
Three-in-one: achieving a robust and effective hydrogen-evolving hybrid material by integrating polyoxometalate, a photo-responsive metal–organic framework, and in situ generated Pt nanoparticles†
Le
Jiao
,
Yuanyuan
Dong
,
Xing
Xin
,
Ruijie
Wang
and
Hongjin
Lv
 *
*
MOE Key Laboratory of Cluster Science, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 102488, P. R. China. E-mail: hlv@bit.edu.cn
First published on 26th May 2021
Abstract
A Wells–Dawson-type polyoxometalate ([P2W18O62]6−, denoted as P2W18), a photo-responsive Zr-based metal–organic framework (MOF, NU-1000), and in situ generated Pt nanoparticles were successfully integrated into a three-in-one hybrid material (P2W18@NU-1000-Pt) using a facile impregnation and subsequent photoreduction method. The resulting three-in-one hybrid material exhibits effective and robust activity towards photocatalytic hydrogen production in a water-compatible system under Xe-lamp irradiation, achieving a hydrogen production of 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 μmol g−1 and a turnover number (TON) of 5484 versus moles of Pt after 5 days of reaction. The post-catalysis three-in-one hybrid composite could be easily recycled at least 5 times without declining catalytic activity. Moreover, such a water-compatible photocatalytic system also reveals potential practical applications for catalyzing hydrogen production under natural sunlight irradiation. A possible photocatalytic mechanism was elucidated based on various photophysical and spectroscopic analyses, proving the importance of synergistic cooperation of the NU-1000 MOF, the P2W18 cluster, and in situ generated Pt NPs in such a three-in-one P2W18@NU-1000-Pt hybrid photocatalyst.
100 μmol g−1 and a turnover number (TON) of 5484 versus moles of Pt after 5 days of reaction. The post-catalysis three-in-one hybrid composite could be easily recycled at least 5 times without declining catalytic activity. Moreover, such a water-compatible photocatalytic system also reveals potential practical applications for catalyzing hydrogen production under natural sunlight irradiation. A possible photocatalytic mechanism was elucidated based on various photophysical and spectroscopic analyses, proving the importance of synergistic cooperation of the NU-1000 MOF, the P2W18 cluster, and in situ generated Pt NPs in such a three-in-one P2W18@NU-1000-Pt hybrid photocatalyst.
Introduction
Energy shortages and environmental pollution caused by fossil fuels are currently a major challenge. Solar energy, as a clean and sustainable energy source, is a very promising alternative solution to the increasingly serious energy crisis. The conversion of solar energy into storable chemical energy via photocatalytic water splitting for hydrogen generation has therefore received widespread attention. Various materials, such as carbon-based materials,1–4 semiconductors,5,6 conjugated polymers,7,8 covalent organic frameworks,9,10 noble metal nanoparticles,11,12 metal organic frameworks,13,14 polyoxometalates,15,16etc., have been investigated for photocatalytic hydrogen production. Polyoxometalates (POMs), as an emerging type of multi-electron-transfer catalyst, have been employed as photocatalysts for water splitting due to their highly tunable physicochemical properties, versatile synthetic methodology and multi-electron-transfer ability. With the assistance of soluble sensitizers such as [Ru(bpy)3]2+ and [Ir(ppy)2(bpy)]+, POMs could exhibit outstanding photocatalytic activity.17–21 In spite of their high TON for hydrogen generation in a homogeneous photocatalytic system, the recycling of POMs is still an urgent problem to be solved. To improve the recyclability of POM photocatalysts, a series of support materials, such as graphene,22 carbon nanotubes,23 metal oxides,24 zeolites,25 porous aromatic frameworks,26 covalent organic frameworks,27 and metal organic frameworks (MOFs),28 have been employed to construct POM-based composites for heterogeneous photocatalytic systems.MOFs, a class of materials composed of metal ions/clusters and organic linkers, have attracted widespread attention for immobilizing POMs due to their porous structures, rich chemical functionality, high surface area, crystallinity and chemical stability.29–35 The resulting POM@MOF composites are thus widely utilized in the photocatalytic hydrogen production system.36,37 For example, Lin and co-workers integrated a Wells–Dawson-type POM [P2W18O62]6− into a Zr-based MOF bearing the [Ru(bpy)x]2+ moiety, and encapsulated a cobalt-containing POM (P2W17Co) and photosensitizer into the pores of MIL-101 to achieve hydrogen production under visible light irradiation, respectively.38,39 Du et al. incorporated a high-nuclear {Cu24I(μ3-Cl)8(μ4-Cl)6}-based POM into ZZULI-1 to construct a dual-functionalized photocatalyst for visible-light-driven H2 and O2 evolution.40 These applications in photocatalytic hydrogen production demonstrate the potential value and diversity of POM@MOF composites. It is worth mentioning that the long-term stability and reusability of these POM@MOF composites are largely determined by the size match between the POM guest molecule and MOF host as well as the physicochemical properties of MOFs. Among various MOF supports, NU-1000, a Zr-based MOF with a prominent photo-response and diverse aperture size (mesopore and micropore), exhibits outstanding physicochemical stability and therefore has been widely employed as the host of composite photocatalysts. For instance, to improve the photogenerated charge separation efficiency, BiOI nanoparticles and CdSe nanocrystals have been successfully incorporated into NU-1000, respectively, and both hybrid composites exhibited excellent photocatalytic activity.41–43 To enhance the photocatalytic stability, Keggin-type H5PV2Mo10O40 and H3PW12O40 have also been immobilized into NU-1000 and the resulting hybrid materials showed efficient catalytic activities for aerobic oxidation of a mustard gas simulant, 2-chloroethyl ethyl sulfide.44,45 However, it is rarely reported about the size matching issue for the immobilization of a POM into NU-1000,37 especially in a solar-driven hydrogen generation system.
Herein, a classical Wells–Dawson-type POM P2W18 is therefore immobilized into NU-1000 to construct the P2W18@NU-1000 composite given the suitable match in size (Scheme 1). We further fabricate a three-in-one P2W18@NU-1000-Pt hybrid photocatalyst through the in situ formation of Pt nanoparticles (Pt NPs) and utilize it for the photocatalytic hydrogen evolution reaction. As a co-catalyst, Pt NPs have been investigated for photocatalytic hydrogen evolution,46–52 including Pt-POM and Pt-MOF composites.53–65 These studies reveal that the photocatalytic efficiency is largely associated with the Pt location relative to the POM and/or MOF. In our work, the in situ generated Pt NPs could be immobilized into the pores of the NU-1000 host or supported on the surface of the P2W18@NU-1000 composite to generate the three-in-one hybrid photocatalyst, which could not only prevent the leaching out of P2W18 guest molecules, but also promotes photogenerated charge separation efficiency to improve the photocatalytic performance. The resulting three-in-one P2W18@NU-1000-Pt photocatalyst therefore exhibits effective and robust photocatalytic activity with a hydrogen production of 6023 μmol g−1 after 12 h illumination that continuously increases to 35100 μmol g−1 after 5 days of reaction.
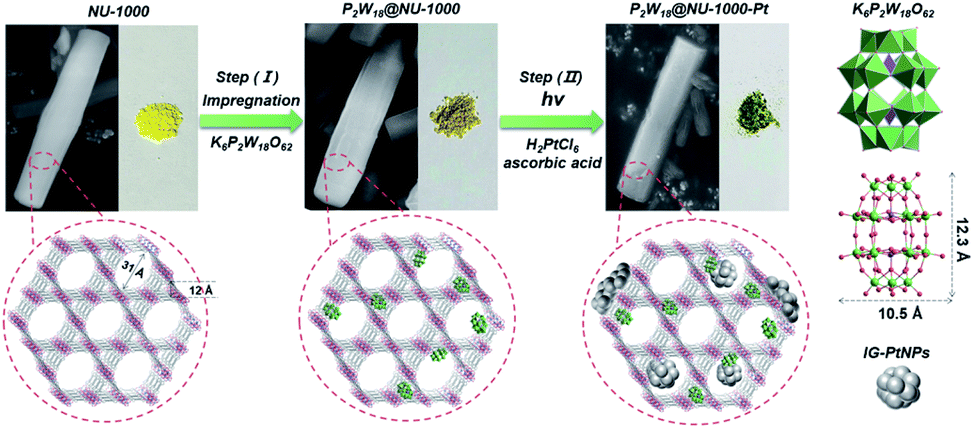 | ||
| Scheme 1 Schematic illustration of the preparation route of the three-in-one P2W18@NU-1000-Pt hybrid composite. The microscopic illustration of the structure is shown in the red circles. | ||
Results and discussion
The three-in-one P2W18@NU-1000-Pt hybrid composite was prepared via a facile impregnation (Step (I)) and subsequent photoreduction (Step (II)) approach as illustrated in Scheme 1. The NU-1000 (yellow powder) was added into the P2W18 aqueous solution, and then the mixture was subjected to ultrasonication, stirring, and post-treatment to obtain the dark yellow P2W18@NU-1000 powder as described in the Experimental section. Subsequently, during the photocatalytic reaction (Step (II)), the NU-1000 host was photoexcited to generate electron–hole pairs where the photogenerated electrons could be transferred to P2W18 and the H2PtCl6 to in situ form the Pt NPs, obtaining the three-in-one P2W18@NU-1000-Pt hybrid composite (dark green powder in Scheme 1). The detailed preparation procedures are described in the Experimental section. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) was employed to determine the loading amount of P2W18 in terms of P2W18 per {Zr6} node in the P2W18@NU-1000 composite (Table 1). The optimal P2W18/{Zr6} ratio was found as 0.34 for the photocatalytic hydrogen evolution reaction, and 0.34-P2W18@NU-1000 was therefore selected as the representative sample for a series of characterization experiments unless otherwise noted. Scanning Electron Microscopy (SEM) images illustrate that the morphology of 0.34-P2W18@NU-1000 still maintains a uniform rod-like structure as that of NU-1000 (Fig. 1a and S1†). The impregnation of the P2W18 guest does not change the smooth surface and morphology of the NU-1000 host. The similar Powder X-ray Diffraction (PXRD) patterns of NU-1000 and 0.34-P2W18@NU-1000 also prove that the immobilization of P2W18 has a negligible effect on the geometrical structure of the NU-1000 host (Fig. S2†). But the absolute intensity of the diffraction signals at the small angles decreases after P2W18 immobilization, which to some extent proves the incorporation of P2W18 clusters into the pores of the NU-1000 host. High-resolution transmission electron microscopy (HR-TEM) images further display the successful incorporation of P2W18 clusters (deep dark dots shown in Fig. 1c) into the NU-1000 host (Fig. 1b).| Compounds | W/P | W/Zr | P2W18/{Zr6} |
|---|---|---|---|
| 0.25-P2W18@NU-1000 | 8.20 | 0.76 | 0.25 |
| 0.29-P2W18@NU-1000 | 8.15 | 0.88 | 0.29 |
| 0.32-P2W18@NU-1000 | 8.09 | 0.95 | 0.32 |
| 0.34-P2W18@NU-1000 | 8.10 | 1.03 | 0.34 |
 | ||
| Fig. 1 Characterization of NU-1000 and the 0.34-P2W18@NU-1000 composite. (a) SEM image of 0.34-P2W18@NU-1000. HR-TEM images of (b) NU-1000 and (c) 0.34-P2W18@NU-1000. (d) The corresponding elemental mapping images of 0.34-P2W18@NU-1000. | ||
In addition, Energy Dispersive X-ray Spectroscopy (EDS) (Fig. S3†) and elements mapping images of 0.34-P2W18@NU-1000 (Fig. 1d) confirm the presence and uniform distribution of Zr and W elements of P2W18 in the NU-1000 host, further providing strong and straight evidence for the successful immobilization of P2W18.
Low temperature N2 adsorption–desorption tests for NU-1000 and 0.34-P2W18@NU-1000 were carried out to further reveal the incorporation and distribution of P2W18 in the NU-1000 host (Fig. 2a). The calculated BET surface areas decreased from 2065.7 m2 g−1 for pristine NU-1000 to 670.6 m2 g−1 for the 0.34-P2W18@NU-1000 composite, revealing the successful incorporation of P2W18 into the pores of NU-1000. The type IV hysteresis loop disappeared in the N2 adsorption–desorption isotherm of NU-1000 after immobilization of P2W18, demonstrating that the typical mesopores have been blocked thereby leading to a decreased adsorbed volume. Meanwhile, the overlapped adsorption and desorption curves imply the dominating role of micropores in the porous structure of 0.34-P2W18@NU-1000. The pore distribution curves in Fig. 2b also confirm the above-mentioned changes in the texture properties of NU-1000 after the immobilization of P2W18. Given the molecular size of P2W18 (Scheme 1), the P2W18 clusters could also be encapsulated into the micropores of NU-1000 as confirmed by the pore distribution curve shown in Fig. 2b.
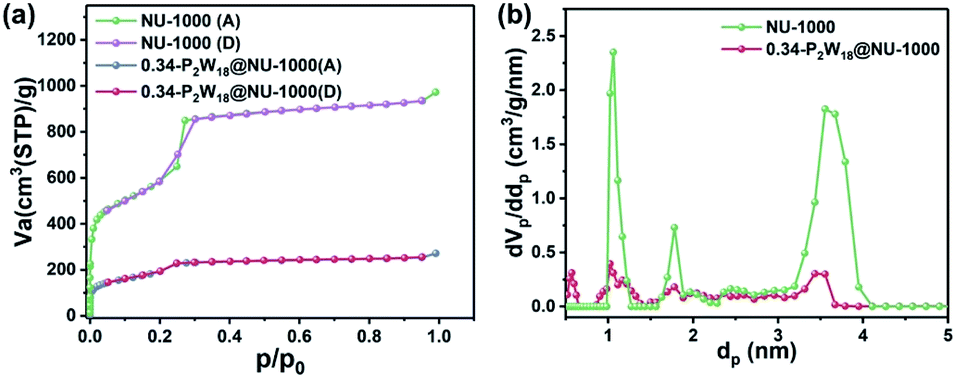 | ||
| Fig. 2 (a) The low temperature N2 adsorption–desorption isotherms and (b) the pore distribution curves of NU-1000 and 0.34-P2W18@NU-1000. Note: A and D represent adsorption and desorption, respectively. | ||
Fourier transform infrared (FT-IR) spectra were employed to determine the physicochemical interactions between the P2W18 guest and NU-1000 host. The characteristic IR bands from 650 to 1200 cm−1 are assigned to the Zr–O vibrational modes and carboxylate stretches in NU-1000, while the peaks from 700 to 1100 cm−1 are attributed to the typical W–O and P–O vibrational bands of P2W18 POM. The presence of W–O and P–O vibrational bands clearly confirms the successful incorporation of P2W18 into the P2W18@NU-1000 composite (Fig. 3a). Additionally, their IR absorption characteristic peaks have a slight red shift upon P2W18 immobilization. For example, the characteristic peaks of P–O and W–O bands shifted from 1091.20 cm−1 and 912.93 cm−1 for bare P2W18 to 1095.48 cm−1 and 907.04 cm−1 for the P2W18@NU-1000 composite, respectively. The slight shifts of IR characterization peaks would be attributed to the electronic interaction between the closely contacted P2W18 guest and NU-1000 host instead of the simple mechanical mixture. UV-Vis diffuse reflectance spectra revealed the changes in optical properties upon immobilization (Fig. 3b). It is observed that the P2W18@NU-1000 composite displays both enhanced absorption ability and an extended absorption edge to a longer wavelength in the visible light region, further demonstrating the electronic interaction between P2W18 and NU-1000.
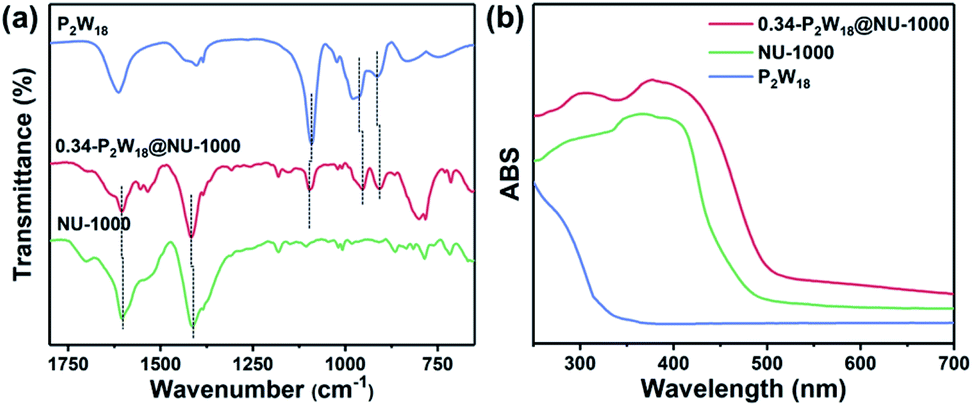 | ||
| Fig. 3 (a) FT-IR spectra and (b) UV-Vis diffuse reflectance spectra of P2W18, NU-1000 and 0.34-P2W18@NU-1000. | ||
The detailed photocatalytic hydrogen production measurements are described in the Experimental section. We first investigated the effects of P2W18 loading quantity, pH value, and the addition amount of Pt to achieve optimal photocatalytic conditions. With the increasing feedstock of P2W18 during the synthesis, the immobilized content of P2W18 into NU-1000 gradually increases (Table 1). Among our tested synthetic samples, the 0.34-P2W18@NU-1000 composite achieves the highest photocatalytic activity, indicating that the presence of P2W18 could facilitate the separation of photogenerated charge carriers in the NU-1000 host and the subsequent electron transfer for hydrogen production (Fig. 4a). The hydrogen production yield exhibits a typical volcano behavior versus pH values (Fig. S4†). The optimal pH value was achieved at 5.5, which could be ascribed to the following reasons: (1) a very low pH value might cause the destruction of the P2W18@NU-1000 structure, (2) the sacrificial electron donor AA shows the best performance at pH 5.5, and (3) an overly high pH value is thermodynamically unfavorable for hydrogen generation. Similarly, the moles of hydrogen production also display the volcano shape versus the added Pt in terms of weight percentage (Fig. 4b). The photocatalytic system of 0.34-P2W18@NU-1000 with 0.25 wt% Pt dosage exhibits the best performance for hydrogen production, achieving 6023 μmol g−1 hydrogen production with an apparent quantum yield of 1.69% (see calculation details in the ESI†) after 12 h illumination under optimized conditions. When the Pt dosage is lower than 0.25 wt%, the synergistic catalysis between P2W18 and Pt NPs is relatively weak; in contrast, addition of too high amount of Pt could not only block the channels of the NU-1000 host but also cause light scattering that are detrimental to efficient hydrogen evolution. Therefore, subsequent studies were performed under optimized conditions (pH = 5.5, 0.25 wt% Pt, 10 mg of 0.34-P2W18@NU-1000 composite) unless otherwise noted.
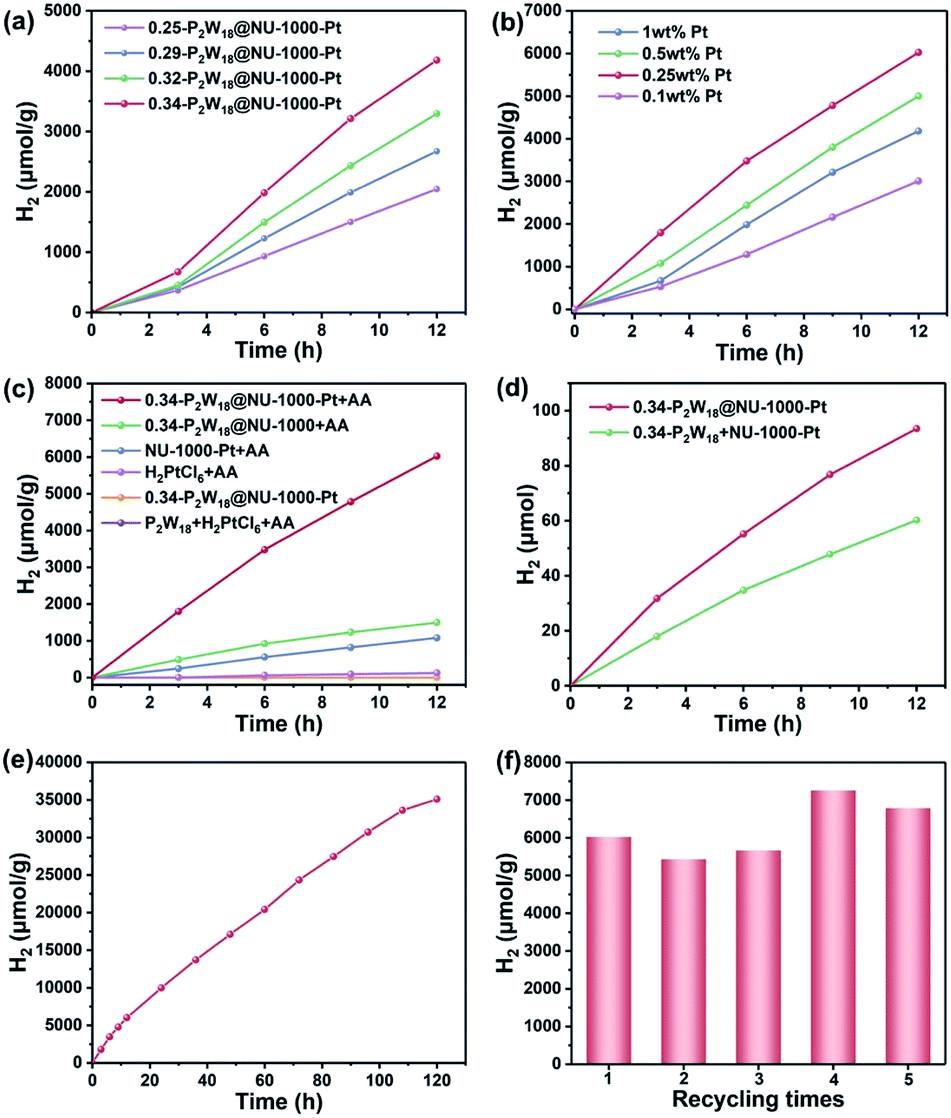 | ||
| Fig. 4 (a) Photocatalytic hydrogen evolution of P2W18@NU-1000 with different molar ratios of P2W18/NU-1000. Conditions: 10 mg catalysts, pH = 4.5, 1 wt% Pt. (b) Photocatalytic hydrogen evolution tests using various feedstocks of Pt. Conditions: 10 mg 0.34-P2W18@NU-1000 photocatalyst, pH = 5.5. (c) Comparison profiles of photocatalytic hydrogen production with different catalysts and reaction conditions (the specific content of Pt contained in the reaction systems is 0.25 wt%). The common conditions: pH = 5.5, 300 W Xe-lamp. (d) The comparison profiles of the photocatalytic hydrogen production for 0.34-P2W18@NU-1000 and P2W18 + NU-1000. Conditions: pH = 5.5, 0.25 wt% Pt. The contents of P2W18 and NU-1000 are equal to that of 0.34-P2W18@NU-1000. (e) Long-term photocatalytic hydrogen production test using the 0.34-P2W18@NU-1000 composite with 0.25 wt% Pt at pH = 5.5. (f) The photocatalytic recycle tests of the 0.34-P2W18@NU-1000 photocatalyst. Conditions: 10 mg 0.34-P2W18@NU-1000 photocatalyst, 0.25 wt% Pt, 300 W Xe-lamp, and 20 mL of 1 M AA aqueous at pH = 5.5. | ||
Based on the above experimental analyses, we can speculate that such superior photocatalytic activity of the three-in-one P2W18@NU-1000-Pt hybrid composite is attributed to the enhanced photo-response, efficient electron transfer between the NU-1000 host and P2W18 guest, and the suitable amount of in situ generated Pt NP co-catalyst. To further prove this hypothesis, we carried out various control experiments under otherwise identical conditions (Fig. 4c). Clearly, NU-1000, the sacrificial donor, P2W18, and the in situ generated Pt NPs are indispensable for efficient photocatalytic hydrogen production. The control experiment in the absence of NU-1000 (purple curve, Fig. 4c) reveals that the absorption of photons by the photo-responsive NU-1000 is a prerequisite for the photocatalytic reaction. The negligible hydrogen production without AA (yellow curve, Fig. 4c) indicates the necessity of a sacrificial agent (AA) in this photocatalytic system. Moreover, the remarkably decreased hydrogen yields for NU-1000-Pt + AA (blue curve, Fig. 4c) and 0.34-P2W18@NU-1000 + AA (green curve, Fig. 4c) systems indicate that the P2W18 clusters and the in situ generated Pt NPs derived from H2PtCl6 reduction are also crucial for efficient photocatalysis. As electron-storage sponge, the outstanding reversible multi-electron-storing ability of P2W18 clusters could facilitate effective separation of photogenerated charge carriers, thereby leading to subsequently efficient hydrogen production.38 The Pt NPs could be in situ generated in the NU-1000-Pt + AA catalytic system, which was confirmed by the high-resolution TEM image (Fig. S5†) and XPS signals of Pt 4f after photocatalysis (Fig. S6†). The Pt 4f XPS signal could be deconvoluted into two species, corresponding to metallic Pt NPs and Pt2+ states, respectively, originating from the photoreduction of H2PtCl6 during photocatalysis. The in situ generated Pt NPs (Step (II) in Scheme 1) could also be evidenced by the EDS analysis and elemental mapping images of 0.34-P2W18@NU-1000-Pt after photocatalysis (Fig. S7 and S8†). Additionally, the low temperature N2 adsorption–desorption isotherms and the changes in pore distribution further proved the in situ formation of Pt NPs during the photocatalytic process (Fig. S9 and S10†). To further evaluate the importance of the in situ formed Pt NPs, we compared the photocatalytic activities using commercially available platinum–carbon (Pt/C) and synthesized cubic Pt NPs under otherwise identical conditions (Fig. S11†). The hydrogen production dramatically decreases using these externally added catalysts, which strongly identifies the importance of close contact between in situ generated Pt NPs, P2W18 clusters, and the NU-1000 host in the three-in-one P2W18@NU-1000-Pt hybrid composite. Moreover, the catalytic system using the mechanical mixture (P2W18 + NU-1000-Pt) shows obviously declined activity, further confirming that the hydrogen production largely depends on the strong electronic interaction between the P2W18 guest and NU-1000 host (Fig. 4d).
To evaluate the stability of the three-in-one hybrid composite 0.34-P2W18@NU-1000-Pt, we have performed a long-term photocatalytic hydrogen evolution test under the optimal experimental conditions (Fig. 4e). Upon irradiation with a full spectrum 300 W Xe-lamp for 120 h, such a three-in-one hybrid photocatalyst maintains a satisfactory photocatalytic activity with hydrogen production as high as 35100 μmol g−1, corresponding to a TON of 5484 versus moles of Pt, which belongs to the top performance compared to similar catalytic systems in the literature (Table S1†). Additionally, the morphology and crystalline structure of the 0.34-P2W18@NU-1000 composite remain largely unchanged after photocatalysis as confirmed by SEM images (Fig. S12†), PXRD patterns (Fig. S13†) and FT-IR spectra (Fig. S14†). We also conducted the recycling stability test of the three-in-one hybrid photocatalyst (Fig. 4f), which could be easily recycled at least 5 times without declining catalytic activity even though there are around 2.1% P2W18 clusters and 0.5% Pt species leaching out into the reaction solution after first-cycle photocatalysis as confirmed by ICP-AES measurements. The slightly increased hydrogen production after 3 cycles could have resulted from the better dispersion of the photocatalyst under a prolonged reaction time, leading to better utilization of absorbed photons. Given the strong absorption of P2W18@NU-1000 composites in the UV light region, it is rational that the photocatalytic activity is better under full-optical irradiation with a 300 W Xe-lamp than under only visible light irradiation (Fig. S15†). But the extended visible light absorption of P2W18@NU-1000 provides decent photocatalytic activity under visible light (>420 nm) compared to that of pristine NU-1000 and P2W18. Such enhanced photo-responsive ability enables the potential of P2W18@NU-1000 to utilize natural sunlight to drive hydrogen evolution. As shown in Fig. S16,† the yield of hydrogen production increases with irradiation time even in a cloudy weather, but it varies with UV light intensity during the daytime. These results demonstrate the potential practical value of our three-in-one P2W18@NU-1000-Pt photocatalyst in the utilization of real natural solar energy.
To deeply elucidate the photocatalytic mechanism, we have conducted a series of characterization studies for NU-1000 and P2W18@NU-1000 composites, respectively. Fig. 5a shows the steady-state photoluminescence (PL) spectra of pristine NU-1000 and the 0.34-P2W18@NU-1000 composite upon excitation with 365 nm laser light. An obvious strong PL signal is found for pristine NU-1000 with the maximum emission at ∼505 nm (red curve, Fig. 5a), while it is effectively quenched after immobilization of P2W18 in the 0.34-P2W18@NU-1000 composite (green curve, Fig. 5a), implying the efficient transfer of photoinduced electrons from the photoexcited NU-1000 host to the P2W18 cluster. The charge carrier transfer dynamics was further determined by time-resolved PL measurements on pristine NU-1000 and 0.34-P2W18@NU-1000 (Fig. 5b). The lifetimes were estimated by fitting the PL decay kinetics data with a three-exponential decay function, and are summarized in Table S2.† Clearly, the immobilization of P2W18 clusters into the NU-1000 host obviously decreases the PL decay lifetime, further proving the effective transfer of photogenerated charge carriers from the excited NU-1000 host to the P2W18 guest.
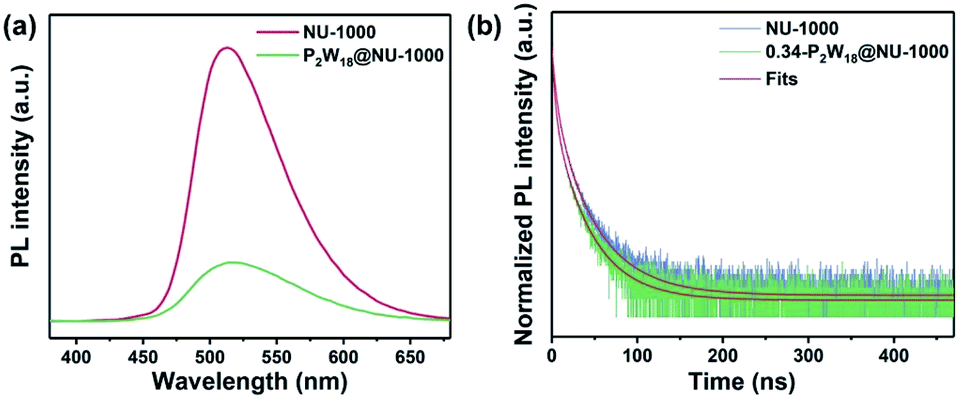 | ||
| Fig. 5 (a) Photoluminescence spectra and (b) normalized time-resolved PL decay kinetics (λem = 505 nm) of NU-1000 and 0.34-P2W18@NU-1000 solid samples upon excitation at 365 nm. | ||
In addition, the XPS technique was further employed to determine the changes in the chemical states of elements for the 0.34-P2W18@NU-1000 composite before and after photocatalysis. The binding energy of Zr in NU-1000 shifts negatively a bit while the binding energy of W shifts positively after incorporation into NU-1000, which could be attributed to the slight electron density movement from P2W18 to the {Zr6} nodes of NU-1000 (Fig. 6a and b). Such experimental evidence confirms the strong electronic interaction between the NU-1000 host and the P2W18 cluster as evidenced by the results of steady-state and time-resolved photoluminescence spectra (Fig. 5). After photocatalysis, both the binding energies of Zr and W shift negatively, indicating the photoreduction of Zr and W centers under a strong reducing environment during photocatalysis (Fig. 6a and b). Moreover, the significant XPS signals of reduced Pt species (Pt0 and Pt2+) also strongly confirmed that the Pt NPs are generated in situ from the reduction of H2PtCl6 during photocatalysis (Fig. 6c). The ICP-AES technique was employed to determine the amount of homogeneous [PtCl6]2− remaining in the supernatant after one-cycle photocatalysis (12 h), and the results revealed that around 0.5% Pt species still remained in the post-reaction solution. The in situ formed Pt NPs are located both on the surface and in the pores of NU-1000 that was confirmed by HR-TEM images of the composite after photocatalysis with the characteristic crystal lattice spacing (0.227 nm) of Pt NPs (Fig. 6d and e). These results also illustrated that in situ generated Pt NPs could work as the active hydrogen-evolving sites during photocatalytic reactions.
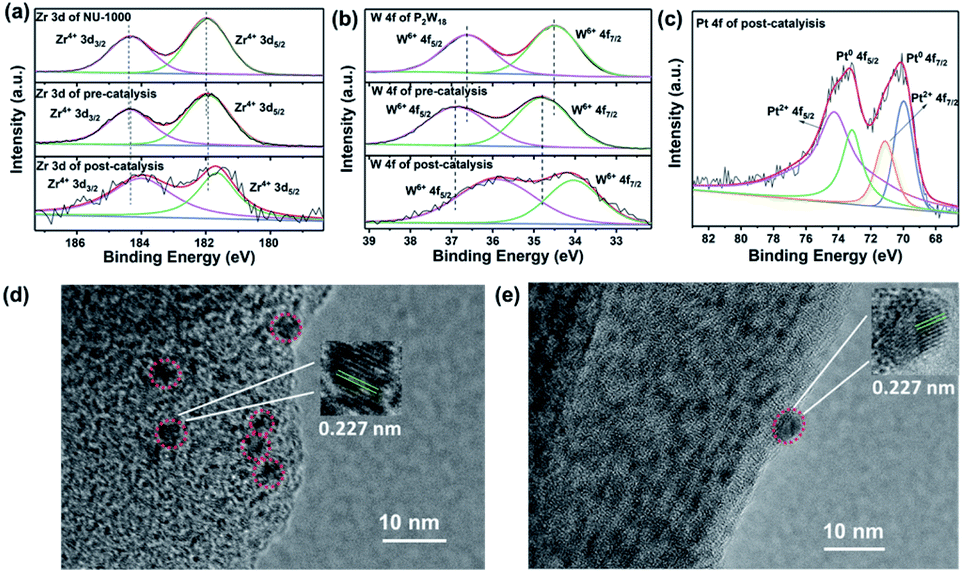 | ||
| Fig. 6 High resolution XPS spectra of (a) Zr 3d and (b) W 4f signals in NU-1000 and 0.34-P2W18@NU-1000 before and after photocatalysis. (c) High resolution XPS spectra of Pt 4f signals in 0.34-P2W18@NU-1000 after photocatalysis. (d and e) HR-TEM images of 0.34-P2W18@NU-1000 after photocatalysis. The in situ formed Pt NPs located into the pores or on the surface of NU-1000 are marked with red circles. | ||
Based on the above observations and analyses in detail, we have drawn a schematic energy level diagram66,67 for photocatalytic hydrogen production using the three-in-one P2W18@NU-1000-Pt photocatalyst (Fig. 7). Herein, the LUMO positions of the P2W18 cluster were measured by using cyclic voltammograms (Fig. S17†), and the potentials at −0.205 V and −0.440 V vs. NHE correspond to the LUMO positions of three- and four-electrons reduced P2W18, respectively, which is supported by the experimental observation of heteropoly blue (reduced P2W18 clusters) as shown in Fig. S16b.† According to the schematic energy level diagram, a systematic photocatalytic mechanism could be speculated as follows. Upon light irradiation, the photo-responsive NU-1000 MOF host can efficiently harvest incident photons to generate photoexcited states. Subsequently, the photoexcited NU-1000 host could be subsequently quenched by transferring photogenerated electrons to the immobilized P2W18 POM cluster to generate the reduced POM (POMred) as indicated by the blue solution color in Fig. S16b.† Meanwhile, the photogenerated electrons can also reduce H2PtCl6 in solution to in situ generate Pt NPs. At the same time, the oxidized states of the NU-1000 MOF could be reduced back to the initial state by the sacrificial reagent (AA). Herein, either P2W18 clusters or Pt NPs alone could catalyze hydrogen production (green and blue curves, Fig. 4c), but the hydrogen production efficiency can be greatly enhanced while the NU-1000 MOF, P2W18 cluster, and Pt NPs work synergistically together as a three-in-one P2W18@NU-1000-Pt photocatalyst (red curve, Fig. 4c). Overall, the outstanding photocatalytic activity of such a three-in-one P2W18@NU-1000-Pt hybrid photocatalyst could be ascribed to the synergistic effect of the good photo-absorbing properties of NU-1000, the reversible multi-electron-transfer ability of the P2W18 catalyst, and the superior catalytic activity of closely contacted Pt NPs generated in situ under turnover conditions.
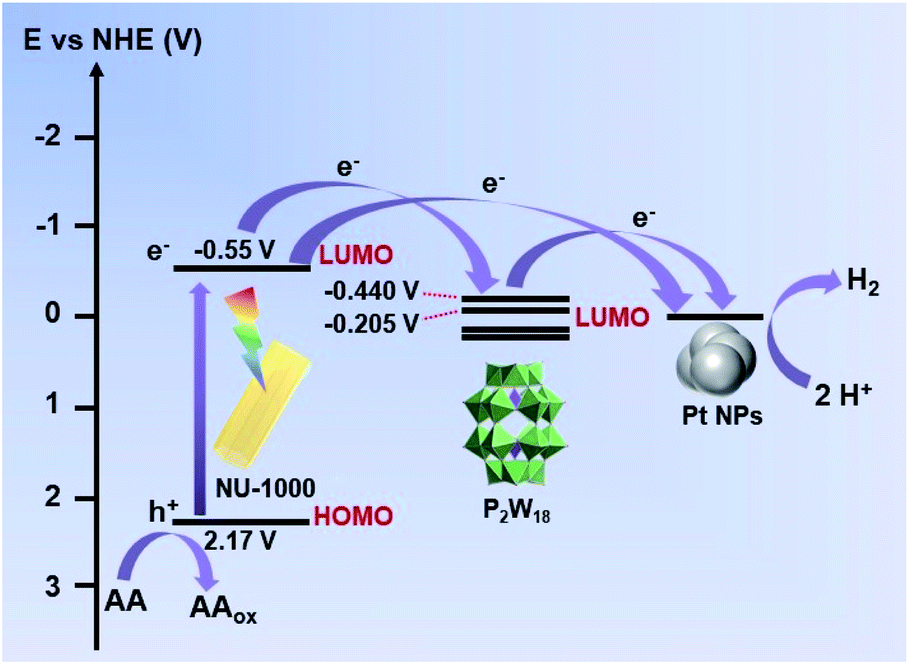 | ||
| Fig. 7 Schematic energy level diagram of photocatalytic hydrogen production using the three-in-one P2W18@NU-1000-Pt photocatalyst. | ||
Conclusions
In summary, we have prepared a three-in-one hybrid material (P2W18@NU-1000-Pt) through the strong host–guest interaction using a facile impregnation and subsequent in situ photoreduction approach. A series of physicochemical techniques confirmed the successful immobilization of P2W18 clusters and the strong electronic interaction between the P2W18 guest and NU-1000 host. Under minimally optimized photocatalytic conditions, the three-in-one 0.34-P2W18@NU-1000-Pt hybrid composite exhibits efficient photocatalytic activity with the hydrogen production of 35100 μmol g−1 after 5 days of reaction. The post-catalysis hybrid composite could be easily recycled at least 5 times without declining catalytic activity. Based on the solid analyses of various spectroscopic and experimental results, we concluded that the outstanding and robust photocatalytic activity of this three-in-one P2W18@NU-1000-Pt hybrid composite could be ascribed to the synergistic effect of the good photo-absorbing properties of NU-1000, the reversible multi-electron-transfer ability of the P2W18 catalyst, and the superior catalytic activity of closely contacted Pt NPs generated in situ under turnover conditions. More importantly, the present water-compatible photocatalytic system can also effectively catalyze hydrogen production under natural sunlight irradiation even in a cloudy weather, demonstrating the potential practical value for natural solar energy utilization. Considering the feasibility of the synthetic strategy, robustness and effectiveness of the resulting three-in-one hybrid materials, many interesting functional materials could be constructed based on POM@MOF architectures by following a similar idea in our work.
Experimental section
Materials and methods
All reagents and solvents are purchased from commercial sources without further purification. EDX spectra and SEM images are obtained on a ZEISS supra55. HRTEM images are obtained on a JEOL JEM-2100. PXRD patterns are collected on a MiniFlex 600 diffractometer with a Cu-Kα X-ray radiation source. Low temperature N2 adsorption–desorption isotherms are measured at 77 K on a Quantachrome Instruments ASiQMVH002-5 after pre-treatment by activating the samples under vacuum at 120 °C for 12 h. UV-Vis spectra are recorded on a Techcomp UV 2600 spectrophotometer. FT-IR spectra are recorded on a Bruker Tensor II with KBr pellets. Fluorescence spectra are recorded with an Edinburgh Instruments Spectrofluorometer FS5. ICP-AES data are obtained with a Spectro Arcos EOP (Axial View Inductively Coupled Plasma Spectrometer). XPS data are collected by using a PHI 5000 VersaProbe III Scanning XPS Microprobe. Gas chromatograph analysis is conducted with a Techcomp GC7900 gas chromatograph equipped with a thermal conductivity detector. Cyclic voltammetry was recorded on a CHI660E Electrochemical potentiostat with a three-electrode setup using a glassy carbon working electrode, saturated calomel electrode (SCE) reference electrode, and Pt wire auxiliary electrode. The working electrode was polished with 0.3 μm alumina, sonicated with deionized water, rinsed with EtOH and then with deionized water before use. The measured potential was calibrated by converting to the normal hydrogen electrode (NHE) scale.Synthetic procedures
Author contributions
Le Jiao: methodology, data curation, formal analysis, investigation, writing – original draft preparation. Yuanyuan Dong: formal analysis, writing – reviewing and editing. Xing Xin: software. Ruijie Wang: data curation. Hongjin Lv: funding acquisition, project administration, conceptualization, supervision, validation, writing – reviewing and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21871025 and 21831001), the Recruitment Program of Global Experts (Young Talents) and the BIT Excellent Young Scholars Research Fund.References
- L. Cui, J. Song, A. F. McGuire, S. Kang, X. Fang, J. Wang, C. Yin, X. Li, Y. Wang and B. Cui, ACS Nano, 2018, 12, 5551–5558 Search PubMed.
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, G. Liu and H. M. Cheng, Adv. Mater., 2015, 27, 4572–4577 Search PubMed.
- A. Indra, A. Acharjya, P. W. Menezes, C. Merschjann, D. Hollmann, M. Schwarze, M. Aktas, A. Friedrich, S. Lochbrunner, A. Thomas and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 1653–1657 Search PubMed.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 Search PubMed.
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 Search PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 Search PubMed.
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, e1907296 Search PubMed.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 Search PubMed.
- A. M. Elewa, M. H. Elsayed, A. F. M. El-Mahdy, C.-L. Chang, L.-Y. Ting, W.-C. Lin, C.-Y. Lu and H.-H. Chou, Appl. Catal., B, 2021, 285, 119802 Search PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 Search PubMed.
- Y. Yamada, T. Miyahigashi, H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 16136–16145 Search PubMed.
- M. Luo, W. Yao, C. Huang, Q. Wu and Q. Xu, J. Mater. Chem. A, 2015, 3, 13884–13891 Search PubMed.
- S. Liu, C. Zhang, Y. Sun, Q. Chen, L. He, K. Zhang, J. Zhang, B. Liu and L.-F. Chen, Coord. Chem. Rev., 2020, 413, 213266 Search PubMed.
- Y. Shi, A.-F. Yang, C.-S. Cao and B. Zhao, Coord. Chem. Rev., 2019, 390, 50–75 Search PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 Search PubMed.
- M. Zhang, H. Li, J. Zhang, H. Lv and G.-Y. Yang, Chin. J. Catal., 2020, 42, 855–871 Search PubMed.
- T. Cui, L. Qin, F. Fu, X. Xin, H. Li, X. Fang and H. Lv, Inorg. Chem., 2021, 60, 4124–4132 Search PubMed.
- H. Lv, Y. Gao, W. Guo, S. M. Lauinger, Y. Chi, J. Bacsa, K. P. Sullivan, M. Wieliczko, D. G. Musaev and C. L. Hill, Inorg. Chem., 2016, 55, 6750–6758 Search PubMed.
- H. Lv, W. Guo, K. Wu, Z. Chen, J. Bacsa, D. G. Musaev, Y. V. Geletii, S. M. Lauinger, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 14015–14018 Search PubMed.
- L. Qin, C. Zhao, L.-Y. Yao, H. Dou, M. Zhang, J. Xie, T.-C. Weng, H. Lv and G.-Y. Yang, CCS Chem., 2021, 3, 651–663 Search PubMed.
- M. Zheng, Y. Ding, X. Cao, T. Tian and J. Lin, Appl. Catal., B, 2018, 237, 1091–1100 Search PubMed.
- X. Gao, J. Wang, Q. Xue, Y.-Y. Ma and Y. Gao, ACS Appl. Nano Mater., 2021, 4, 2126–2135 Search PubMed.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 Search PubMed.
- B. Chakraborty, G. Gan-Or, M. Raula, E. Gadot and I. A. Weinstock, Nat. Commun., 2018, 9, 4896 Search PubMed.
- S. Pourbeyram, M. Moosavifar and V. Hasanzadeh, J. Electroanal. Chem., 2014, 714–715, 19–24 Search PubMed.
- P. Wang, L. Jiang, X. Zou, H. Tan, P. Zhang, J. Li, B. Liu and G. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 25910–25919 Search PubMed.
- H. Ma, B. Liu, B. Li, L. Zhang, Y. G. Li, H. Q. Tan, H. Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 Search PubMed.
- C.-Y. Sun, S.-X. Liu, D.-D. Liang, K.-Z. Shao, Y.-H. Ren and Z.-M. Su, J. Am. Chem. Soc., 2009, 131, 1883–1888 Search PubMed.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 Search PubMed.
- G. Li, K. Zhang, C. Li, R. Gao, Y. Cheng, L. Hou and Y. Wang, Appl. Catal., B, 2019, 245, 753–759 Search PubMed.
- M. Samaniyan, M. Mirzaei, R. Khajavian, H. Eshtiagh-Hosseini and C. Streb, ACS Catal., 2019, 9, 10174–10191 Search PubMed.
- P. Yang, W. Zhao, A. Shkurenko, Y. Belmabkhout, M. Eddaoudi, X. Dong, H. N. Alshareef and N. M. Khashab, J. Am. Chem. Soc., 2019, 141, 1847–1851 Search PubMed.
- X. H. Li, Y. W. Liu, Y. Lu, Z. Zhang, H. R. Tian, S. M. Liu and S. X. Liu, Chem. Commun., 2020, 56, 1641–1644 Search PubMed.
- L. Zeng, X. Guo, C. He and C. Duan, ACS Catal., 2016, 6, 7935–7947 Search PubMed.
- X. J. Kong, Z. Lin, Z. M. Zhang, T. Zhang and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 6411–6416 Search PubMed.
- J. Tian, Z. Y. Xu, D. W. Zhang, H. Wang, S. H. Xie, D. W. Xu, Y. H. Ren, H. Wang, Y. Liu and Z. T. Li, Nat. Commun., 2016, 7, 11580 Search PubMed.
- L. Jiao, Y. Dong, X. Xin, L. Qin and H. Lv, Appl. Catal., B, 2021, 291, 120091, DOI:10.1016/j.apcatb.2021.120091.
- Z. M. Zhang, T. Zhang, C. Wang, Z. Lin, L. S. Long and W. Lin, J. Am. Chem. Soc., 2015, 137, 3197–3200 Search PubMed.
- H. Li, S. Yao, H.-L. Wu, J.-Y. Qu, Z.-M. Zhang, T.-B. Lu, W. Lin and E.-B. Wang, Appl. Catal., B, 2018, 224, 46–52 Search PubMed.
- D. Shi, R. Zheng, C. S. Liu, D. M. Chen, J. Zhao and M. Du, Inorg. Chem., 2019, 58, 7229–7235 Search PubMed.
- I. Choudhuri and D. G. Truhlar, J. Phys. Chem. C, 2020, 124, 8504–8513 Search PubMed.
- X. Li, K. Gao, B. Mo, J. Tang, J. Wu and H. Hou, Inorg. Chem., 2021, 60, 1352–1358 Search PubMed.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2016, 8, 20675–20681 Search PubMed.
- C. T. Buru, M. C. Wasson and O. K. Farha, ACS Appl. Nano Mater., 2019, 3, 658–664 Search PubMed.
- C. T. Buru, P. Li, B. L. Mehdi, A. Dohnalkova, A. E. Platero-Prats, N. D. Browning, K. W. Chapman, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 5174–5181 Search PubMed.
- S. Cao, J. Jiang, B. Zhu and J. Yu, Phys. Chem. Chem. Phys., 2016, 18, 19457–19463 Search PubMed.
- J. F. Guayaquil-Sosa, B. Serrano-Rosales, P. J. Valadés-Pelayo and H. de Lasa, Appl. Catal., B, 2017, 211, 337–348 Search PubMed.
- P. J. Kulesza, M. Chojak, K. Karnicka, K. Miecznikowski, B. Palys, A. Lewera and A. Wieckowski, Chem. Mater., 2004, 16, 4128–4134 Search PubMed.
- Y. Liu, Y. Li, F. Peng, Y. Lin, S. Yang, S. Zhang, H. Wang, Y. Cao and H. Yu, Appl. Catal., B, 2019, 241, 236–245 Search PubMed.
- M. Luo, P. Lu, W. Yao, C. Huang, Q. Xu, Q. Wu, Y. Kuwahara and H. Yamashita, ACS Appl. Mater. Interfaces, 2016, 8, 20667–20674 Search PubMed.
- E. Park, J. Jack, Y. Hu, S. Wan, S. Huang, Y. Jin, P. C. Maness, S. Yazdi, Z. Ren and W. Zhang, Nanoscale, 2020, 12, 2596–2602 Search PubMed.
- I. Vamvasakis, B. Liu and G. S. Armatas, Adv. Funct. Mater., 2016, 26, 8062–8071 Search PubMed.
- J. He, J. Wang, Y. Chen, J. Zhang, D. Duan, Y. Wang and Z. Yan, Chem. Commun., 2014, 50, 7063–7066 Search PubMed.
- M. Lan, R.-M. Guo, Y. Dou, J. Zhou, A. Zhou and J.-R. Li, Nano Energy, 2017, 33, 238–246 Search PubMed.
- P. Ling, J. Lei, L. Jia and H. Ju, Chem. Commun., 2016, 52, 1226–1229 Search PubMed.
- Y. Liu, Z. Liu, D. Huang, M. Cheng, G. Zeng, C. Lai, C. Zhang, C. Zhou, W. Wang, D. Jiang, H. Wang and B. Shao, Coord. Chem. Rev., 2019, 388, 63–78 Search PubMed.
- X. Ma, L. Wang, Q. Zhang and H. L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12175–12179 Search PubMed.
- M. Sun, C. Sun, X. Wang and Z. Su, Catal. Commun., 2020, 137, 105930 Search PubMed.
- C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 Search PubMed.
- T. Hsu-Yao, K. P. Browne, N. Honesty and Y. J. Tong, Phys. Chem. Chem. Phys., 2011, 13, 7433–7438 Search PubMed.
- U. Lee, H. C. Joo, K. M. Park, S. S. Mal, U. Kortz, B. Keita and L. Nadjo, Angew. Chem., Int. Ed., 2008, 47, 793–796 Search PubMed.
- S. Li, S. Liu, S. Liu, Y. Liu, Q. Tang, Z. Shi, S. Ouyang and J. Ye, J. Am. Chem. Soc., 2012, 134, 19716–19721 Search PubMed.
- B. Zhang, G. Sun, S. Ding, H. Asakura, J. Zhang, P. Sautet and N. Yan, J. Am. Chem. Soc., 2019, 141, 8185–8197 Search PubMed.
- Z. Zhang, Q. Lin, S. T. Zheng, X. Bu and P. Feng, Chem. Commun., 2011, 47, 3918–3920 Search PubMed.
- W. Guo, H. Lv, Z. Chen, K. P. Sullivan, S. M. Lauinger, Y. Chi, J. M. Sumliner, T. Lian and C. L. Hill, J. Mater. Chem. A, 2016, 4, 5952–5957 Search PubMed.
- F. Bozheyev, F. Xi, P. Plate, T. Dittrich, S. Fiechter and K. Ellmer, J. Mater. Chem. A, 2019, 7, 10769–10780 Search PubMed.
- P. P. Bag, X. Wang, P. Sahoo, J. Xiong and R. Cao, Catal. Sci. Technol., 2017, 7, 5113–5119 Search PubMed.
- I.-M. Mbomekalle, Y. W. Lu, B. Keita and L. Nadjo, Inorg. Chem. Commun., 2004, 7, 86–90 Search PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 Search PubMed.
- T. C. Wang, N. A. Vermeulen, I. S. Kim, A. B. Martinson, J. F. Stoddart, J. T. Hupp and O. K. Farha, Nat. Protoc., 2016, 11, 149–162 Search PubMed.
- C. M. Sánchez-Sánchez, J. Solla-Gullón, F. J. Vidal-Iglesias, A. Aldaz, V. Montiel and E. Herrero, J. Am. Chem. Soc., 2010, 132, 5622–5624 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02792a |
| This journal is © The Royal Society of Chemistry 2021 |