
Conjugated molecule based 2D perovskites for high-performance perovskite solar cells†
Tao
Zhu
a,
Lening
Shen
a,
Hanlin
Chen
a,
Yongrui
Yang
a,
Luyao
Zheng
a,
Rui
Chen
 a,
Jie
Zheng
b,
Junpeng
Wang
*a and
Xiong
Gong
*a
a,
Jie
Zheng
b,
Junpeng
Wang
*a and
Xiong
Gong
*a
aSchool of Polymer Science and Polymer Engineering, The University of Akron, Akron, OH 44325, USA. E-mail: xgong@uakron.edu; jwang6@uakron.edu; Fax: +1 330 9723406
bDepartment of Chemical, Biomolecular and Corrosion Engineering, College of Engineering and Polymer Science, The University of Akron, Akron, OH 44325, USA
First published on 26th August 2021
Abstract
Conjugated molecules have been typically utilized as either hole or electron extraction layers to boost the device performance of perovskite solar cells (PSCs), formed from three-dimensional (3D) perovskites, due to their high charge carrier mobility and electrical conductivity. However, the passivating role of conjugated molecules in creating two-dimensional (2D) perovskites has rarely been reported. In this study, we report novel conjugated aniline 3-phenyl-2-propen-1-amine (PPA) based 2D perovskites and further demonstrate efficient and stable PSCs containing a (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film (where MA is CH3NH3+). The (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film possesses superior crystallinity and passivated trap states, resulting in enhanced charge transport and suppressed charge carrier recombination compared to those of a 3D MAPbI3 thin film. As a result, PSCs containing the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibit a power conversion efficiency (PCE) of 21.98%, which is approximately a 25% enhancement compared to that of the MAPbI3 thin film. Moreover, un-encapsulated PSCs containing the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film retain 50% of their initial PCE after 1200 hours in an ambient atmosphere (25 °C, and 30 ± 10 humidity), whereas PSCs with the 3D MAPbI3 thin film show significant degradation after 100 hours and a degradation of more than 50% of their original PCE after 500 hours. These results demonstrate that the incorporation of conjugated molecules as organic spacer cations to create 2D perovskites on top of 3D perovskites is an effective way to approach high-performance PSCs.
1. Introduction
Three-dimensional (3D) organic–inorganic metal halide hybrid perovskites are promising photovoltaic materials due to their unique optoelectronic properties.1–11 A state-of-the-art perovskite solar cell (PSC) has been certified to exhibit an astonishing power conversion efficiency (PCE) of 25.5%.12 However, 3D perovskites are quite sensitive to moisture and oxygen, resulting in significant degradation in ambient atmospheres. Thus, the long-term stability of PSCs by 3D perovskites is a major bottleneck for their practical applications.13,14 In comparison with 3D perovskites, two-dimensional (2D) perovskites have been developed to create stable PSCs.15 Studies have indicated that the utilization of a small amount of large organic cations to replace regular smaller cations in the perovskite precursors to form a 2D perovskite passivation layer on the top of a 3D perovskite is a facile way to boost both the efficiency and stability of PSCs.16–21 Using phenethylammonium iodide (PEAI) to treat the surface of a 3D perovskite, You et al. found out that PEAI-based 2D perovskites could reduce surface defects and suppress surface non-radiative recombination, resulting in enhanced PCEs.19 Chen et al.21 demonstrated that 2-thiophenemethylammonium cations can be successfully embedded into 3D perovskites, resulting in enlarged crystallinity and orientated 2D/3D perovskite bilayer thin films. Recently, we reported novel propargylamine-based quasi-2D perovskite thin films and further demonstrated efficient and stable PSCs with dramatically suppressed photocurrent hysteresis.15 While impressive device performance has been reported for PSCs with 2D/3D perovskite bilayer thin films, 2D perovskites based on insulating organic molecules possess poor charge transport, restricting the device performance of PSCs.22–28Conjugated organic molecules have received considerable attention as the hole and/or electron transport layers in PSCs with 3D perovskite thin films due to their high charge carrier mobilities and electrical conductivities compared to those of insulating organic molecules.29,30 But the use of conjugated organic molecules to create 2D perovskites has rarely been reported. Hence, it is reasonable to develop conjugated organic molecule-based 2D perovskites for boosting the device performance of PSCs with 2D/3D perovskite bilayer thin films. The advantages of the high charge carrier mobility of conjugated molecules and the high stability of 2D perovskites are highly imperative to ultimately balance the efficiency and stability of PSCs with 2D/3D perovskite bilayer thin films.
In this work, we report efficient and stable PSCs with a 2D/3D perovskite bilayer thin film, where the 2D perovskite thin layer is created using an innovative conjugated aniline 3-phenyl-2-propen-1-amine (PPA) as the organic space cation. The (PPA)x(MAPbI3)1−x/MAPbI3 perovskite bilayer thin film (where MA is CH3NH3+) possesses higher crystallinity and passivated trap states, leading to enhanced charge transport and suppressed charge carrier recombination compared to those of the 3D MAPbI3 thin film. As a result, PSCs with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibit a PCE of 21.98% and significantly boosted stability in an ambient atmosphere. Our results demonstrate that the incorporation of conjugated molecules as organic spacer cations to create a 2D perovskite on top of a 3D perovskite is an effective way to develop high-performance PSCs.
2. Experimental section
2.1 Materials
Gamma-butyrolactone (GBL, 99%), methylamine (MA) (33 wt% in absolute ethanol), anhydrous acetonitrile (ACN, 99.8%), poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA), dimethylformamide (DMF, anhydrous, 99.8%), dimethyl sulfoxide (DMSO, anhydrous, 99.9%), toluene (anhydrous, 99.8%), bathocuproine (BCP, 99.99%), aluminum (Al) and silver (Ag) slugs were purchased from Sigma Aldrich. Lead iodide (PbI2, 99.9985% metals basis) was purchased from Alfa Aesar. C60 (99.95% carbon powder) was purchased from Purec60OliveOil. Methyl-ammonium iodide (MAI) was purchased from Greatcell Solar. All materials were used as received without any further treatment. 3-Phenyl-2-propen-1-amine (PPA) was synthesized in our laboratory. The details of the synthesis procedures and characterization of PPA are described in the ESI† (SI 1).2.2 Preparation of the precursor solution
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were dissolved in GBL and stirred at 80 °C for over 2 hours (h) to form a 1.2 M solution. The solution was filtered into a vial and placed into an oil bath at 110 °C for several h until black crystals precipitated at the bottom of the vial. The black MAPbI3 crystals were washed with isopropanol and dried three times. Then, a certain amount of MAPbI3 crystals was placed in a vial, which was then sealed into a bottle containing methylamine solution and kept for over 2 h. Methylamine gas in the bottle diffused into the vial and reacted with the MAPbI3 crystals, resulting in a liquid perovskite intermediate (MAPbI3). Subsequently, MAPbI3 crystals were dissolved into ACN to form a MAPbI3 ACN solution.
:1) were dissolved in GBL and stirred at 80 °C for over 2 hours (h) to form a 1.2 M solution. The solution was filtered into a vial and placed into an oil bath at 110 °C for several h until black crystals precipitated at the bottom of the vial. The black MAPbI3 crystals were washed with isopropanol and dried three times. Then, a certain amount of MAPbI3 crystals was placed in a vial, which was then sealed into a bottle containing methylamine solution and kept for over 2 h. Methylamine gas in the bottle diffused into the vial and reacted with the MAPbI3 crystals, resulting in a liquid perovskite intermediate (MAPbI3). Subsequently, MAPbI3 crystals were dissolved into ACN to form a MAPbI3 ACN solution.
2.3 Preparation and characterization of the perovskite thin film
The pre-cleaned quartz (or glass) substrates were treated by UV–ozone plasma for about 20 minutes (min). The MAPbI3 thin film or (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film was prepared by the spin-cast method with a spin speed of 4000 RPM for 30 seconds (s) from the corresponding solution described above. After that, all of the thin films were thermally annealed at 100 °C for 10 min and then cooled down to room temperature (RT) naturally.The X-ray diffraction (XRD) patterns were obtained by a Rigaku Smartlab. The UV-visible absorption spectra were characterized by a Lambda 900 UV-Vis-NIR spectrophotometer (PerkinElmer, Waltham, MA, USA). The photoluminescence (PL) spectra were obtained from a QuantaMaster 2361 (HORIBA). The scanning electron microscopy (SEM) images were obtained using a field emission scanning electron microscope (model JEOL-7401). The film thicknesses were measured using a Bruker DektakXT Stylus Profilometer with a scan rate of 0.03 mm s−1.
2.4 Fabrication and characterization of the PSCs
The pre-cleaned ITO/glass substrates were treated by UV-ozone plasma for about 20 min, where ITO is indium tin oxide. Then, a ∼20 nm PTAA thin film was spin-casted on the top of the ITO/glass substrates with a spin speed of 6000 RPM for 30 s from 2 mg mL−1 PTAA toluene solution, followed by thermal annealing at 100 °C for 10 min. After the PTAA coated substrates were cooled down to RT, either MAPbI3 or (PPA)x(MAPbI3)1−x/MAPbI3 thin films were prepared by the methods described above. Afterward, ∼40 nm C60 and ∼8 nm BCP were sequentially thermally deposited on the top of either the MAPbI3 thin film or the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film in a vacuum system with a base pressure of 4 × 10−6 Torr. The PSCs were completed by the thermal deposition of ∼100 nm Al on the top of the BCP layer in a vacuum system. The device area was measured to be 0.043 cm2.The current density versus voltage (J–V) characteristics of the PSCs were tested by a Keithley model 2400 source unit. The light source was a Newport Air Mass 1.5 Global (AM1.5G) full spectrum simulator with a light intensity of 100 mW cm−2, which was calibrated by utilizing a mono-silicon detector (with a KG-5 visible color filter) from the National Renewable Energy Laboratory to reduce the spectral mismatch. The external quantum efficiency (EQE) spectrum was obtained by a solar cell quantum efficiency measurement system (QEX10). The impedance spectroscopy (IS) was conducted using a HP 4194A impedance/gain-phase analyzer under illumination and in the dark, at the voltage close to the open-circuit voltage (VOC) of the PSCs. The frequency was varied from 5 Hz to 105 Hz. Capacitance–voltage (C–V) testing was also performed using the same setup. The transient photocurrent (TPC) measurement was done using a homemade setup in our laboratory.5,15,31,32
3. Results and discussion
PPA, as shown in Fig. 1a, is a conjugated molecule used to create a discrete 2D (PPA)x(MAPbI3)1−x thin layer on the top of 3D MAPbI3 thin film to generate the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin films. Fig. 1b presents the UV-visible absorption spectra of the MAPbI3 thin film and the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film. Due to the quantum confinement, a slightly blue-shifted absorption spectrum is observed from the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film compared to that of the MAPbI3 thin film. Moreover, a blue-shifted PL is also observed from the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film (774 nm) compared to that (780 nm) from the MAPbI3 thin film (Fig. 1c and d). In addition, an emission peak located at 722 nm observed for the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film indicates the existence of (PPA)x(MAPbI3)1−x perovskite due to multiple n values coexisting in the system. All of these results indicate that (PPA)x(MAPbI3)1−x is formed as a 2D perovskite structure.33–35 In addition, compared to 3D MAPbI3, an obviously increased PL intensity is observed for the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film (Fig. S1†), which indicates that the non-radiative recombination within the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film was significantly suppressed. | ||
| Fig. 1 (a) The molecular structure of PPA. (b) The absorption spectra of the MAPbI3 and (PPA)x(MAPbI3)1−x/MAPbI3 thin films. The PL spectra of the (c) MAPbI3 and (d) (PPA)x(MAPbI3)1−x/MAPbI3 thin films from the front side and back side. | ||
The XRD patterns of the MAPbI3 thin film and the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film are shown in Fig. 2a. The (110) and (220) peaks at 2θ of ∼14° and ∼28°, respectively, demonstrate that both the MAPbI3 thin film and the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film possess the tetragonal (I4/mcm) crystal structure.36 However, a peak located at a 2θ of ∼6.6°, corresponding to the (002) plane, is observed for the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film, which further confirms that (PPA)x(MAPbI3)1−x is formed as a 2D perovskite structure.33–35Fig. 2b shows the patterns of the (110) peaks for the MAPbI3 thin film and the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film. It is found that the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film possesses a smaller full width at half maximum (FWHM) (0.201°) compared to that (0.309°) observed for the MAPbI3 thin film, indicating that the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibits greater crystallinity.37 This greater crystallinity is attributed to the progressive trap healing at the grain boundaries and the passivated surface of the thin film.38,39
 | ||
| Fig. 2 (a) XRD patterns of the MAPbI3 and (PPA)x(MAPbI3)1−x/MAPbI3 thin films and (b) magnified (110) peak of the XRD patterns. Top-view SEM images of the (c) MAPbI3 and (d) (PPA)x(MAPbI3)1−x/MAPbI3 thin films, and the current density versus voltage characteristics of the diodes fabricated with the (e) MAPbI3 and (f) (PPA)x(MAPbI3)1−x/MAPbI3 thin films, respectively. | ||
Fig. 2c and d present the SEM images of the MAPbI3 thin film and the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film, respectively. It is found that patches of a new phase with bright grain spots are observed for the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film compared to the MAPbI3 thin film. These bright grain spots originate from the lower electrical conductivity of the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film compared to that of the MAPbI3 thin film. All of these characteristics further confirm that (PPA)x(MAPbI3)1−x is indeed formed as a 2D perovskite structure.40
To verify that the conjugated molecule, PPA, could facilitate charge transport, the charge carrier mobility of the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film is investigated through the characterization of both electron-only and hole-only diodes, based on the space charge limited current (SCLC) method, according to the Mott–Gurney law.41–44 The electron-only diode, with a device structure of ITO/SnO2/active layer/C60/Al, and the hole-only diode, with a device structure of ITO/PEDOT:PSS/active layer/MoO3/Ag, where the active layer is either the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film or the MAPbI3 thin film, are fabricated and characterized. As shown in Fig. 2e and f, the J–V characteristics can be divided into the ohmic region (J ∝ V) under the low bias and the SCLC region (J ∝ V2) under high bias. The J–V curve in the SCLC region could be well-fitted by the Mott–Gurney law: 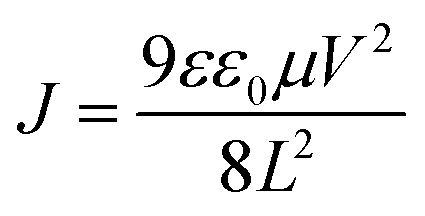 (where μ is the charge carrier mobility, V is the external bias, L is the thickness of the perovskite thin film, ε0 is the vacuum permittivity and ε is the dielectric constant for the perovskite thin film).41–44 Based on the capacitance versus frequency (C–V) characteristics of the perovskite thin films (Fig. S2†), the ε values are calculated to be 26.6 and 25.4 for the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film and the MAPbI3 thin film, respectively. Therefore, the charge carrier mobilities of the MAPbI3 thin film are calculated to be 6.52 × 10−4 cm2 V−1 s−1 (electron mobility) and 1.43 × 10−3 cm2 V−1 s−1 (hole mobility). These values are consistent with the reported values.45,46 Furthermore, these charge carrier mobility values indicate that the charge transport is unbalanced (μe/μh = 0.46) within the MAPbI3 thin film. The charge carrier mobilities for the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film are calculated to be 7.52 × 10−3 cm2 V−1 s−1 (electron mobility) and 8.24 × 10−3 cm2 V−1 s−1 (hole mobility). Both the hole mobility and electron mobility of the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film are higher than those of the MAPbI3 thin film. Such boosted charge carrier mobilities are attributed to the conjugated chain of PPA as compared with the insulating organic spacers.22–28 Moreover, these charge carrier mobility values indicate that the charge transport tends to be balanced (μe/μh = 0.90). As a result, an enhanced JSC is expected to be observed from the PSCs with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film.
(where μ is the charge carrier mobility, V is the external bias, L is the thickness of the perovskite thin film, ε0 is the vacuum permittivity and ε is the dielectric constant for the perovskite thin film).41–44 Based on the capacitance versus frequency (C–V) characteristics of the perovskite thin films (Fig. S2†), the ε values are calculated to be 26.6 and 25.4 for the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film and the MAPbI3 thin film, respectively. Therefore, the charge carrier mobilities of the MAPbI3 thin film are calculated to be 6.52 × 10−4 cm2 V−1 s−1 (electron mobility) and 1.43 × 10−3 cm2 V−1 s−1 (hole mobility). These values are consistent with the reported values.45,46 Furthermore, these charge carrier mobility values indicate that the charge transport is unbalanced (μe/μh = 0.46) within the MAPbI3 thin film. The charge carrier mobilities for the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film are calculated to be 7.52 × 10−3 cm2 V−1 s−1 (electron mobility) and 8.24 × 10−3 cm2 V−1 s−1 (hole mobility). Both the hole mobility and electron mobility of the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film are higher than those of the MAPbI3 thin film. Such boosted charge carrier mobilities are attributed to the conjugated chain of PPA as compared with the insulating organic spacers.22–28 Moreover, these charge carrier mobility values indicate that the charge transport tends to be balanced (μe/μh = 0.90). As a result, an enhanced JSC is expected to be observed from the PSCs with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film.
The photovoltaic properties of the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film are investigated through the characterization of the PSCs with a device structure of ITO/PTAA/perovskite/C60/BCP/Al, where ITO acts as the anode, PTAA acts as the hole extraction layer (HEL), C60 is used as the electron extraction layer (EEL) and the hole blocking layer (HBL), BCP is used as another HBL and Al acts as the cathode. Fig. 3a presents the statistics of the PCEs of the PSCs with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin films with different PPA concentrations. The PCE values of the PSCs are increased and then decreased with the increased concentration of PPA. When the concentration of PPA is at 0.5%, the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibits the best PCE. The J–V characteristics of the PSCs with either the MAPbI3 thin film or the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film (x = 0.5%) are shown in Fig. 3b. The PSC with the MAPbI3 thin film exhibits a short-circuit current density (JSC) of 21.41 mA cm−2, a VOC of 1.04 V and a fill factor (FF) of 78%, with a corresponding PCE of 17.37%. These device performance parameters are consistent with the reported values.47 The PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibits a JSC of 25.92 mA cm−2, a VOC of 1.06 V, a FF of 80% and a corresponding PCE of 21.98%, which is approximately a 25% enhancement compared to that with the MAPbI3 thin film. This boosted PCE is ascribed to the conjugated PPA organic spacer with better charge transport properties in comparison with insulating molecules such as butyl amine and benzene-based amines.19–28Fig. 3c shows the EQE spectra of the PSCs. The PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibits an enhanced EQE value compared to that with the MAPbI3 thin film. The integrated current densities are 20.89 mA cm−2 and 25.24 mA cm−2 for the PSC with the MAPbI3 thin film and the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film, respectively. These integrated current densities are in good agreement with the JSC values obtained from the J–V characteristics (Fig. 3b).
 | ||
| Fig. 3 (a) The statistics of the PCE of the PSCs with (PPA)x(MAPbI3)1−x/MAPbI3 thin films with different PPA concentrations. (b) The J–V characteristics and (c) EQE spectra of PSCs with either the MAPbI3 or (PPA)x(MAPbI3)1−x/MAPbI3 thin films. (d) The stabilities of the PSCs with either the MAPbI3 thin film or the (PPA)x(MAPbI3)1−x/MAPbI3 thin film. | ||
The stability of the un-encapsulated PSC measured in the ambient atmosphere (25 °C and 30 ± 10 humidity) is shown in Fig. 3d. The PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film retains 50% of its initial PCEs after 1200 hours (h), whereas the PSC with the MAPbI3 thin film shows a significant degradation after 100 h and more than 50% of the original PCE is degraded after 500 h. The conjugated long-chain cations of PPA could anchor on the surface of the perovskite grain boundary, which could passivate the trap states, resulting in a robust (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film which can withstand moisture and oxygen. In addition, the enhanced stability of the PSC is attributed to the pinhole-free thin film of the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer.
To understand the underlying physics of the enhanced JSC, TPC measurements are carried out to probe the photogenerated charge transport rather than to just measure the photocurrent.32Fig. 4a presents the TPC curves of the PSCs. As an external bias of −1.0 V is applied across the PSCs to minimize the charge carrier recombination, the charge carrier extraction time is estimated by extrapolating the photocurrent density to zero from the linear regime.32 Based on the normalized TPC curves, the charge carrier extraction times are estimated to be ∼90 ns and ∼70 ns for the PSC with the MAPbI3 thin film and the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film, respectively. A short charge carrier extraction time indicates that an efficient charge extraction process takes place in solar cells.32 Thus, the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibits a higher JSC.
 | ||
| Fig. 4 (a) The normalized transient photocurrent (TPC) curve of the PSCs with either the MAPbI3 thin film or the (PPA)x(MAPbI3)1−x/MAPbI3 thin film. (b) VOC and JSCversus light intensity of the PSCs with either the MAPbI3 thin film or the (PPA)x(MAPbI3)1−x/MAPbI3 thin film. (c) Nyquist plots of the PSCs with either the MAPbI3 thin film or the (PPA)x(MAPbI3)1−x/MAPbI3 thin film, measured in the dark, and (d) the Mott–Schottky analysis of the PSCs. | ||
The light intensity-dependent JSC and VOC are investigated to visualize the charge carrier recombination loss mechanisms in the PSCs. As indicated in Fig. 4b, both PSCs follow the relationship of VOC ∝ Sln (I)48 (where S = nkBT/q, kB is the Boltzmann constant, T is the absolute temperature, q is the elementary charge and I is the light intensity). The S = 1.56kBT/q observed from the PSC with the MAPbI3 thin film indicates that both bimolecular charge carrier recombination and trap-assisted charge carrier recombination occurred in the PSC; whereas the S = 1.11kBT/q observed from the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film indicates that the trap-assisted charge carrier recombination within PSCs is suppressed. Furthermore, based on JSC ∝ Iα (where I is the light intensity and α is the coefficient),48 both PSCs exhibit a power-law dependence of JSC on the light intensity. As α = 1, all charge carriers are swept out before recombination. α of 0.83 and 0.94 are observed for the PSC with the MAPbI3 thin film and the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film, respectively. A large α suggests that the bimolecular recombination is suppressed. Thus, the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibit a boosted JSC compared to that with the MAPbI3 thin film.
IS is applied to investigate the charge transport and recombination processes within the PSCs.49 As indicated in Fig. 4c, the charge carrier recombination resistances (Rrec) for the PSC with the MAPbI3 thin film and the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film, measured in the dark, are estimated to be ∼440 Ω, and ∼720 Ω, respectively. A high Rrec indicates that weak charge carrier recombination takes place in the PSC. Thus, the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibits a higher JSC. The charge transfer resistance (RCT) values are further calculated from the IS spectra at low frequencies, measured under white light illumination (Fig. 4c). The RCT for the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film is calculated to be ∼80 Ω, whereas a RCT of ∼130 Ω is calculated for the PSC with the MAPbI3 thin film. A small RCT indicates that the charge transport properties of the PSC are boosted, thereby resulting in an enhanced JSC.50
Mott–Schottky analysis, based on the C–V measurements, is further carried out to investigate the built-in potentials (Vbi) of the PSCs.50 As shown in Fig. 4d, under the forward bias, the Vbi of the PSCs are extracted from  48 (where ε is the dielectric constant, ε0 is the vacuum permittivity and A is the active area) by extrapolating the linear fitting line to the intercept on the x-axis. It is found that a Vbi of ∼1.15 V is observed for the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film, whereas a Vbi of ∼1.09 V is observed for the PSC with the MAPbI3 thin film. Therefore, as expected, the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibits a larger VOC compared to that with the MAPbI3 thin film.
48 (where ε is the dielectric constant, ε0 is the vacuum permittivity and A is the active area) by extrapolating the linear fitting line to the intercept on the x-axis. It is found that a Vbi of ∼1.15 V is observed for the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film, whereas a Vbi of ∼1.09 V is observed for the PSC with the MAPbI3 thin film. Therefore, as expected, the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibits a larger VOC compared to that with the MAPbI3 thin film.
4. Conclusions
In conclusion, an innovative conjugated aniline PPA to create a 2D (PPA)x(MAPbI3)1−x thin layer and to further form the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film to develop efficient and stable perovskite solar cells was reported in this study. Systematic studies indicated that the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film possessed higher crystallinity and passivated trap states, resulting in improved charge carrier transport and suppressed charge carrier recombination with respect to those of the MAPbI3 thin film. As a result, the PSC with the (PPA)x(MAPbI3)1−x/MAPbI3 bilayer thin film exhibited a PCE of 21.98% and a significantly boosted stability in an ambient atmosphere. Our results suggested that the utilization of conjugated molecules as organic spacer cations to create 2D perovskites and further form 2D/3D perovskite bilayer thin films is an effective way to develop high-performance PSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the National Science Foundation (ECCS/EPMD1903303) and the Air Force Office of Scientific Research (AFOSR) (through the Organic Materials Chemistry Program, grant number: FA9550-15-1-0292, Program Manager, Dr Kenneth Caster) for the financial support.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051, DOI:10.1021/ja809598r.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647, DOI:10.1126/science.1228604.
- J. H. Heo, S. H. Im, J. H. Noh, T. N. Mandal, C.-S. Lim, J. A. Chang, Y. H. Lee, H.-J. Kim, A. Sarkar and M. K. Nazeeruddin, Nat. Photonics, 2013, 7, 486–491, DOI:10.1038/nphoton.2013.80.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630, DOI:10.1021/jz4020162.
- K. Wang, L. Zheng, T. Zhu, L. Liu, M. L. Becker and X. Gong, Nano Energy, 2020, 67, 104229, DOI:10.1016/j.nanoen.2019.104229.
- K. Wang, L. Zheng, T. Zhu, X. Yao, C. Yi, X. Zhang, Y. Cao, L. Liu, W. Hu and X. Gong, Nano Energy, 2019, 61, 352–360, DOI:10.1016/j.nanoen.2019.04.073.
- W. Xu, L. Zheng, T. Zhu, L. Liu and X. Gong, ACS Appl. Mater. Interfaces, 2019, 11, 34020–34029, DOI:10.1021/acsami.9b12346.
- L. Dou, Y. M. Yang, J. You, Z. Hong, W.-H. Chang, G. Li and Y. Yang, Nat. Commun., 2014, 5, 5404, DOI:10.1038/ncomms6404.
- C. Liu, K. Wang, P. Du, E. Wang, X. Gong and A. J. Heeger, Nanoscale, 2015, 7, 16460–16469, 10.1039/C5NR04575D.
- T. Zhu, Y. Yang, L. Zheng, L. Liu, M. L. Becker and X. Gong, Adv. Funct. Mater., 2020, 30, 1909487, DOI:10.1002/adfm.201909487.
- W. Xu, Y. Guo, X. Zhang, L. Zheng, T. Zhu, D. Zhao, W. Hu and X. Gong, Adv. Funct. Mater., 2018, 28, 1705541, DOI:10.1002/adfm.201705541.
- N. NREL, Best Research-Cell Efficiency Chart, US Department of Energy, 2019, https://www.nrel.gov/pv/cell-efficiency.html Search PubMed.
- G. Niu, X. Guo and L. Wang, J. Mater. Chem. A, 2015, 3, 8970–8980, 10.1039/C4TA04994B.
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5, eaav0693, DOI:10.1126/sciadv.aav0693.
- T. Zhu, Y. Yang, K. Gu, C. Liu, J. Zheng and X. Gong, ACS Appl. Mater. Interfaces, 2020, 12, 51744–51755, DOI:10.1021/acsami.0c16514.
- Y. Bai, S. Xiao, C. Hu, T. Zhang, X. Meng, H. Lin, Y. Yang and S. Yang, Adv. Energy Mater., 2017, 7, 1701038, DOI:10.1002/aenm.201701038.
- Q. He, M. Worku, L. Xu, C. Zhou, H. Lin, A. J. Robb, K. Hanson, Y. Xin and B. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 1159–1168, DOI:10.1021/acsami.9b17851.
- J. Y. Ye, J. Tong, J. Hu, C. Xiao, H. Lu, S. P. Dunfield, D. H. Kim, X. Chen, B. W. Larson, J. Hao, K. Wang, Q. Zhao, Z. Chen, H. Hu, W. You, J. J. Berry, F. Zhang and K. Zhu, Sol. RRL, 2020, 4, 2000082, DOI:10.1002/solr.202000082.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466, DOI:10.1038/s41566-019-0398-2.
- F. Wang, W. Geng, Y. Zhou, H. H. Fang, C. J. Tong, M. A. Loi, L. M. Liu and N. Zhao, Adv. Mater., 2016, 28, 9986–9992, DOI:10.1002/adma.201603062.
- T. Zhou, H. Lai, T. Liu, D. Lu, X. Wan, X. Zhang, Y. Liu and Y. Chen, Adv. Mater., 2019, 31, 1901242, DOI:10.1002/adma.201901242.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190, DOI:10.1021/jacs.8b10851.
- L. Mao, W. Ke, L. Pedesseau, Y. Wu, C. Katan, J. Even, M. R. Wasielewski, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 3775–3783, DOI:10.1021/jacs.8b00542.
- N. Zhou, B. Huang, M. Sun, Y. Zhang, L. Li, Y. Lun, X. Wang, J. Hong, Q. Chen and H. Zhou, Adv. Energy Mater., 2020, 10, 1901566, DOI:10.1002/aenm.201901566.
- Y. Zhang, J. Chen, X. Lian, M. Qin, J. Li, T. R. Andersen, X. Lu, G. Wu, H. Li and H. Chen, Small Methods, 2019, 3, 1900375, DOI:10.1002/smtd.201900375.
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 7843–7850, DOI:10.1021/jacs.5b03796.
- J. Qiu, Y. Xia, Y. Zheng, W. Hui, H. Gu, W. Yuan, H. Yu, L. Chao, T. Niu, Y. Yang, X. Gao, Y. Chen and W. Huang, ACS Energy Lett., 2019, 4, 1513–1520, DOI:10.1021/acsenergylett.9b00954.
- F. Zhang, D. H. Kim, H. Lu, J.-S. Park, B. W. Larson, J. Hu, L. Gao, C. Xiao, O. G. Reid, X. Chen, Q. Zhao, P. F. Ndione, J. J. Berry, W. You, A. Walsh, M. C. Beard and K. Zhu, J. Am. Chem. Soc., 2019, 141, 5972–5979, DOI:10.1021/jacs.9b00972.
- K. Wang, J. Liu, J. Yin, E. Aydin, G. T. Harrison, W. Liu, S. Chen, O. F. Mohammed and S. De Wolf, Adv. Funct. Mater., 2020, 30, 2002861, DOI:10.1002/adfm.202002861.
- M. Zhang, S. Dai, S. Chandrabose, K. Chen, K. Liu, M. Qin, X. Lu, J. M. Hodgkiss, H. Zhou and X. Zhan, J. Am. Chem. Soc., 2018, 140, 14938–14944, DOI:10.1021/jacs.8b09300.
- T. Zhu, L. Zheng, Z. Xiao, X. Meng, L. Liu, L. Ding and X. Gong, Sol. RRL, 2019, 3, 1900322, DOI:10.1002/solr.201900322.
- J. Seifter, Y. Sun and A. J. Heeger, Adv. Mater., 2014, 26, 2486–2493, DOI:10.1002/adma.201305160.
- J. Sun, N. Chandrasekaran, C. Liu, A. D. Scully, W. Yin, C. K. Ng and J. J. Jasieniak, ACS Appl. Energy Mater., 2020, 3, 8205–8215, DOI:10.1021/acsaem.0c00553.
- G. Liu, X.-X. Xu, S. Xu, L. Zhang, H. Xu, L. Zhu, X. Zhang, H. Zheng and X. Pan, J. Mater. Chem. A, 2020, 8, 5900–5906, 10.1039/C9TA14139A.
- Z. Wang, Q. Lin, F. P. Chmiel, N. Sakai, L. M. Herz and H. J. Snaith, Nat. Energy, 2017, 2, 17135, DOI:10.1038/nenergy.2017.135.
- T. Oku, Solar Cells-New Approaches and Reviews, 2015. DOI:10.5772/59284.
- V. Pecharsky and P. Zavalij, Fundamentals of Powder Diffraction and Structural Characterization of Materials, Springer Science & Business Media, 2008 Search PubMed.
- D.-Y. Son, J.-W. Lee, Y. J. Choi, I.-H. Jang, S. Lee, P. J. Yoo, H. Shin, N. Ahn, M. Choi, D. Kim and N.-G. Park, Nat. Energy, 2016, 1, 16081, DOI:10.1038/nenergy.2016.81.
- N. Phung, A. Al-Ashouri, S. Meloni, A. Mattoni, S. Albrecht, E. L. Unger, A. Merdasa and A. Abate, Adv. Energy Mater., 2020, 10, 1903735, DOI:10.1002/aenm.201903735.
- D. Kim, H. J. Jung, I. J. Park, B. W. Larson, S. P. Dunfield, C. Xiao, J. Kim, J. Tong, P. Boonmongkolras, S. G. Ji, F. Zhang, S. R. Pae, M. Kim, S. B. Kang, V. Dravid, J. J. Berry, J. Y. Kim, K. Zhu, D. H. Kim and B. Shin, Science, 2020, 368, 155–160, DOI:10.1126/science.aba3433.
- R. H. Bube, J. Appl. Phys., 1962, 33, 1733–1737, DOI:10.1063/1.1728818.
- E. A. Duijnstee, J. M. Ball, V. M. Le Corre, L. J. A. Koster, H. J. Snaith and J. Lim, ACS Energy Lett., 2020, 5, 376–384, DOI:10.1021/acsenergylett.9b02720.
- X. Zhang, X. Ren, B. Liu, R. Munir, X. Zhu, D. Yang, J. Li, Y. Liu, D.-M. Smilgies, R. Li, Z. Yang, T. Niu, X. Wang, A. Amassian, K. Zhao and S. F. Liu, Energy Environ. Sci., 2017, 10, 2095–2102, 10.1039/C7EE01145H.
- P. Murgatroyd, J. Phys. D: Appl. Phys., 1970, 3, 151, DOI:10.1088/0022-3727/3/2/308.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480, DOI:10.1038/nmat3911.
- L. M. Herz, ACS Energy Lett., 2017, 2, 1539–1548, DOI:10.1021/acsenergylett.7b00276.
- A. Mahapatra, D. Prochowicz, M. M. Tavakoli, S. Trivedi, P. Kumar and P. Yadav, J. Mater. Chem. A, 2020, 8, 27–54, 10.1039/C9TA07657C.
- F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Sero and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083–9118, 10.1039/C0CP02249G.
- T. Zhu, Y. Yang, Y. Liu, R. Lopez-Hallman, Z. Ma, L. Liu and X. Gong, Nano Energy, 2020, 78, 105397, DOI:10.1016/j.nanoen.2020.105397.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207, DOI:10.1103/PhysRevB.82.245207.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta05934c |
| This journal is © The Royal Society of Chemistry 2021 |