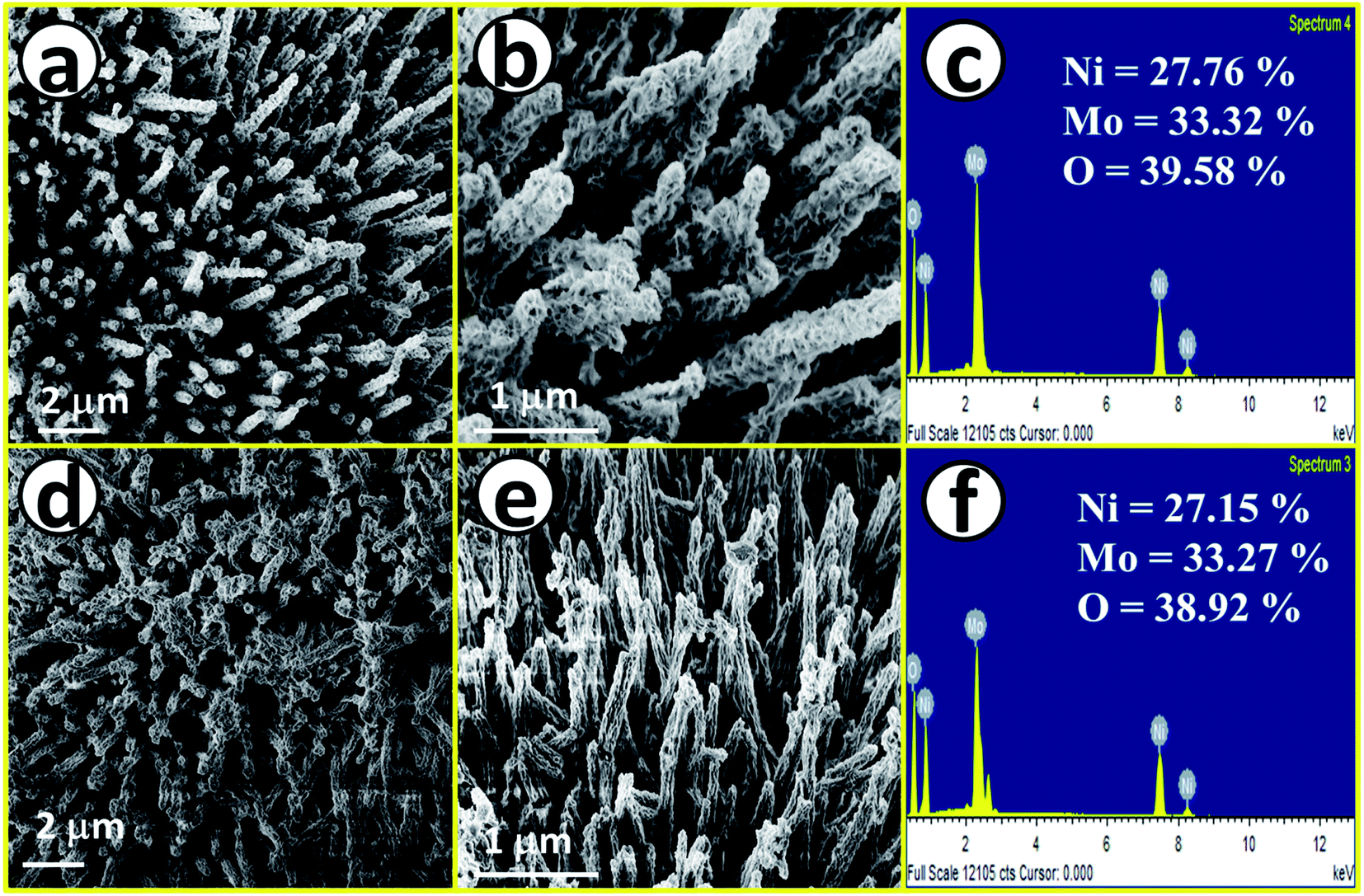
After having proper knowledge about the structural characterization, morphological analysis was first performed through FE-SEM analysis and the corresponding results are given in Fig. 2. Fig. 2a and b shows FE-SEM images of NiMoO4, from a lower to higher magnification value revealing rod-like morphology. Fig. 2c displays the EDS spectrum of NiMoO4 in the FE-SEM mode with quantification of all the expected elements present in the lattice, also suggesting purity. Fig. 2d and e shows FE-SEM images of NiMoO4(Vo) from a lower to higher magnification value and it shows the same rod-like structure that was retained even after the treatment of NiMoO4 with NaBH4 solution. Fig. 2f shows the corresponding EDS spectrum with the quantification of all elements that further reveal the pure nature of the prepared material. The decrease in the amount of the elemental distribution after treatment with NaBH4 solution may have been caused by the loss of the loosely bound particles during the long-term immersion of the electrode materials. Another possible reason behind the decrease in the quantity of the elements is that when NiMoO4 coated on NF was immersed, huge bubbling with oxygen liberation was observed, and the mechanical force associated with the bubbles might have also helped to leach a certain quantity of NiMoO4 from NF.
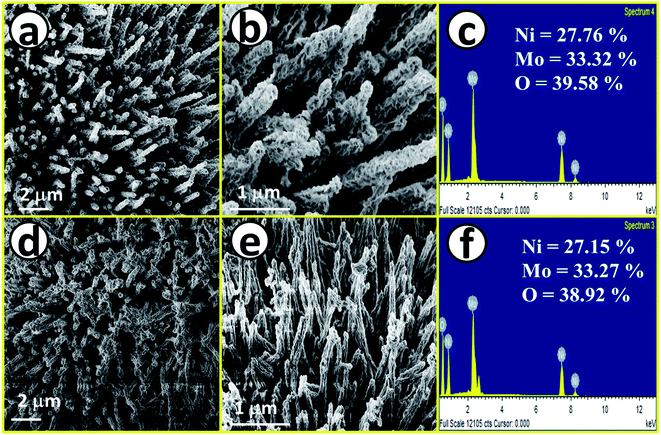 |
| | Fig. 2 (a) and (b) FE-SEM images of NiMoO4 nano-rods, (c) EDS spectrum of NiMoO4 in the FE-SEM mode, (d) and (e) FE-SEM images of NiMoO4(Vo) nanorod and (f) EDS spectrum of NiMoO4(Vo) in the FE-SEM mode. | |
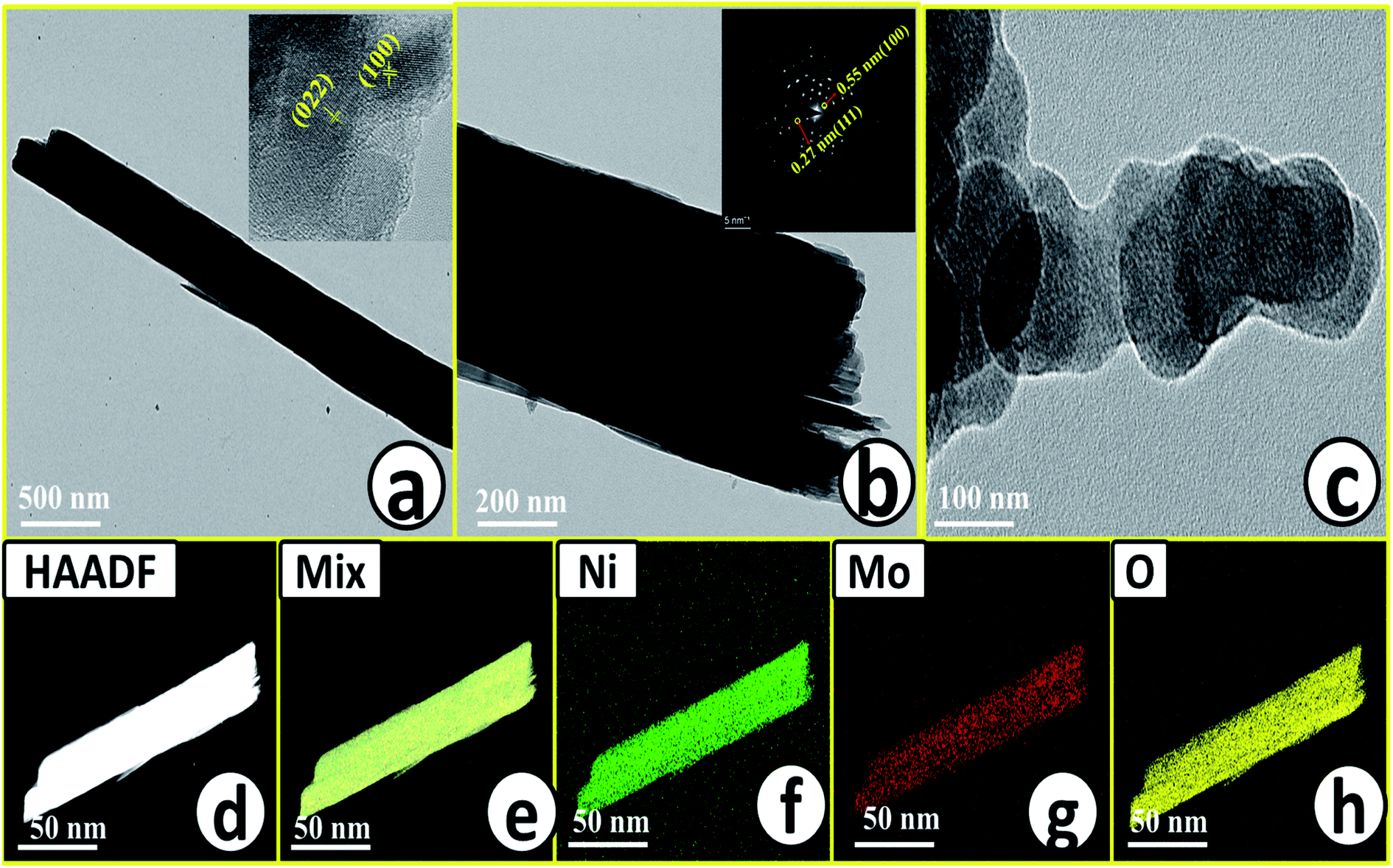
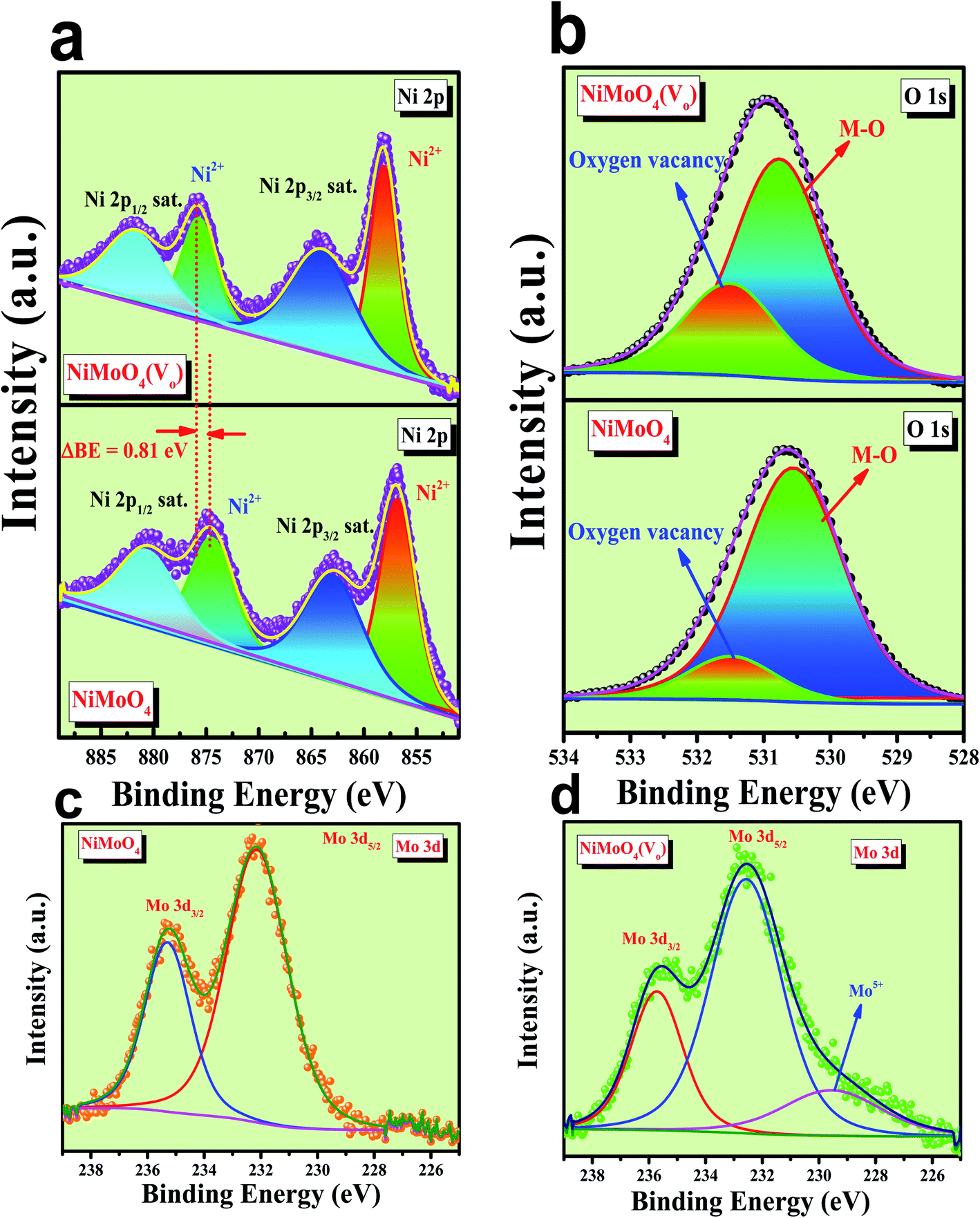
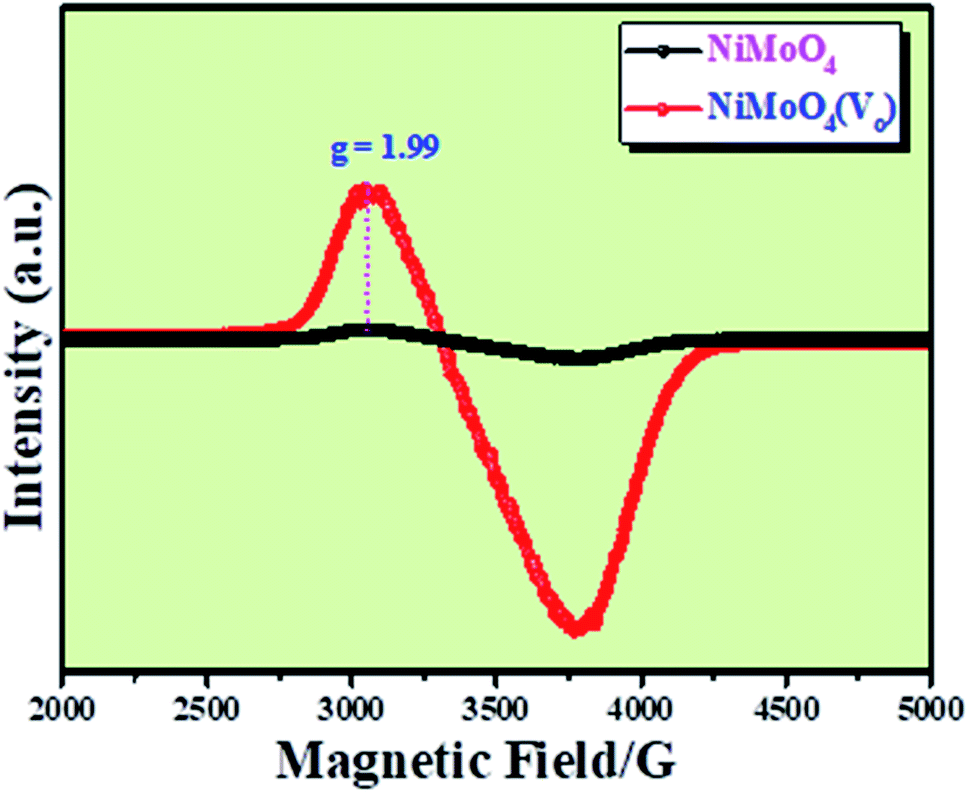
Microstructural analysis of NiMoO4 was conducted using HR-TEM and the corresponding images are shown in Fig. 3. Fig. 3a–c shows the low to high magnification HR-TEM images of NiMoO4 and it demonstrates rod-like microstructural outcomes for NiMoO4. The inset of Fig. 3b shows the SAED pattern of NiMoO4, which shows the polycrystalline nature of the particles and it reveals the diffraction of (111) and (112) planes that match with the XRD pattern shown in Fig. 1a. Lattice fringe pattern is given as an inset in Fig. 3c that shows the d spacing value of 0.45 nm corresponding to the (100) plane of NiMoO4 that is consistent with the ICDD card no: 00-016-0291. To confirm the presence and distribution of all the expected elements in the lattice, high-angle annular dark-field (HAADF) colour-mapping analysis was carried out and the corresponding HAADF area chosen for colour-mapping analysis is shown in Fig. 3d. Colour mapping results of all elements such as Ni, Mo and O are shown in Fig. 3e–h, and it shows a uniform distribution for the elements. Similarly, microstructural analysis of NiMoO4(Vo) is provided in Fig. 4. Fig. 4a–c shows low to high magnification HR-TEM images of NiMoO4(Vo), which reveal the same rod-like morphology of the particles that is consistent with the FE-SEM results shown in Fig. 2. After having a proper understanding of the structural and morphological features of both the materials, their chemical nature was analysed by XPS analysis. Fig. 5a represents high-resolution XPS spectra of Ni 2p orbitals of NiMoO4 and NiMoO4(Vo). It shows doublet peaks for NiMoO4 at binding energies of 856.3 and 874.3 eV, which corresponded to Ni 2p3/2 and Ni 2p1/2, respectively, and confirming the presence of nickel in the +2 oxidation state.24–26 The other two peaks at binding energies 860.4 and 880.6 eV correspond to Ni 2p3/2 and Ni 2p1/2 satellite peaks. XPS high-resolution spectra for Ni 2p of NiMoO4(Vo) are similar to those of NiMoO4 but close analysis reveals that there is a shifting of all maxima towards higher binding energy by 0.81 eV.27 This shifting is indicating the depletion of electron density in the vicinity of the nickel ions. Fig. 5b shows the deconvoluted XPS spectra of O 1s orbital, which show two spin–orbit coupling originated peaks. The peak at a binding energy of 530.6 eV is attributed to the metal–oxygen (M–O) bond and the other peak at a binding energy of 531.4 eV corresponds to low coordinated M–O bonds, which confirmed the formation of oxygen vacancy in the lattice.10,21,28 But the interesting fact is that the area of the second peak becomes 1.85 times larger in the case of NiMoO4(Vo) than that of NiMoO4 suggesting the increase in oxygen vacancy concentration after treatment with NaBH4 solution. Fig. 5c shows the deconvoluted XPS spectra of Mo 3d orbitals of NiMoO4 showing the presence of two peaks originating from spin–orbit coupling mechanism at binding energies of 232.18 and 235.3 eV which are corresponding to Mo 3d5/2 and Mo 3d3/2 orbitals, respectively, suggesting the presence of molybdenum in +6 oxidation state.21 In the case of NiMoO4(Vo) the same XPS pattern can be observed except for one additional peak at a binding energy of 229.6 eV, which is due to the presence of molybdenum in the +5 oxidation state, which might be originating due to the reduction of molybdenum from the +6 oxidation state. Although XPS spectra of O 1s orbital qualitatively provide information about the oxygen vacancy it is not strong enough, because at the same binding energy, many more functional groups can show the existence of the same peaks. To provide strong evidence for the presence of oxygen vacancy, EPR spectra were acquired for both NiMoO4 and NiMoO4(Vo) and the corresponding results are shown in Fig. 6. It displayed an increase in structural defects after treatment with NaBH4 as compared to bare NiMoO4. A large increase in the peak intensity at g = 1.99 inferred that the structural defects originated from the creation of oxygen vacancy, which is in accordance with the result reported earlier.13 To provide more information on the exact quantification by percentile ratio of the metal components, X-ray fluorescence (XRF) studies were carried out (Fig. S1a and b‡) for both NiMoO4 and NiMoO4(Vo) at 10 different places with a total surface area of about 0.5 × 1 cm2 and the average was taken for both metals and the ratio of Ni to Mo was almost the same as that obtained from EDS analysis in the FE-SEM mode i.e., 0.85 ± 0.1.
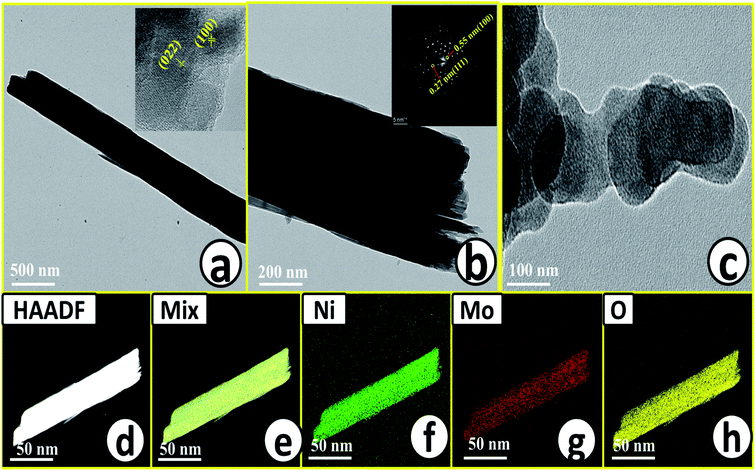 |
| | Fig. 3 (a) and (c) Low to high magnification HR-TEM images of NiMoO4, inset of (a) and (b) are corresponding lattice fringes and SAED pattern respectively, (d) is the HAADF image of rod-like NiMoO4 taken for the mapping analysis, (e)–(h) are the characteristic colour mapping results of mix, Ni K, Mo K and O K respectively. | |
 |
| | Fig. 4 (a)–(c) Low to high magnification HR-TEM images of NiMoO4(Vo); (d) is the HAADF image of rod-like NiMoO4(Vo) taken for the mapping analysis; (e)–(h) are the characteristic mapping results of mix, Ni K, Mo K, O K and respectively. | |
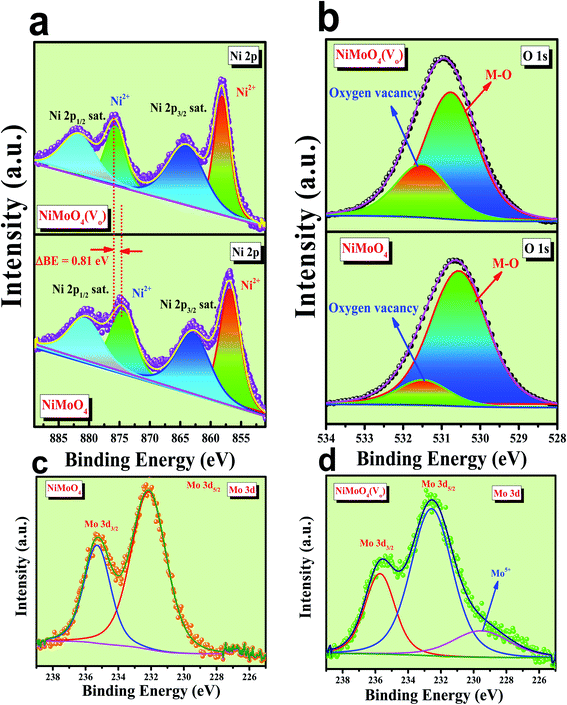 |
| | Fig. 5 (a) Combined high-resolution XPS spectra of Ni 2p orbitals for NiMoO4 and NiMoO4(Vo), (b) deconvoluted XPS spectrum of O 1s orbitals, (c) XPS spectra of Mo 3d orbital of NiMoO4 and, (d) XPS spectra of Mo 3d orbital of NiMoO4(Vo). | |
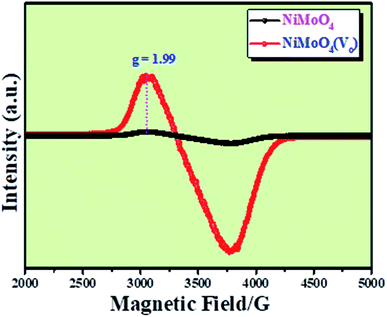 |
| | Fig. 6 EPR spectra of NiMoO4 and NiMoO4(Vo) at room temperature. | |
Electrocatalytic OER activity of NiMoO4 and NiMoO4(Vo) in 1 M KOH
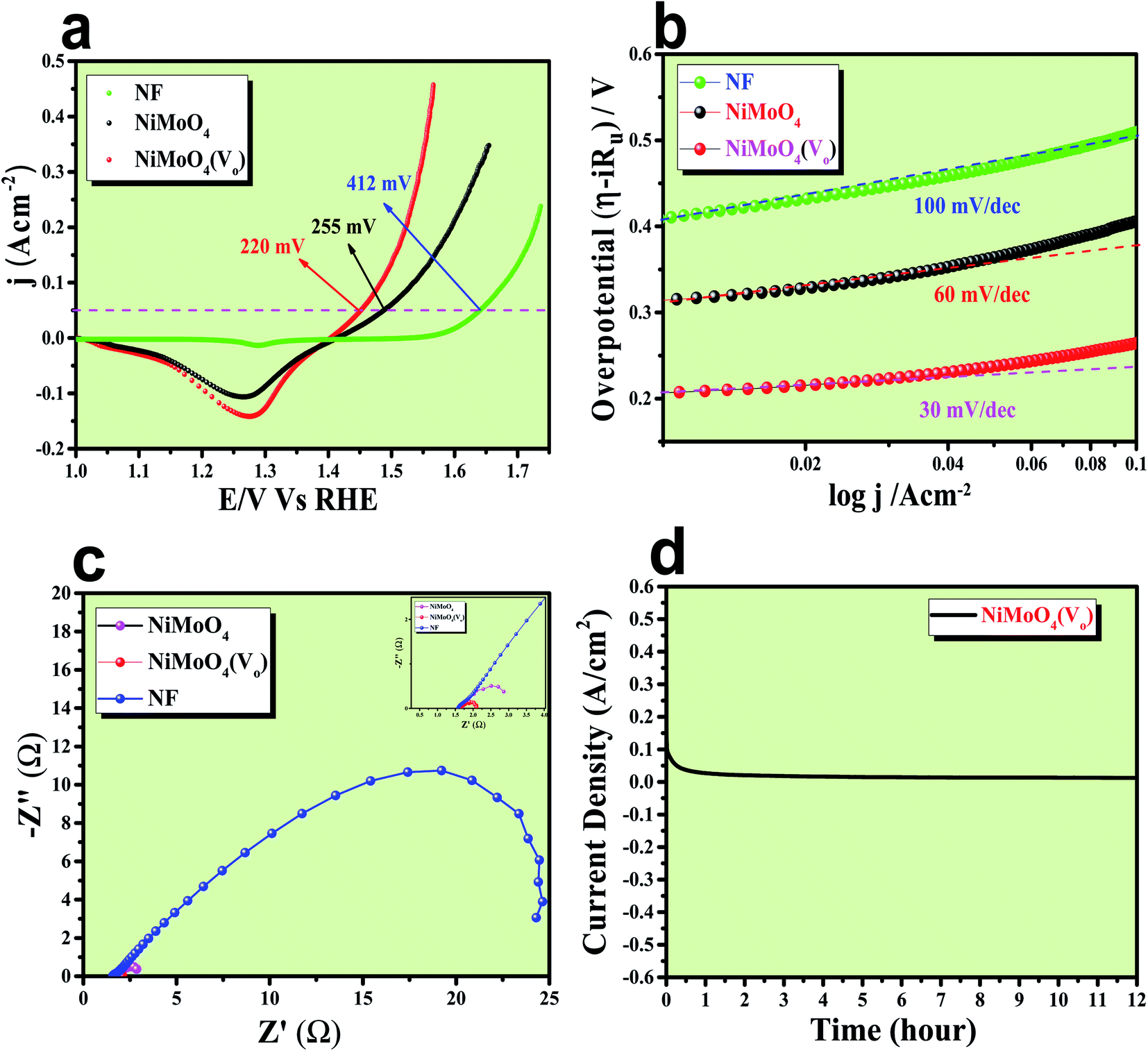
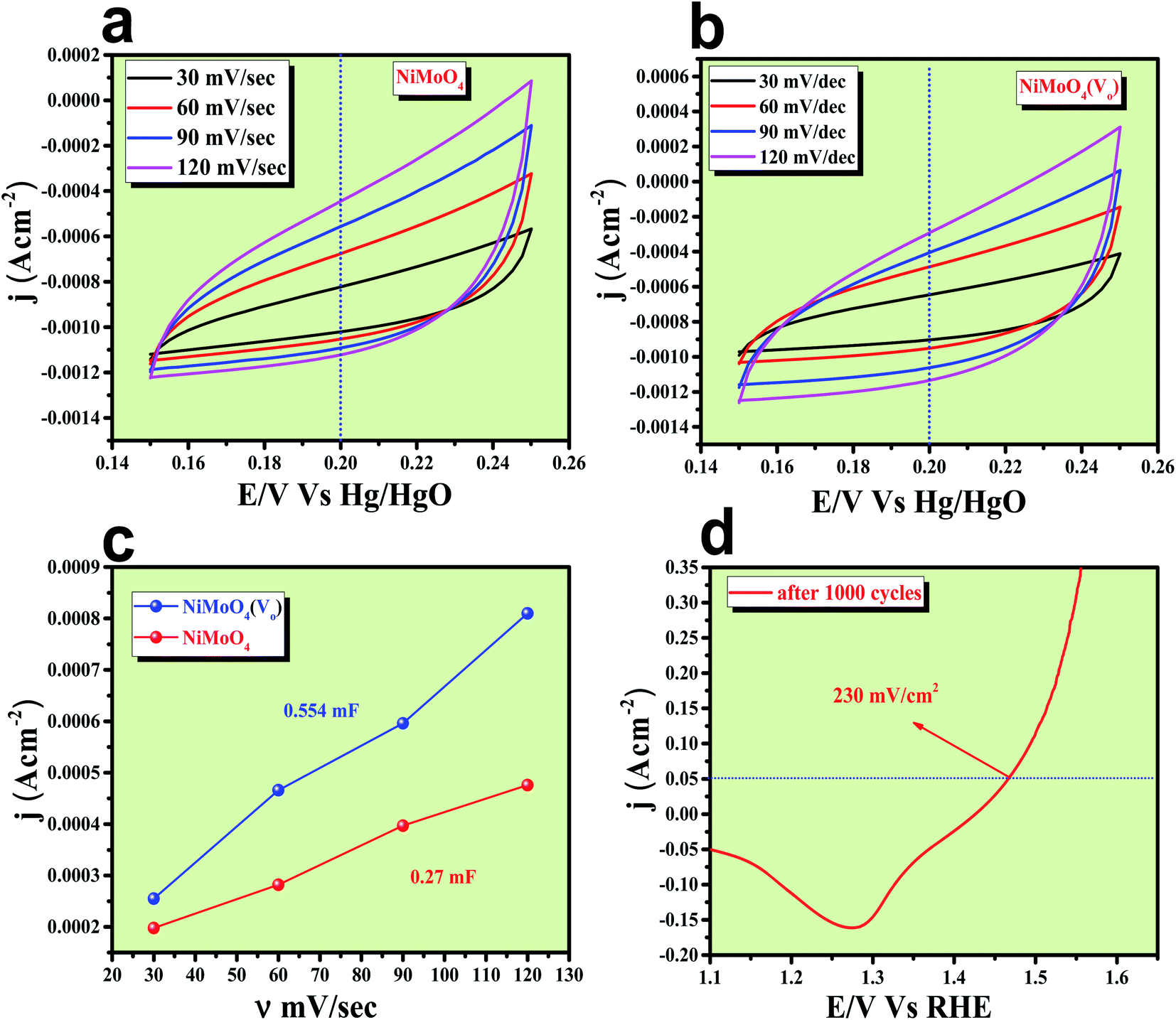
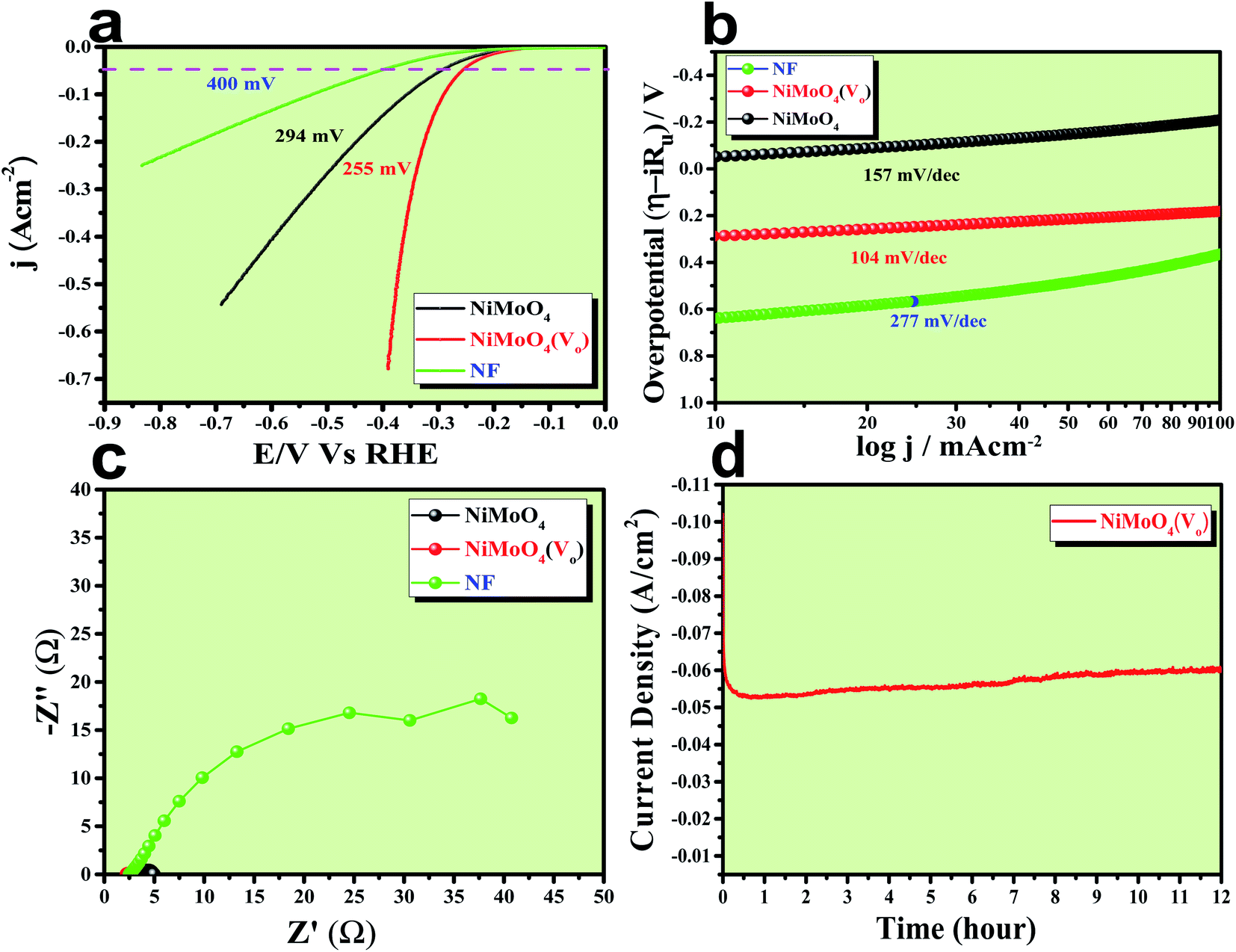
After having the exact knowledge of structural and morphological aspects of NiMoO4 and NiMoO4(Vo), both the materials were subjected to OER and HER applications in ‘Fe-free’ 1 M KOH solution. Due to the large prevalence of oxidation peaks at a potential of around 1.35 V (vs. RHE) with an excess current in the forward CV, to have an expressive identification of the activity, the backward CV was considered to precisely calculate the overpotential. For comparing the activity of all catalysts, we calculated the required overpotential values at 50 mA cm−2 current density. It can be observed from Fig. 7a that to reach the above-mentioned current density, NiMoO4(Vo) demands only 220 mV overpotential, whereas bare NiMoO4 required 255 mV overpotential to reach the same current density. Overpotential values required to achieve current densities of 10, 50 and 100 mA cm−2 were calculated and represented by a bar diagram as given in Fig. S2,‡ which shows the superior activity of NiMoO4(Vo) compared to that of bare NiMoO4. To gain knowledge about the charge transfer kinetics at the electrode–electrolyte interfaces, Tafel slope analysis was conducted from the iR drop free LSV curves and the corresponding Tafel plot is shown in Fig. 7b. NiMoO4(Vo) possesses a Tafel slope value of 30 mV dec−1, whereas NiMoO4 and NF possess the Tafel slope values of 60 mV dec−1 and 100 mV dec−1, respectively. Observed Tafel slope values indicating the highest rate of electron transfer at the electrode–electrolyte interfaces containing NiMoO4(Vo) as a catalyst. Charge transfer resistance was calculated from EIS spectra at an overpotential value of 300 mV and corresponding results are shown in Fig. 7c. NiMoO4(Vo) shows the lowest Rct value of 0.47 Ω, whereas bare NiMoO4 and NF display Rct values of 1.15 and 22.85 Ω, respectively, in consistence with high rated electron transfer in the case of NiMoO4(Vo). The chronoamperometric study was carried conducted out at a current density of 50 mA cm−2 for 12 h and it was observed that the catalyst possessed very high stability. To look more inside into the cause of the high activity of vacancy-enriched NiMoO4, electrochemical active surface areas (ECSA) were calculated in the potential region of 0.15 to 0.25 V (vs. Hg/HgO) and the corresponding CV curves are shown in Fig. 8a and b. The observed Cdl values (Fig. 8c) were 0.554 and 0.27 mF for NiMoO4(Vo) and NiMoO4, respectively, revealing the exposure of a more number of surface-active sites after vacancy creation on the surfaces. To know the robustness of the catalyst accelerated degradation (AD) studies were carried out at a scan rate of 200 mV s−1 for 1000 cycles and it showed a constant activity, as shown in Fig. 8d, even after such a high scan rate potential application for long cycles. The turn over frequency (TOF) values were calculated for NiMoO4 and NiMoO4(Vo) at an overpotential value of 310 mV and the obtained values are 1.37 × 10−4 and 4.016 × 10−4 s−1, respectively, showing the superior activity of the vacancy-enriched NiMoO4 for OER. A comparison table (Table S1 in ESI section‡) for our catalyst with recently reported similar types of materials shows that our synthesized catalyst has superior and highest activity as compared to others.
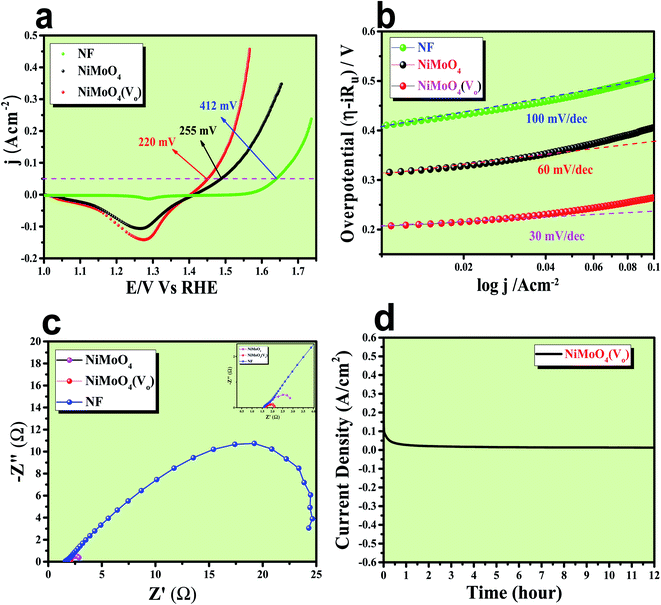 |
| | Fig. 7 (a) Is the backward CV of rod like NiMoO4(Vo) and NiMoO4 at a scan rate of 5 mV s−1, (b) corresponding Tafel plot, (c) impedance spectra obtained at 300 mV overpotential and (d) chronoamperometric outcomes for 12 h. | |
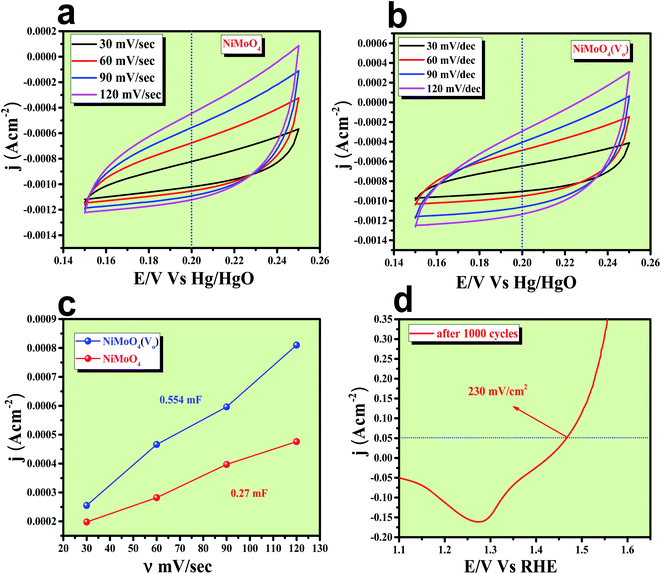 |
| | Fig. 8 (a) and (b) Are the CVs recorded for NiMoO4 and NiMoO4(Vo) respectively in a non-faradaic region with increasing scan rate for the determination of ECSA from its double layer capacitance; (c) are the corresponding plots to the measure Cdl; (d) backward CV after 1000 cycles of AD study. | |
Electrocatalytic HER activity of NiMoO4 and NiMoO4(Vo) in 1 M KOH
The electrocatalytic HER application was studied in a 1 M KOH solution and all the corresponding results are given in Fig. 9. First of all, an LSV polarization study was carried out to evaluate the HER activity trends in terms of calculation of overpotential at a current density of 50 mA cm−2 and the obtained result is shown in Fig. 9a. It was observed that NiMoO4(Vo) demanded only 255 mV overpotential, whereas, bare NiMoO4 and NF required 294 and 400 mV of overpotentials, respectively, to reach the same current density, showing the improvements in catalytic activity as a result of oxygen vacancy creation. Tafel slope analysis was taken from the iR drop free LSV curve and is shown in Fig. 9b. The lowest Tafel slope value was observed for NiMoO4(Vo), which is about 104 mV dec−1, whereas, bare NiMoO4 and NF possess the Tafel slope values of 157 and 277 mV dec−1, respectively, revealing fast charge transfer kinetics for NiMoO4(Vo) at electrode–electrolyte interfaces. EIS was carried out at an overpotential value of 300 mV to know the charge transfer resistance at the electrode surfaces and the corresponding spectra are given in Fig. 9c. NiMoO4(Vo) shows the lowest Rct value of 0.43 Ω whereas bare NiMoO4 and NF display Rct values of 1.28 and 37.5 Ω, respectively, that is consistent with highly-rated electron transfer in the case of NiMoO4(Vo). The chronoamperometric study was carried out at a current density of 50 mA cm−2 for 12 hours and it was observed that the catalyst possesses a very high stability with an increase in the activity as shown in Fig. 9d. Overpotential values required to achieve 10, 50 and 100 mA cm−2 current density values were calculated and represented as a bar diagram in Fig. S3a,‡ which shows the superior activity of NiMoO4(Vo) compared to that of bare NiMoO4. To ensure the robustness of the catalyst, the AD study was carried out at a scan rate of 200 mV s−1 for 1000 cycles and the LSV study was carried out after the cycling. Fig. S3b‡ shows the corresponding LSV curve and it can be observed that the overpotential value was decreased to 219 mV suggesting an increase in the activity of the catalyst and the result is consistent with chronoamperometric results shown in Fig. 9d. Here also the turn over frequency (TOF) values were calculated for NiMoO4 and NiMoO4(Vo) at an overpotential value of 310 mV and the obtained values are 1.63 × 10−4 and 4.06 × 10−4 s−1, respectively, showing the superior activity of the vacancy-enriched NiMoO4 for HER.
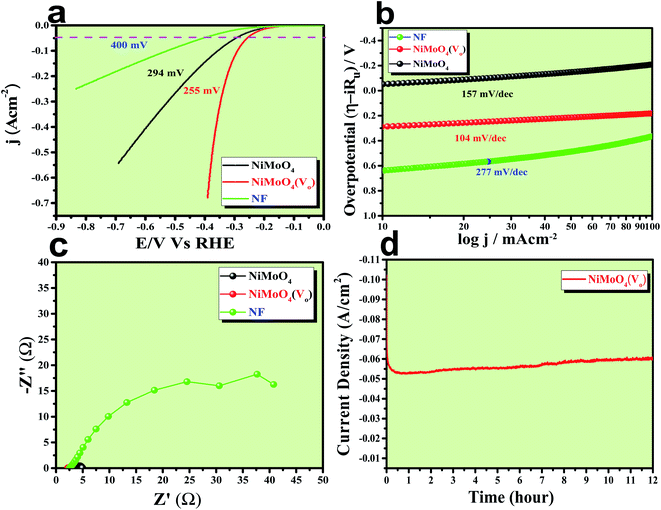 |
| | Fig. 9 (a) LSV curve of rod like NiMoO4(Vo) and NiMoO4 at a scan rate of 5 mV s−1, (b) corresponding Tafel plot, (c) impedance spectra obtained at 250 mV overpotential and (d) chronoamperometric outcomes for 12 hours. | |
The total water splitting study of NiMoO4(Vo) as both anode and cathode
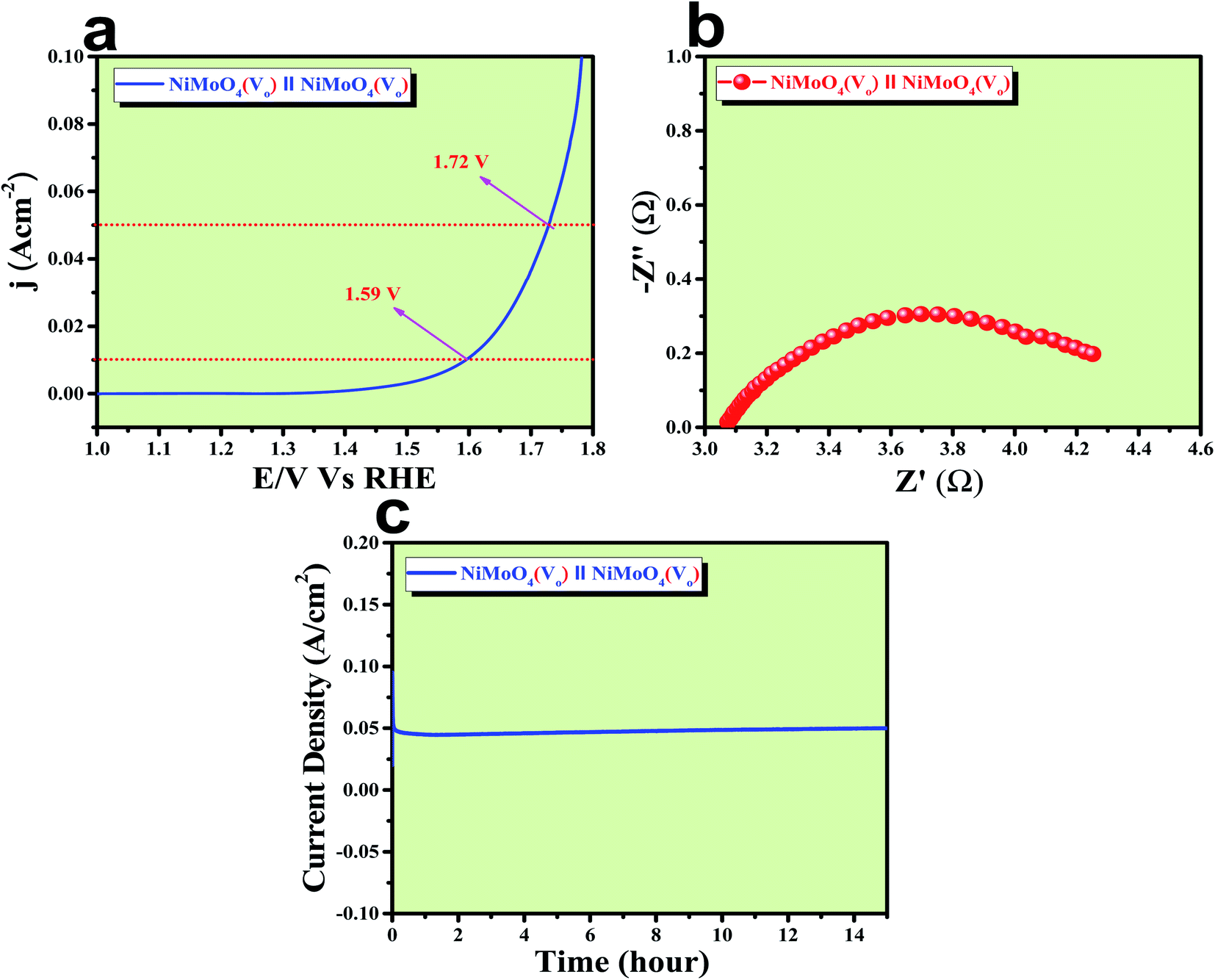
After confirming the outstanding activity and stability of NiMoO4(Vo) under OER and HER potential regions under alkaline conditions, NiMoO4(Vo) coated electrodes were applied in a two-electrode system for visualizing the real activities of large-scale production of H2 and O2, as shown in Fig. 10. For that, NiMoO4(Vo) was used as both anode and cathode and delivered a substantial water splitting activity by demanding 490 mV of overpotential to reach 50 mA cm−2 of current density (Fig. 10a). Further, EIS was performed to know the charge transfer resistance at the electrode–electrolyte interface and the observed Rct value was 1.43 Ω (Fig. 10b). To show the stable nature of this two-electrode set-up, a chronoamperometric study was carried out for 15 hours at an overpotential of 490 mV and it showed a constant current density, revealing the stable nature of the catalyst (Fig. 10c). From these studies, it is clear that NiMoO4(Vo) could be a potential candidate for delivering large-scale production of both hydrogen and oxygen and that too with the very low applied input of energy by delivering concomitant activities.
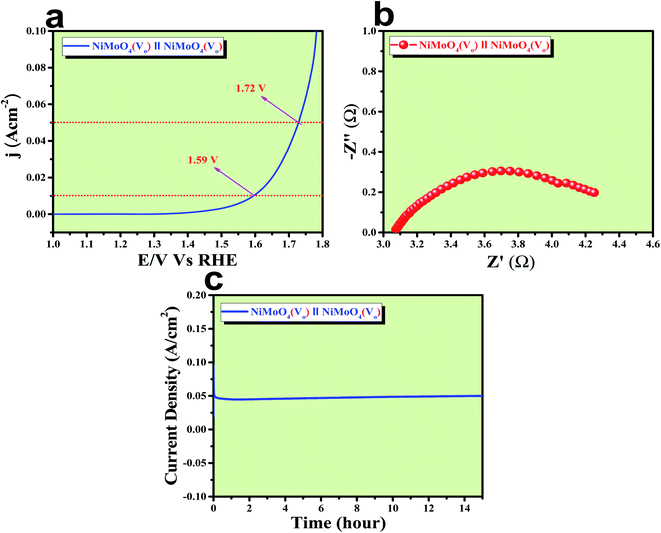 |
| | Fig. 10 (a) LSV curve of rod like NiMoO4(Vo) as an anode and cathode in a two electrode system at a scan rate of 2 mV s−1, (b) impedance spectra obtained at 400 mV overpotential, and (c) chronoamperometric outcomes for 15 hours. | |
Role of vacancies from NiMoO4(Vo) in electrocatalytic water splitting activity
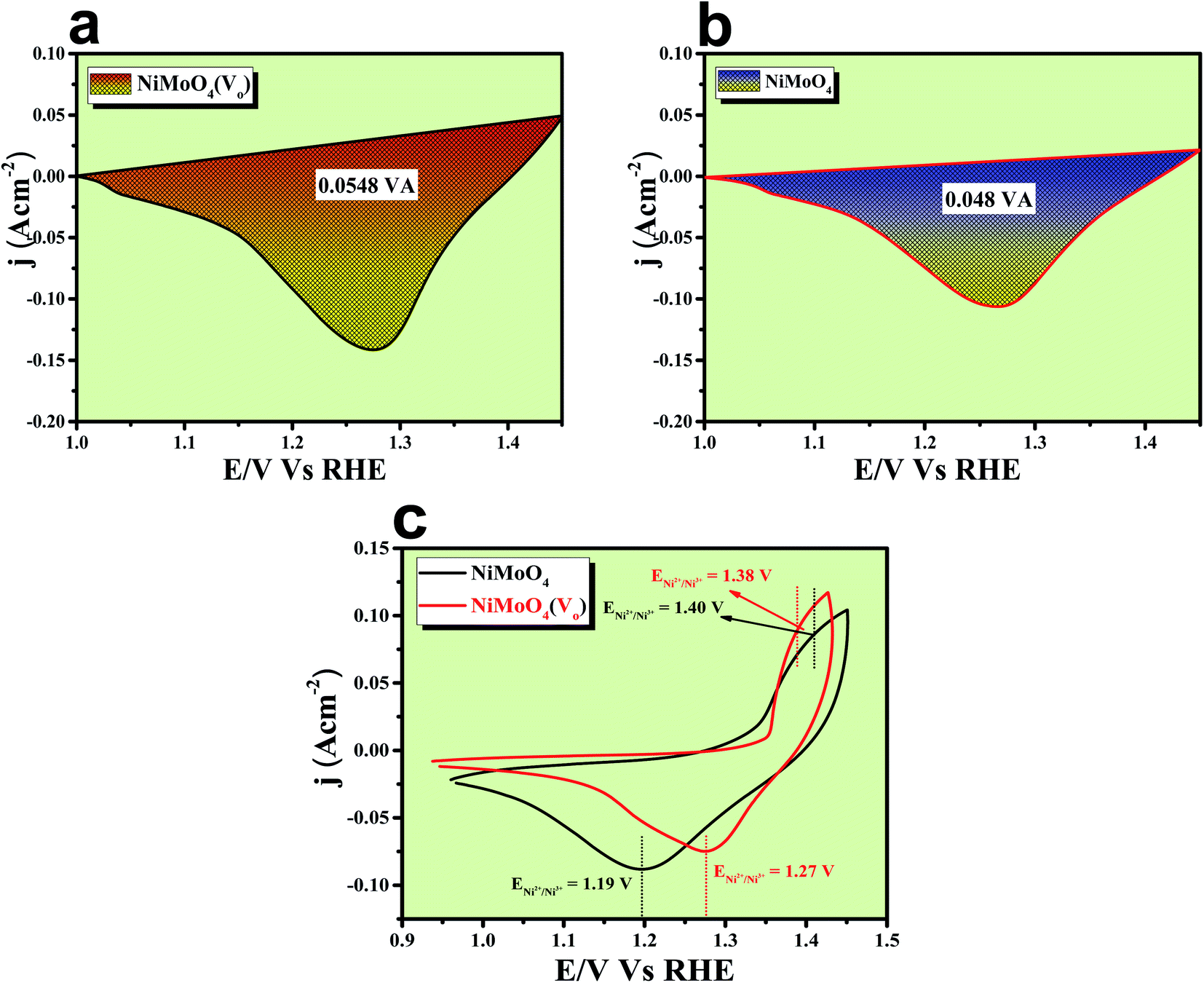
The formation of unsaturated metal ions on the surface of the catalyst would always lead to an increase in surface energy of the particles, which in turn results in an increase in catalytic activity. Vacancies in a lattice can be of three types viz. (1) cation vacancy; (2) anion vacancy and (3) multiple vacancies.27,29 Out of all these type of vacancies, the ones formed by missing one or several anions in the lattice are the most studied and characterized ones. Internal oxygen vacancy creation is one of the most pronounced defect formations in the lattice, which can drastically improve the electrocatalytic activity of a catalyst. Oxygen vacancy can be formed by means of different ways like solvent exfoliation,28 plasma etching,30 high-temperature annealing at reducing environments, annealing at oxygen atmosphere and wet chemical reduction by using different reducing agents. Out of various available methods, wet chemical reduction by using a suitable reducing agent is the easiest and acceptable one because of its easy handling in achieving plenty amounts of oxygen vacancy formation. The formation of oxygen vacancy takes place via the following two steps mechanism.27where, the *, + and − denote nominal neutral, positive and negative charges, respectively. It is well established that a lower coordination number in the catalytically active site would offer a greater extent of dangling bonds on the surface of the catalyst, which results in an increase in the activity of the catalyst.31,32 As we aware of the fact that OER is a multistep process that proceeds via the formation of different intermediates towards the forward direction by forming and breaking of various metal–oxygen bonds. So, to proceed in the forward direction, it is necessary to optimize the metal–oxygen bond strength. The most important step for OER is the formation of oxyhydroxide formation via the oxidation of metal ions with the O–O bond formation, which is formed by the hydroxide ions attack on M–O bonds.31,32 Creation of oxygen vacancy close to the metal–oxygen bonds leads to the extraction of electron density from metal ions and there will be an increase in electron deficiency on the metal, which further leads to favour the nucleophilic attack of H2O/OH− to form an oxyhydroxide derivative. With the increase in the extent of attacking from hydroxide ions to metal oxide bonds, there will be a greater chance for the formation of oxyhydroxide derivative with lower applied potentials. The extent of hydroxide ion interaction at active sites can indirectly be identified via the redox-featured area calculation from CV by choosing the proper area of either oxidation or reduction peaks of different catalyst materials.33 The corresponding areas for reduction peaks for NiMoO4(Vo) and NiMoO4 are shown in Fig. 11a and b, respectively, and the calculated areas are 0.0548 VA and 0.048 VA, respectively, that reveals the fact that the formation of oxygen vacancy could direct the increase in the surface active sites for OER. To gain information on the redox behaviour of metal ions, CV cycling was carried out in the potential region of 1 to 1.5 V vs. RHE, and the corresponding CV plot is shown in Fig. 11c. It can be observed that due to the vacancy formation, there is a large shift in the redox behaviour of the catalyst materials, such as the oxidation peak for Ni2+/Ni3+ redox couple for NiMoO4(Vo) is shifted to lower potentials (1.38 V vs. RHE) as compared to that of NiMoO4 (1.40 V vs. RHE), suggesting the feasibility of the active intermediate formation in NiMoO4(Vo) as compared to the bare one. Suntivich et al. reported the electrocatalytic performance of transition metal-based compounds which were highly dependable on electron occupancy of crystal field which was split as eg orbitals.8 Highest catalytic activity would be shown by a catalyst that has eg orbital occupancy close to unity. Considering crystal field splitting electronic arrangements of Ni2+ (t2g6eg2) and Ni3+ (t2g6eg1), the latter one has more chances of having an electron occupancy close to unity and thus leads to more OER activity. As we have discussed before, the formation of oxygen vacancy led to the creation of electron deficiency in the Ni2+ sites and hence favoured the Ni3+ oxidation state. The electronic arrangement of NiMoO4(Vo) in the Ni centre can be seen in Scheme 2, which displays that oxidation of Ni2+ that results in the electron occupancy in the eg orbital to be close to unity and favouring OER with lower overpotential.13 Apart from the electronic effect, one more important factor is the bond strength of metal oxide formed during the course of the reaction. The formation of metal oxide bonds through the hydroxide ion attacks should be optimal, i.e., neither too strong nor very weak. If we consider the molecular orbital theory, the bond strength of particular bonds primarily depends on the electron density on the antibonding orbitals. Based on the electronic origin of the bond strength descriptor given by Tao et al., here, we propose the model shown in Scheme 3 based on the molecular orbital and band structure theory.34 Being the second highest electronegative element, the oxygen atom always behaves as an acceptor during bond formation with metal ions. Here, we will consider the interaction between O and transition metal ions (3d or 4d transition metals) that would contribute to allowing the effective coupling between O 2p and highest occupied d states and that coupling would result in the formation of bonding and antibonding states. Generally, bonding states are staying far below the Fermi level (EF) and are fully filled.27,35–38 On the other hand, filling of higher energy-antibonding state largely depends on the energy difference between metal d bands and Fermi energy levels, and hence, higher the energy difference lower will be the electron occupancy in that antibonding orbitals. For stoichiometric oxide materials, the placement of the d band is usually far below that of EF, which results in lower energy of the leading to the antibonding states, resulting in completely filled states of antibonding orbitals and thereby formation of weak metal–oxygen bonds. In the case of nonstoichiometric oxide, i.e., enriched with a large number of oxygen vacancies, they can create a new electronic state called band gap states (BGS), which arise due to the availability of a large number of unpaired d electrons at higher energies that keep them above Fermi levels. This particular increase in the energy of d bands elevating the antibonding states above EF with considerable enhancements in energy leads to partial filling of antibonding states, which assists the formation of metal bonds with optimum bond energy indirectly. So, in conclusion, the formation of oxygen vacancies is highly advantageous not only favouring the incoming of electroactive species but also reduces the Fermi-level energy by keeping antibonding states at higher energies. This fruitful application of vacancy creations ensured favourable electronic arrangement, redox features of metal ions in the less applied overpotentials (Fig. 11c) and ensured bonding capability of oxygen with rich electroactive sites for having more improvements in OER activity.
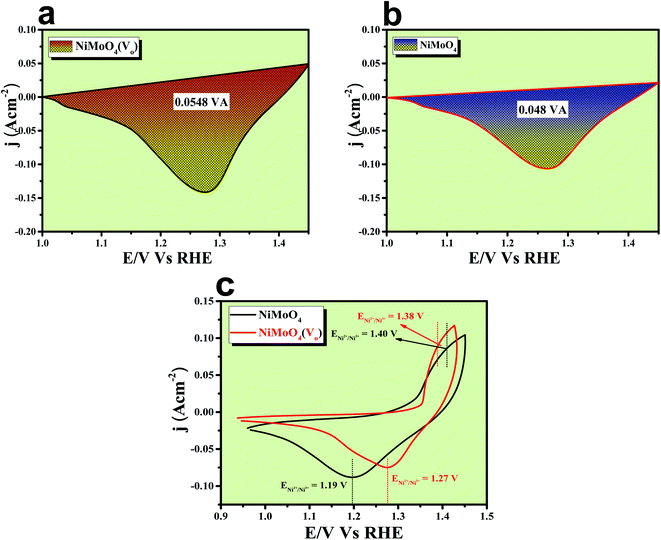 |
| | Fig. 11 (a) and (b) Reduction peak area of rod-like NiMoO4(Vo) and NiMoO4; (c) CV cycles for NiMoO4(Vo) and NiMoO4. | |
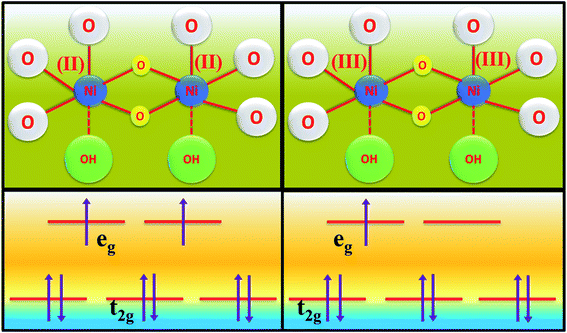 |
| | Scheme 2 Scheme of electron occupancy in different crystal field splitted orbitals of the Ni centre. | |
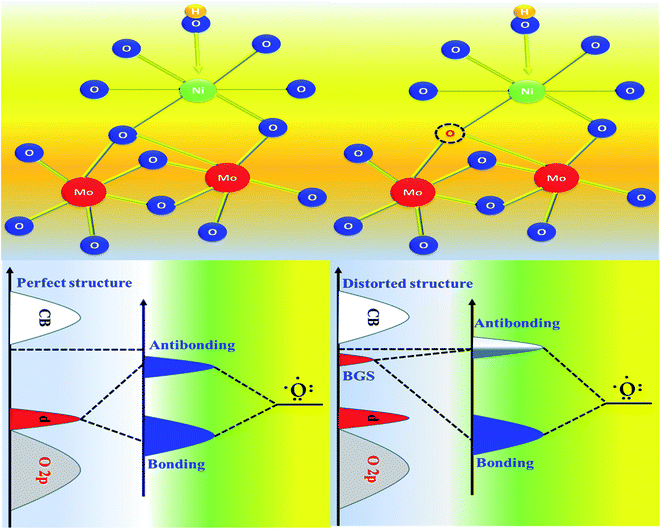 |
| | Scheme 3 Schematic representation of molecular orbital and band structure, for the origination of high electrocatalytic activity of NiMoO4(Vo) through vacancy formation. | |
Post characterization study of NiMoO4(Vo)
It is indeed necessary to provide information about the morphological and chemical changes that have occurred during the harsh anodic potential application. For this purpose, first, we carried out FE-SEM colour-mapping, HR-TEM and XPS analysis of NiMoO4(Vo) after the OER study. First of all, to confirm the presence of all the elements even after harsh anodic potential application, FE-SEM colour mapping analysis was carried out and the corresponding results are shown in Fig. S4,‡ and it shows a uniform distribution of O, Ni and Mo, which confirmed the exceptional stability of the catalyst under harsh anodic condition for long times.
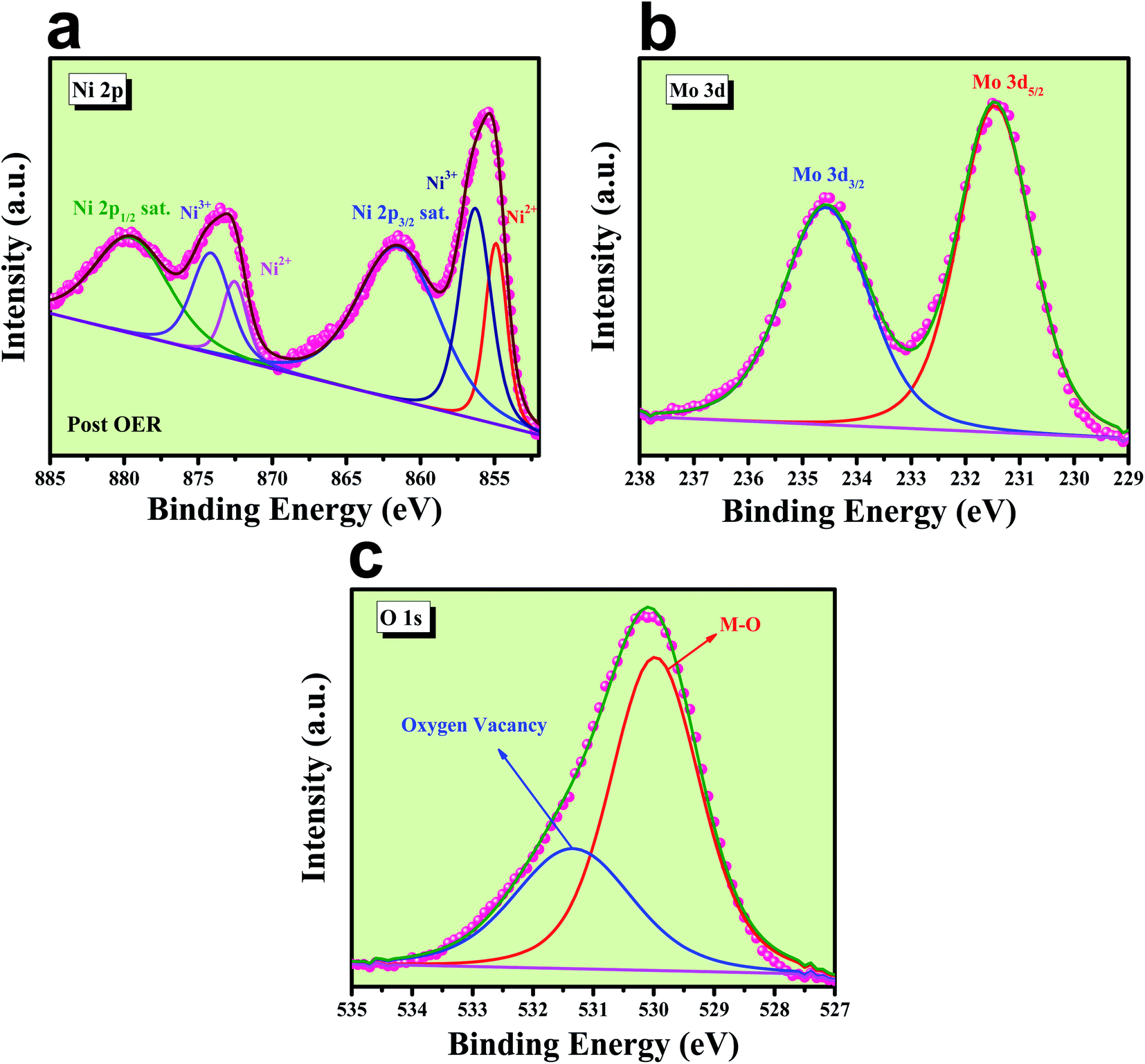
EDS analysis after OER was carried out at the FE-SEM mode and is given in Fig. S5.‡ It shows the presence of all the expected elements along with a new peak for K, which arises from the electrolyte used during electrochemical OER studies. Post OER, the microstructural outcome was observed from HR-TEM analysis and the corresponding images are shown in Fig. S6a–c‡ from low to high magnification, respectively, and displays the same rod-like morphology for the particles. The SAED pattern (inset of Fig. S6a‡) shows the polycrystalline nature of the particles with the diffraction of the particles and mainly showed (001) and (111) planes. HAADF colour mapping was also performed for confirming the elemental distribution of all the elements and is shown in Fig. S6d–h,‡ and it shows a uniform distribution of all the expected elements like Ni, Mo and O all over the surface. The chemical nature of all the elements was analysed using XPS spectroscopy, results of which are shown in Fig. 11. The deconvoluted XPS spectrum of Ni 2p orbitals shown in Fig. 12a indicates the same pattern as it was in the pre-catalyst, except that one new peak arises at a binding energy of 856.03 eV due to the presence of nickel ions in the +3 oxidation state, which was formed by the prolonged anodic potential application on the working electrode with the active site being NiOOH during OER. The XPS spectra of Mo 3d orbitals shown in Fig. 12b indicate two maxima at binding energies of 231.48 and 234.58 eV for Mo 3d5/2 and 3d3/2 orbitals, respectively, revealing the presence of Mo in +6 oxidation states. High-resolution XPS spectra of O 1s orbital are shown in Fig. 12c displaying the existence of two spin–orbit couplings originating peaks at the same binding energies as that was in pre-catalysts. The areas of the oxygen vacancy peaks for both pre and post-OER catalyst materials were calculated and the obtained ratio was about 1.125, which revealed that the concentration of the oxygen vacancy decreases after the long-term application of anodic potential. The origin of this decrease in oxygen vacancy concentration might be arising due to oxidation of the surfaces under anodic conditions. From the cooperative post characterization results, even after the application of harsh anodic potentials, the prepared rod-shaped oxygen vacancy-enriched NiMoO4 catalysts were found to be highly stable in nature and also the morphological outcomes were retained and hence can be employed for large scale water splitting in the future.
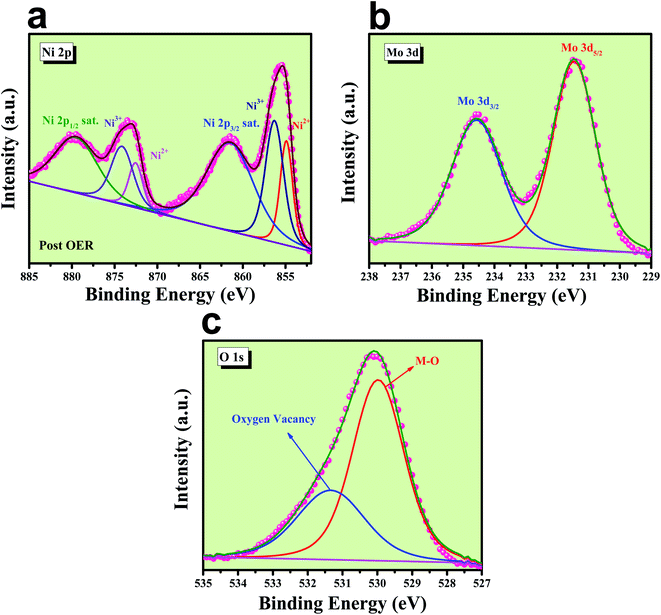 |
| | Fig. 12 Post XPS characterization of NiMoO4(Vo): (a) high-resolution XPS spectra of Ni 2p orbitals, (b) XPS spectra of Mo 3d orbital and (c) deconvoluted XPS spectrum of O 1s orbitals. | |
Conclusion
To summarize, this work highlighted for the very first time, the fast formation of vacancy-enriched NiMoO4 nanorods via microwave heating. The formation of oxygen vacancy-enriched NiMoO4 nanorods led to improvements in the electrocatalytic performance of the catalyst under alkaline conditions with very low overpotentials in OER and HER as well as in the total water splitting study in 1 M KOH. XPS and EPR analysis confirmed the presence of oxygen vacancy formation that ensured improvisation of the surface electronic structure. This favourable-vacancy enriched NiMoO4(Vo) nanorods show huge improvements in activities when compared with bare NiMoO4 nanorods. Overall, NiMoO4(Vo) delivered excellent OER and HER performances reached 50 mA cm−2 of current density, the vacancy-enriched NiMoO4(Vo) required 220 and 255 mV of overpotentials for OER and HER conditions, respectively. After realizing, excellent OER and HER performances, NiMoO4(Vo) was used as both, anode and cathode, in the two-electrode system to reach a current density of 10 mA cm−2, at a overpotential of 360 mV only. This precise approach to vacancy creation is certainly one of the best ways of increasing the electrocatalytic OER/HER performance with highly exposed active sites. Moreover, the idea of the formation of oxygen vacancy-enriched NiMoO4 catalysts can also be extended successfully in other catalytic systems such as different chalcogenides and oxides-based materials in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We wish to acknowledge Dr N. Kalaiselvi, Director, CSIR-CECRI for her constant support and encouragement. Arun Karmakar wishes to acknowledge CSIR-New Delhi for the award of a Junior Research Fellowship (JRF). K. Karthick and Sam Sankar wish to acknowledge UGC for the SRF award. Sangeetha Kumaravel and Madhu Ragunath wish to acknowledge DST inspire fellowships. Subrata Kundu wishes to acknowledge the Department of Science and Technology (DST) for HCF (Hydrogen and Fuel Cell) research funding of number #DST/TMD/HFC/2K18/60 on 7th October 2019 with institute OM number 18-29-03/(31/19)-TTBD-CSIR-CECRI on 24/10/2019.
References
- J. O. M. Bockris, Int. J. Hydrogen Energy, 2002, 27, 731–740 CrossRef CAS.
- I. Dincer, Int. J. Hydrogen Energy, 2012, 37, 1954–1971 CrossRef CAS.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- T. Audichon, T. W. Napporn, C. Canaff, C. Morais, C. Comminges and K. B. Kokoh, J. Phys. Chem. C, 2016, 120, 2562–2573 CrossRef CAS.
- R. R. Rao, M. J. Kolb, N. B. Halck, A. F. Pedersen, A. Mehta, H. You, K. A. Stoerzinger, Z. Feng, H. A. Hansen, H. Zhou, L. Giordano, J. Rossmeisl, T. Vegge, I. Chorkendorff, I. E. L. Stephens and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2626–2637 RSC.
- R. L. Tichenor, Ind. Eng. Chem., 1952, 44, 973–977 CrossRef CAS.
- M. Yu, G. H. Moon, R. G. Castillo, S. DeBeer, C. Weidenthaler and H. Tüysüz, Angew. Chem., Int. Ed., 2020, 59, 16544–16552 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, M. Ragunath and S. Kundu, Inorg. Chem., 2021, 60, 2680–2693 CrossRef CAS PubMed.
- A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9, 1314–1352 RSC.
- S. Anantharaj and S. Noda, Small, 2020, 16, 1–24 CrossRef PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- A. Karmakar, K. Karthick, S. Kumaravel, S. S. Sankar and S. Kundu, Inorg. Chem., 2021, 60, 2023–2036 CrossRef CAS PubMed.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- Y. Zhu, H. C. Chen, C. S. Hsu, T. S. Lin, C. J. Chang, S. C. Chang, L. D. Tsai and H. M. Chen, ACS Energy Lett., 2019, 4, 987–994 CrossRef CAS.
- J. Joo, T. Kim, J. Lee, S. Il Choi and K. Lee, Adv. Mater., 2019, 31, 1–23 CrossRef PubMed.
- S. Kumaravel, P. Thiruvengetam, S. R. Ede, K. Karthick, S. Anantharaj, S. Sam Sankar and S. Kundu, Dalton Trans., 2019, 48, 17117–17131 RSC.
- N. Han, F. Zhao and Y. Li, J. Mater. Chem. A, 2015, 3, 16348–16353 RSC.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. G. De Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- K. Karthick, A. B. Mansoor Basha, A. Sivakumaran and S. Kundu, Catal. Sci. Technol., 2020, 10, 3681–3693 RSC.
- X. Zhang, H. Su and X. Du, New J. Chem., 2020, 44, 8176–8182 RSC.
- Z. Zhang, X. Ma and J. Tang, J. Mater. Chem. A, 2018, 6, 12361–12369 RSC.
- J. Chen, G. Zhao, Y. Chen, K. Rui, H. Mao, S. X. Dou and W. Sun, Chem.–Eur. J., 2019, 25, 280–284 CrossRef CAS PubMed.
- P. R. Jothi, K. Shanthi, R. R. Salunkhe, M. Pramanik, V. Malgras, S. M. Alshehri and Y. Yamauchi, Eur. J. Inorg. Chem., 2015, 2015, 3694–3699 CrossRef CAS.
- Q. Liu, J. Huang, Y. Zhao, L. Cao, K. Li, N. Zhang, D. Yang, L. Feng and L. Feng, Nanoscale, 2019, 11, 8855–8863 RSC.
- M. Arif, G. Yasin, M. Shakeel, M. A. Mushtaq, W. Ye, X. Fang, S. Ji and D. Yan, J. Energy Chem., 2021, 58, 237–246 CrossRef.
- K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- P. Zhou, Y. Wang, C. Xie, C. Chen, H. Liu, R. Chen, J. Huo and S. Wang, Chem. Commun., 2017, 53, 11778–11781 RSC.
- M. Q. Yang, J. Wang, H. Wu and G. W. Ho, Small, 2018, 14, 1–24 Search PubMed.
- R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1–7 Search PubMed.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- C. Hao, Y. Wu, Y. An, B. Cui, J. Lin, X. Li, D. Wang, M. Jiang, Z. Cheng and S. Hu, Mater. Today Energy, 2019, 12, 453–462 CrossRef.
- K. Karthick, S. Anantharaj, P. E. Karthik, B. Subramanian and S. Kundu, Inorg. Chem., 2017, 56, 6734–6745 CrossRef CAS PubMed.
- H. B. Tao, L. Fang, J. Chen, H. Bin Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed.
- M. García-Mota, A. Vojvodic, H. Metiu, I. C. Man, H. Y. Su, J. Rossmeisl and J. K. Nørskov, ChemCatChem, 2011, 3, 1607–1611 CrossRef.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- A. C. Papageorgiou, N. S. Beglitis, C. L. Pang, G. Teobaldi, G. Cabailh, Q. Chen, A. J. Fisher, W. A. Hofer and G. Thornton, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2391–2396 CrossRef PubMed.
- S. Wendt, P. T. Sprunger, E. Lira, G. K. H. Madsen, Z. Li, J. Hansen, J. Matthiesen, A. Blekinge-Rasmussen, E. Lægsgaard, B. Hammer and F. Besenbacher, Science, 2008, 320, 1755–1759 CrossRef CAS PubMed.
Footnotes |
| † CSIR-CECRI MS reference number #CECRI/PESVC/Pubs./2021-039. |
| ‡ Electronic supplementary information (ESI) available: Scheme of synthesis, electrode fabrications, material preparation for different characterizations, data for OER, comparative electrocatalysis data. See DOI: 10.1039/d1ta02165f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Kannimuthu
Karthick
ab,
Kannimuthu
Karthick