
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oligoene and cyanine features of tetracyano quinoidal oligothiophenes†
Sergio
Moles Quintero
a,
José Luis
Zafra
a,
Keitaro
Yamamoto
b,
Yoshio
Aso
 b,
Yutaka
Ie
bc and
Juan
Casado
*a
b,
Yutaka
Ie
bc and
Juan
Casado
*a
aDepartment of Physical Chemistry, University of Malaga, Campus de Teatinos s/n, 29071 Malaga, Spain. E-mail: casado@uma.es
bThe Institute of Scientific and Industrial Research (ISIR), Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan
cInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 19th May 2021
Abstract
This paper interprets a palette of spectroscopic data on tetracyano thienoquinoidal molecules of different sizes oriented towards the elucidation of their electronic behavior and other aspects of interest in materials chemistry. It also provides a contextualization of these properties between those of even oligoenes (carotene-like) and those of odd cyanines as a function of the reduction state. A complete UV-Vis-NIR electronic absorption, fluorescence emission and infrared spectroscopic study in part helped by quantum chemical calculations in the framework of the DFT theory has been carried out. The study encompasses the neutral and charged states. The optical properties of these compounds in the neutral state are controlled by the competition of bright (electric dipole–dipole allowed) and dark (electric dipole–dipole forbidden) singlet excited states. In the anionic state, the trimeric compounds disclose behaviors compatible with being either charge localized or delocalized mixed valence systems. Interestingly, we show the π-dimerization of a radical anion of a thienoquinoidal compound forming a π-dimer dianion as a manifestation of the delocalization of the charge in the charged monomer. The hexamer compound in the neutral state discloses incipient diradical character which permits the detection of a NIR emission band from the dark state at energies well below 1 eV. As thiophene benzo-annulation minimizes the diradical character, it forces the confinement of the charge in the vicinity of the terminal dicyanomethylene groups in the dianion. The oxidized species of the compounds have been also analyzed as models of charge localization states. The competition between doublet and quartet high spin states in the radical cations has been explored with the objective of visualizing potential molecules able to produce SOMO–HOMO energy inversion. This paper adds an interesting perspective of the electronic structure of quinoidal molecules with respect to their interest in the field of organic electronics.
I. Introduction
Aromatic oligothiophenes,1 AOTs in Scheme 1, are well-known in the field of organic electronics, with hexathiophene2 being, together with pentacene,3 one of the first organic π-conjugated compounds used as electrically active semiconductors in organic field-effect transistor (OFET) devices. These demonstrated to be p-type semiconductors4 as expected for organic materials with an electron rich nature which prefer to stabilize and conduct positive charges (i.e., being oxidized). Aromatic hexathiophene can be viewed as a cis–trans oligoene with 12 olefins where the cis conformation is fixed within the thiophene ring (Scheme 1). However, the aromatic character of thiophene makes these oligothiophenes deviate from the behavior of oligoenes, a detrimental aspect regarding the modulation of some properties such as π-electron delocalization and electronic and photonic properties based on it. Medium-long oligoenes are characterized by a first singlet excited state of dark nature5 (forbidden by the dipole–dipole electric selection rules) which promotes two-photon absorption activity and, parallelly, fully quenches the molecular fluorescence. The presence of this dark excited state is also beneficial for applications such as in singlet-exciton fission6 because it is a precursor of the two entangled triplet states.7 | ||
| Scheme 1 Examples of the chemical structures of oligoenes (with four olefins), aromatic oligothiophenes (AOTs, bithiophene), and quinoidal oligothiophenes (QOTs, quinoidal bithiophene). | ||
Quinoidal oligothiophenes,8–11 QOTs in Scheme 1, revert this situation. The extension of the electron wavefunction into the exocyclic bonds between the thiophenes disrupts its localization in the five membered rings by aromaticity. As a result, QOTs gain more oligoene-like character than their aromatic parents, thus recovering some of the beneficial properties of the carotenoid-like systems, such as the stabilization of the dark singlet excited state as the first energy lying singlet; in fact, this has enabled the appearance of intramolecular singlet exciton fission in one of the members of the QOTs.12–14 The efficient inter-annular extension of π-electron delocalization in QOTs also gives rise to the appearance of the electronic absorption bands well displaced into the Vis-NIR region, indicating them to be NIR absorbing molecules. These optical properties of QOTs are accompanied by rich electrochemical properties in the cyclic voltammetry experiments showing reduction potentials at relatively low values and, in the case of the long oligomers, even reversible oxidations conferring amphoteric redox behavior.15,16 This rich redox activity of QOTs has been exploited in organic field effect transistors with high electron mobility and ambipolar behavior in OFET devices.17,18 More recently, tetracyanoquinoidal oligothiophenes have been studied by treatment with an electron donor to produce n-doped substrates which have been demonstrated to be efficient conductors for thermoelectric devices.19
AOTs with as many as 96 thiophenes20 have been prepared. The longest QOTs, however, are hexamers.8,16 These known hexamers are formed by dicyanomethylene encapsulation of the quinoidal core, a substitution that further contributes to the extension of π-electron delocalization, to the outstanding electron acceptor properties and to the intense Vis-NIR absorption. QOTs have been distinctively studied in recent years as stable diradicaloids.8,16 This is based on the tendency to recover the aromaticity of the non-aromatic rings in the neutral state which for a certain molecular length (number of thiophenes) is able to stabilize the homolytic rupture of one double bond forming an open-shell diradicaloid.8,16 Given the balance between aromaticity energy gain and the energy required to break a double bond, QOT diradicals are formed starting from the pentamer with the tetramer being in the borderline of the quinoidal → aromatic transition. Diradicaloids have appeared in the field of organic electronics given their promise to act as spintronic substrates based on their very low energy lying thermally accessible high spin triplet states which can act as spin polarizers or spin filters of current.21,22 Diradicaloids and diradicals have been also demonstrated to be good non-linear optical chromophores23,24 with large two-photon cross sections and large third order molecular hyperpolarizability, etc.
To the best of our knowledge, only a few tetracyano thienoquinoidal hexamers have been prepared and studied.8,16 After more than 15 years since the first studies of these systems, herein we present the study of the electronic properties of a new tetracyano thienoquinoidal hexamer (B2-6TQ in Fig. 1) in the light of new spectroscopic properties as well as in comparison with shorter tetracyanothienoquinoidal parent compounds (3TQ, B-3TQ, B3-3TQ and 2TQ in Fig. 1). A study of the electronic and vibrational properties of the tetracyanoquinoidal hexamer in connection with its diradicaloid, oligoene-like, and electron-acceptor characters is proposed. For this hexamer, the possibility to display SOMO–HOMO energy level inversion is also explored. On the other hand, the smaller trimeric compounds are analyzed from the point of view of their mixed valence properties and in the context of the properties of cyanines.
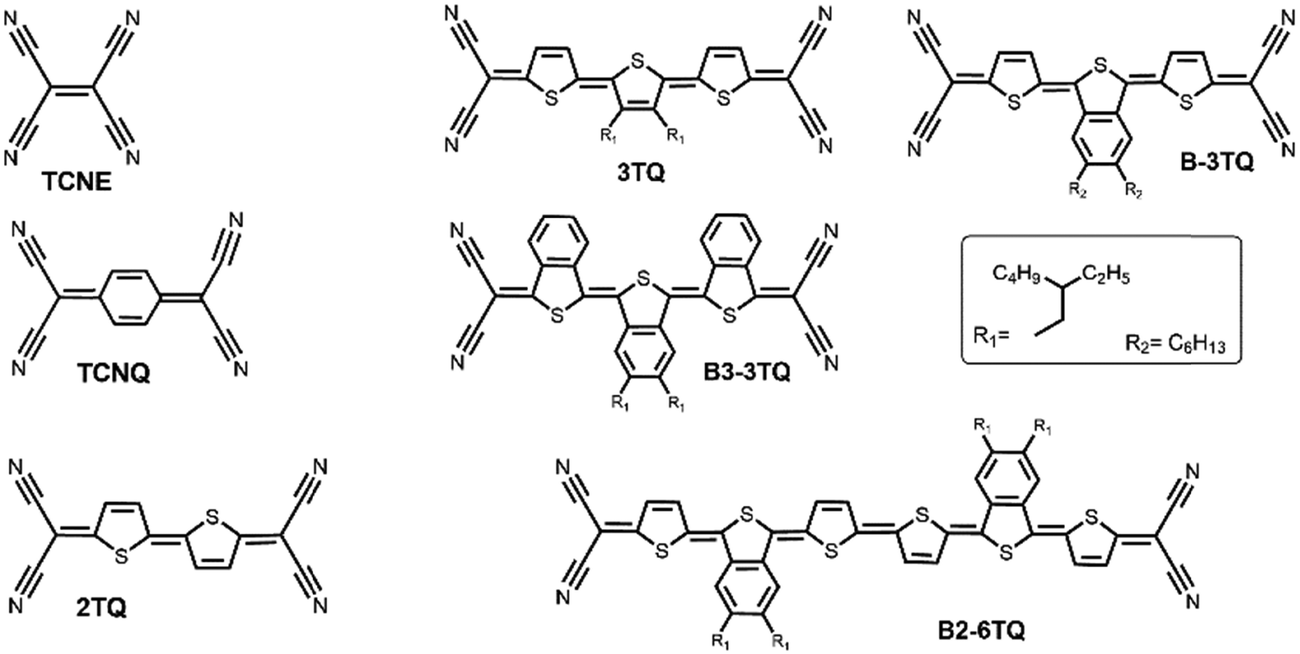 | ||
| Fig. 1 Chemical structures of the QOTs studied and those of the known TCNE, TCNQ and 2TQ used as reference compounds. | ||
II. Experimental and theoretical methods
The syntheses of the benzothienoquinoidal compounds, 3TQ, B-3TQ, B3-3TQ and B2-6TQ, have been recently reported.25,26 On the other hand, the preparation and characterization of thienoquinoidal parent compound 2TQ were published some years ago.8In situ infrared and UV-Vis-NIR spectroelectrochemical studies were conducted on a VERTEX 70 FT-IR spectrometer and on a Varian Cary 5000 UV-Vis-NIR spectrophotometer, respectively. A C3 epsilon potentiostat from BASi was used for the electrolysis using a thin layer cell from a demountable omni cell from Specac. In this cell a three electrode system was coupled to conduct in situ spectroelectrochemistry. A Pt gauze was used as the working electrode, a Pt wire was used as the counter electrode, and an Ag wire was used as the pseudo-reference electrode. The corresponding cyclic voltammetry data and related energy levels are shown in Table 1 and are taken from ref. 25 and 26. The spectra were collected using constant potential electrolysis and the potentials were changed in intervals of 10 mV. The electrochemical medium used was 0.1 M tetrabutyl ammonium hexafluorophosphate, Bu4NPF6, in fresh distilled dichloromethane, at room temperature with a sample concentration of 10−3 M. Low temperature absorption and emission measurements were carried out in a cryostat OPTISTAT cell from Oxford Instruments.Emission fluorescence spectra were obtained using a JASCO V-570 spectrometer. The measurements were carried out in degassed/anhydrous dichloromethane. All solvents used were of spectroscopic grade purchased from Aldrich. The emission spectra in the NIR region were obtained either by using a Bruker Senterra Raman spectrometer equipped with a microscope with excitation up to 785 nm by averaging spectra during 10 min with a resolution of 3–5 cm−1 or by using a RAMII FT-Raman module of a VERTEX 70 FT-IR spectrometer with excitation at 1064 nm. The Raman and emission scattering radiation was collected in a back-scattering configuration with a standard spectral resolution of 4 cm−1. 500 scans were averaged for each spectrum.
Quantum chemical calculations were done with the Gaussian16 suite of programs.27 Molecular geometry optimizations were performed with the B3LYP functional and the 6-31G** standard basis set.28,29 Energy optimizations were performed by allowing all geometric parameters to vary independently. For the analysis of the open-shell diradicals, the broken-symmetry approximation in the unrestricted DFT methodology (UDFT) was assumed. The singlet–triplet energy gaps were calculated as the energy difference between those of the optimized geometries of the singlet-open shell (broken symmetry) and triplet electronic states. The UDFT methodology was also used for the theoretical study of the energies and geometries of monoradical systems. Vertical transition energies were computed with the time-dependent version of DFT (TDDFT) at the same level of theory.30,31 Some additional basic theoretical data such as the energy values of the frontier molecular orbitals are available in the corresponding articles where the molecules were reported.
III. Results and discussion
III.1. Electronic structure of the neutral compounds
Fig. 2 displays the electronic absorption spectra of the three quinoidal trimers, 3TQ, B-3TQ and B3-3TQ. In 3TQ the main absorption band appears at 670 nm, as in other reported quinoidal trimers. This band blue-shifts upon incorporation of one and three benzothiophene units up to 643 nm in B-3TQ and to 613 nm in B3-3TQ. These bands correspond to the respective HOMO → LUMO one electron transitions according to TD-DFT calculations which predict them at 624 nm in 3TQ, at 599 nm in B-3TQ and 568 nm in B3-3TQ in nice agreement with the experimental data and trends (Fig. S1, ESI†). This wavelength maxima behavior is a consequence of the destabilization of the LUMO (Fig. S2, ESI†) on 3TQ → B-3TQ → B3-3TQ due to the increasing number of nodes in the wavefunction upon inclusion of lateral benzenes (i.e., between the thiophene carbon atoms and those of the benzenoid rings). This is nicely reflected in the electrochemical reduction potentials of the three compounds which evolve to more negative values of −0.58 V in 3TQ, −0.74 V in B-3TQ and −1.02 V in B3-3TQ.25,26 Interestingly, despite the cathodic displacement of the potentials, the reduction waves all correspond to the transference of two electrons each suggesting that the destabilization of the LUMO is accompanied by a progressive localization of the extra charge in the reduced species in two separated domains. This would make the electron transfer of two electrons happen at similar potentials (not distinguished in the cyclic voltammetry experiment). Conversely to the cyclic voltammetry experiment, in the spectroelectrochemical cell during reduction, we will describe below the distinctive and subsequent formation of a radical anion and of a dianion in the three smaller quinoidals. On the other hand, the HOMO energy of the three trimeric compounds (Fig. S2, ESI†) is scarcely affected in the series owing to the non-bonding character of the connecting atoms between those of the thiophenes and of the benzenes explaining the very similar oxidation potentials for the generation of their radical cations.25,26 The overall result is an enlargement of the HOMO–LUMO gap and of the optical gap upon increasing numbers of benzothiophenes.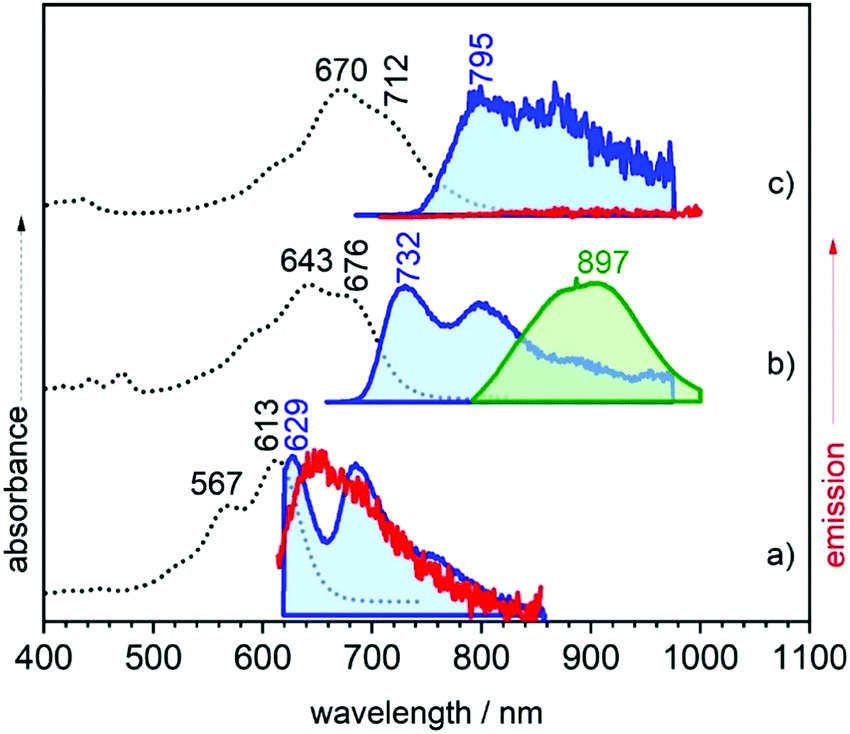 | ||
| Fig. 2 UV-Vis-NIR absorption (black dotted lines) and emission (red and green lines at 298 K and blue lines at 80 K) spectra of (a) B3-3TQ, (b) B-3TQ, and (c) 3TQ. | ||
The emission properties of the studied compounds have been analyzed upon excitation of the main absorption bands. The three quinoidal trimers show very weak or no emission at room temperature in the range up to 1100 nm. In this regard, to complement these data, Fig. 2 also shows the spectra at 80 K. There are significant changes from molecule to molecule in the low temperature spectra: whereas B3-3TQ displays an emission associated with the optically pumped excitation band with a small Stokes shift (i.e., 415 cm−1), B-3TQ shows an emission band with a larger Stokes shift (i.e., 1130 cm−1) and, finally, 3TQ exhibits a broad band (not resolved at room temperature) featuring a Stokes shift of ≈1600 cm−1. Interestingly, upon excitation at 785 nm of the low energy part of the intense absorption of B-3TQ, a new emission feature is detected at room temperature that is not detected upon excitation on the peak maxima (643/676 nm) of the intense band under the same temperature conditions. This new band shows a similar shape to the main absorption band with a maximum at 897 nm thus representing a very large Stokes shift of >4500 cm−1. This makes us likely to ascribe this emission to a radiative process from a different excited electronic state which is lower in energy compared to that linked with the main band of the absorption spectrum. The low temperature spectra of B3-3TQ and B-3TQ clearly disclose vibronic structures related with the increment of torsional mobility in the excited state upon partial relaxing of the planarity between the thiophene rings imparted by the quinoidal shape in the ground electronic state.
It is known for quinoidal compounds that there is a dark singlet excited state12,13 (i.e., non-allowed by the electric dipole selection rules, denoted as S2 in Fig. 3) close in energy to the optical state (i.e., bright state, denoted as S1 in Fig. 3) that must be considered to explain their photophysical properties. This excited state distribution in quinoidal compounds is very similar to that described in oligoenes and carotenoid-like compounds.5 In B3-3TQ, the situation is that the S1 is lower in energy than S2 by which we detect the emission only from S1, or S1 → S0 at 629 nm, thus accounting for its small Stokes shift. In B-3TQ, the situation is that the energies of the S1 and S2 are very close, consequently promoting S1 → S2 conversion through a conical intersection which has been described in similar quinoidal compounds;13 the overall result in B-3TQ is the detection of the two S1 → S0 and S2 → S0 emissions (a wrong very large Stokes shift is found if we consider the absorption by the S1 and the emission from the S2). Finally, in 3TQ, the situation is that S1 → S2 is apparently blocked, by which none of them are detected at room temperature due to the smallest gaps (the S1 → S0 is weakly detected at low temperature). TD-DFT calculations predict the bright singlet excited state always as the S0 → S1 absorption with the S0 → S2 excitation lying always higher in energy for the three compounds. It is well known, however, that the relative disposition of these two excited states is strongly dependent on electron correlation which is not fully considered in our simple TD-DFT approach; thus, we are confident that if these correlation effects would be taken into account in calculations, such as already reported for a homologue 2TQ quinoidal compound,8,16 the most likely evolution of the energy of the S2 state in the three compounds is such as that in Fig. 3 (note that high level theoretical calculations are beyond the objective of the present work).
 | ||
| Fig. 3 Qualitative excited state energy ordering of the bright (S1) and dark (S2) states of the studied compounds: (a) B3-3TQ, (b) B-3TQ, (c) 3TQ and (d) B2-6TQ. The broken arrows denote the absorption/excitation peaks and the solid arrows represent the emission either from the bright state (blue) or from the bark state (green). | ||
III.2. Electronic structure of the charged species in the context of mixed valence behavior
As already mentioned, in the spectroelectrochemical experiment, the spectra of the anion radicals and of the dianions are distinguished. Given the electron withdrawing character of the cyano groups, these drain the added negative charge; the point is whether the charge in the radical anion is preferentially stabilized in one of the dicyanomethylenes or stabilized by semi-contributions of the two and by the bridge. In the framework of the Robin–Day classification32 of mixed valence systems,33 these two situations correspond to a charge localized class II mixed valence system and to a class III full delocalized mixed valence system, respectively. Recently, the interesting cases of mixed valence systems that behave in the frontier between class II and class III transiting between them depending on the external stimuli has also been established.34 The starting point in this discussion is the case of the radical anions of the reference compounds TCNE and TCNQ (Fig. 1), which have been described as class III mixed valence systems with full charge delocalization between the two respective dicyanomethylenes.35 In comparison with these, the discussion of the electronic absorption and infrared spectra of the charged species of 3TQ, B-3TQ and B3-3TQ is presented.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching mode already revealing the intervention of the terminal cyano groups in this excitation, an effect absent in the spectrum of the 3TQ anion (i.e., one vibronic progression of 859 cm−1). The weakness of the bands of the anion radical of B3-3TQ precludes the evaluation of the vibronic structure.
N stretching mode already revealing the intervention of the terminal cyano groups in this excitation, an effect absent in the spectrum of the 3TQ anion (i.e., one vibronic progression of 859 cm−1). The weakness of the bands of the anion radical of B3-3TQ precludes the evaluation of the vibronic structure.
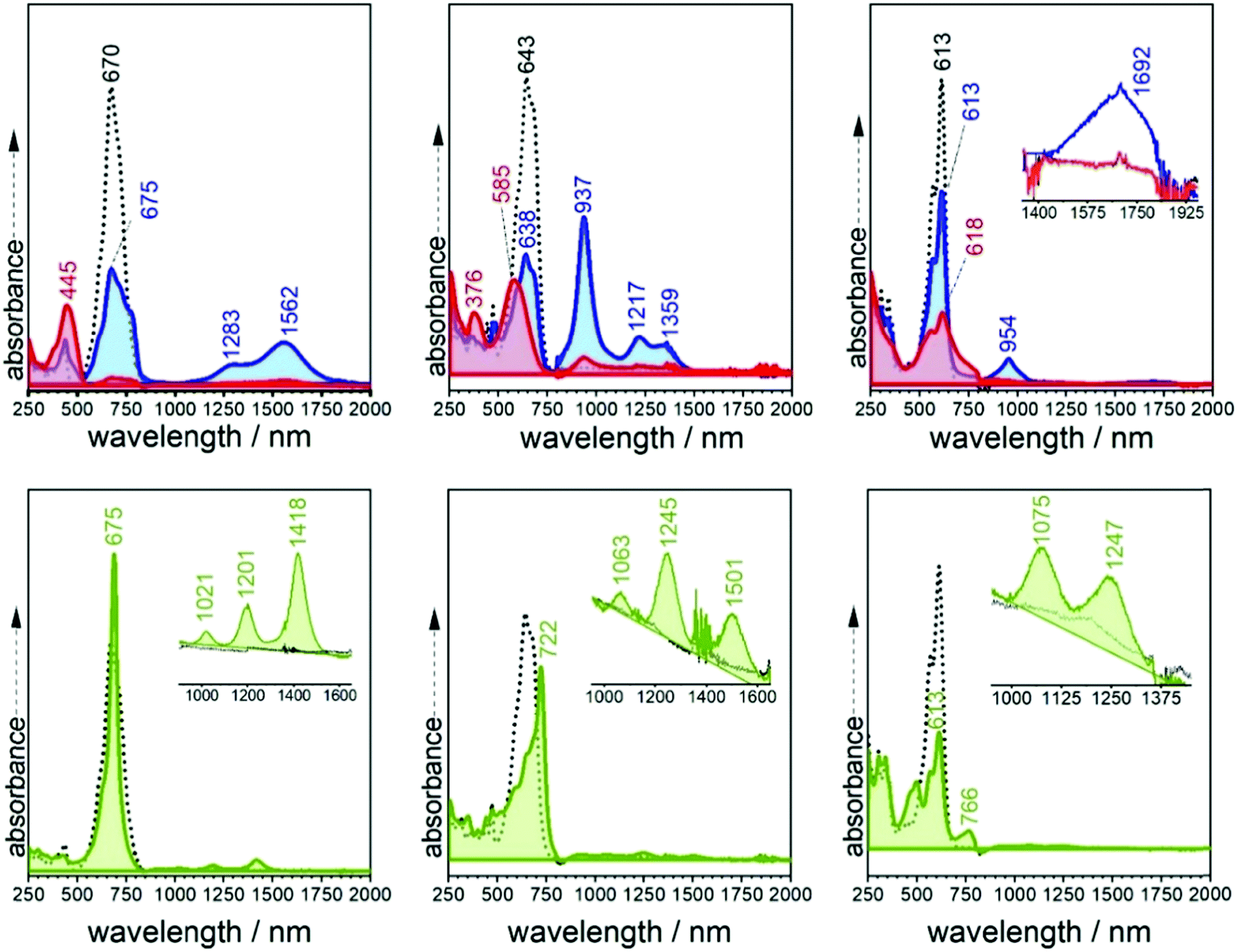 | ||
| Fig. 4 Top (bottom): UV-Vis-NIR electronic absorption spectra taken during the spectroelectrochemical reduction (oxidation) in 0.1 M TBPF6 in CH2Cl2 at room temperature of 3TQ (left), B-3TQ (middle) and B3-3TQ (right). Neutral (broken lines), radical anions (blue shaded), dianions (red shaded) and radical cations (green shaded). | ||
TD-DFT quantum chemical calculations were carried out for the three radical anions. For that of 3TQ, theory predicts two main excitations at 675 nm correlated with the experimental one at 660 nm and another transition at 1192 nm corresponding with the experimental bands at 1217/1359 cm−1. Though calculations of the neutral compounds and of the radical anion of 3TQ reproduce well the experiments, the same level calculations in the other two radical anions do not work well. The UV-Vis-NIR absorption spectra of the dianions of the three thienoquinoidal trimers display one main band which contrarily to the case of the radical anions red-shifts from 3TQ (i.e., at 445 nm), to B-3TQ (i.e., at 585 nm), and to B3-3TQ (i.e., at 618 nm).
The three compounds can be also reversibly oxidized to the radical cations whose UV-Vis-NIR absorption spectra are collected also in Fig. 4 and Fig. S4 (ESI†). The spectra of the three cationic species share similar patterns which consist of two features: (i) moderate/strong/weak visible bands at 675, 722 and 766 nm in 3TQ, B-3TQ and B3-3TQ, respectively, which (ii) are accompanied in the three cases by very weak NIR absorption at 1201/1418 (i.e., spaced by 1274 cm−1), 1245/1501 nm (i.e., spaced by 1370 cm−1), and 1075/1247 nm (i.e., spaced by 1283 cm−1) in the three compounds, respectively. The similar energy spacing of the three progressions reveals the confinement of the charge in similar molecular parts. These spectra typically correspond to species with highly localized charge defects, that is, extraction of the charge in the parent neutral compound takes place far apart of the electron withdrawing cyano groups, very likely in the molecular center around the electron rich sulfur atom(s) of the central thiophene(s). The discussion of these features in the cationic absorption spectra allows us to compare radical anions and radical cations of B3-3TQ: both monovalent species have a common spectral fingerprint related with the presence of very weak absorption NIR bands due to the existence in both of localized charged defects. The difference between the two is that in the anion the charge confinement is on/around the cyano moieties while for the cation the charge is preferentially accommodated closer to the central thiophenes.
Contrary to the cases of the benzothiophene quinoidals, in the anion radical of 3TQ there are indications that suggest that this can be assigned to a delocalized class III mixed valence species. These are: (i) the UV-Vis-NIR absorption spectra of the radical anion and dianion of the homologue 2TQ have been obtained (Fig. S5, ESI†). The cyclic voltammetry of 2TQ38 shows two well resolved and distinctive one-electron reductions due to the univocal formation of an anion radical followed by a dianion. The radical anion discloses an absorption spectrum with bands at 629 and 1062 nm (with a shoulder at 922 nm spaced by 1430 cm−1), and thus, with a very similar pattern to that of the radical anion of 3TQ in line with both radical anions sharing the same class III mixed valence property. (ii) Overall, the large differences in the shapes of the radical anion spectra of the three trimers suggest these to possess different electron distributions. (iii) Finally, the existence of a cyano based vibronic progression in the radical anion of B-3TQ further supports this to be of localized character around these groups.
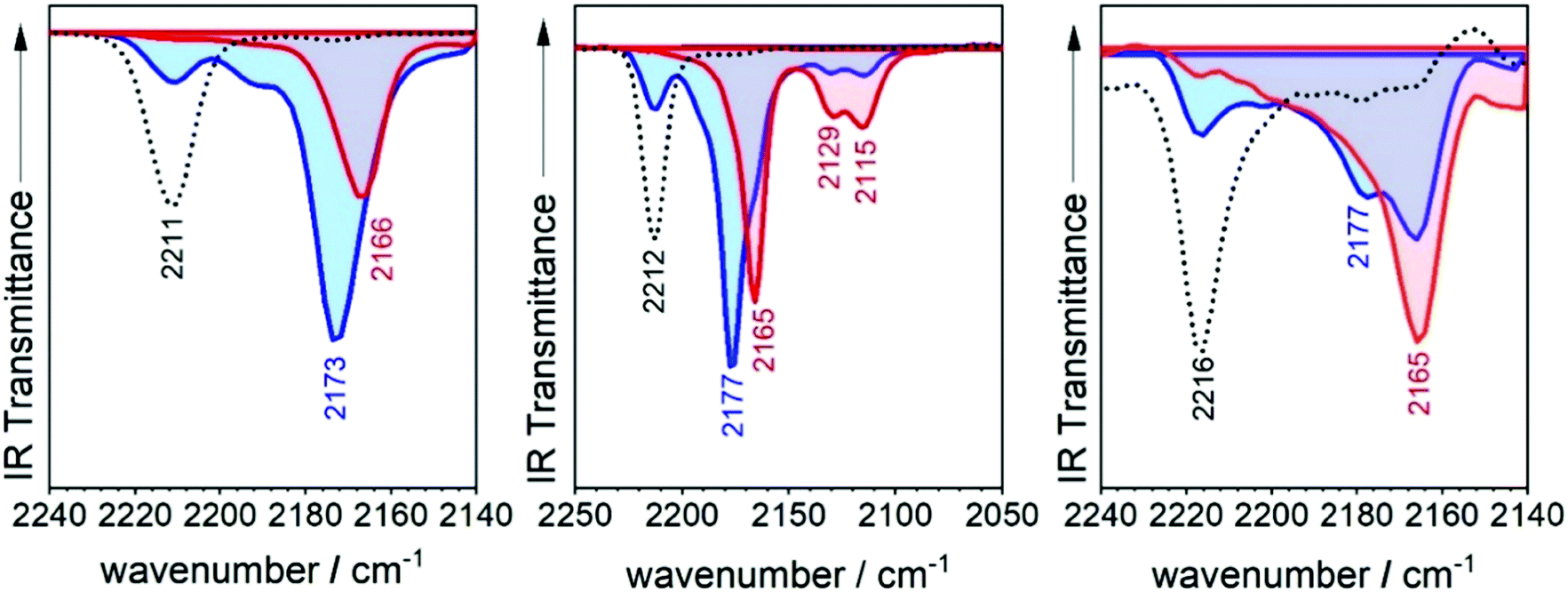 | ||
| Fig. 5 Infrared spectra taken during the spectroelectrochemical reduction in 0.1 M TBPF6 in CH2Cl2 at room temperature of 3TQ (left), B-3TQ (middle) and B3-3TQ (right). Neutral (broken lines), radical anions (blue shaded) and dianions (red shaded). | ||
The infrared spectrum of 3TQ in the anion radical state is characterized by a main band due to the CN stretching vibration at 2173 cm−1 which evolves from that at 2211 cm−1 in the neutral species. Subsequent reduction of this to the dianion gives rise to a further wavenumber downshift to 2166 cm−1. The wavenumber downshift of the CN stretching infrared band, Δν(CN), is a result of the antibonding character of the LUMO orbitals in the cyano groups so that their occupation upon reduction enlarges the CN distance and decreases the wavenumber of the associated stretching band. This process is progressive up to 2166 cm−1 on going from the radical anion to the anion of 3TQ due to the double occupancy of the initial LUMO. Reduction of B-3TQ also produces a change from 2212 cm−1 in the neutral species to 2177 cm−1 in the radical anion and 2165 cm−1 in the dianion, whereas in B3-3TQ, the ν(CN) infrared band is at 2216 cm−1 which moves up to 2177 cm−1 in the radical anion and to 2165 cm−1 in the dianion. The evolution of the wavenumber values in all neutrals, radical anions and dianions of all tetracyano compounds in this work is presented in Fig. 6. The Δν(CN) is a very sensitive measure of the charge excess installed in the cyano groups upon reduction39 with the prototypical example of tetracyanoethylene (i.e., TCNE in Fig. 1) and tetracyanoquinodimethane (i.e., TCNQ) whose Δν(CN) is also shown in Fig. 6. For the smaller TCNE, the Δν(CN) values from the neutral to the anion radical and from this to the dianion are similar, spectroscopic changes in line with the charge of the radical anion of TCNE to be fully delocalized among the four cyano groups (i.e., approx. 1/4 of the charge on each cyano in the anion). In its dianion, driven by electrostatic repulsion, one charge is on each dicyanomethylene group (i.e., 1/2 of charge on each cyano). As a result, the addition of one electron on neutral → anion and on anion → dianion generates similar changes in the ν(CN) bands (each cyano group doubles its charge in each reduction step). For TCNQ, the Δν(CN) from the neutral (at 2223 cm−1) to the anion radical (at 2180 cm−1), by 43 cm−1, is greater than that from the anion to the dianion (at 2152 cm−1), by 28 cm−1. The larger shift in the neutral → anion step might reveal that the distribution of the charge in the TCNQ anion differs from that of the anion radical of TCNE. Furthermore, the infrared spectra of the anionic species of 2TQ have been obtained and shown in Fig. S5 (ESI†) which, for the radical anion, display a main ν(CN) band at 2177 cm−1 (neutral one at 2216 cm−1) disclosing a Δν(CN) in this first step of 39 cm−1. Going to the dianion, the ν(CN) band of 2TQ moves to 2159 cm−1 yielding a Δν(CN) value of 18 cm−1 reinforcing the dissymmetry of the electron distribution in the two reduction steps.
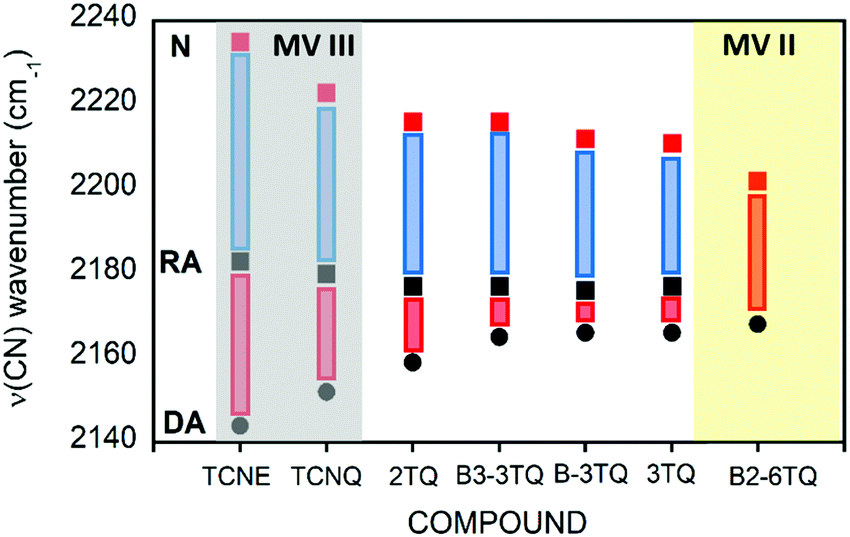 | ||
| Fig. 6 Representation of the values of the ν(CN) bands from the infrared spectra of the neutrals (red squares), radical anions (black squares) and dianions (black circles) of the tetracyano compounds studied in this work. The blue and red bars display the Δν(CN) values in the two reduction steps. | ||
The radical anions of the thienoquinoidal trimers display (i) very similar Δν(CN) shifts from the neutral to the anion: 38 cm−1 in 3TQ, 35 cm−1 in B-3TQ and 39 cm−1 in B3-3TQ indicating similar affectation of the CN moieties upon single charging; and (ii) from the anion to the dianion, the shifts are much smaller than in the previous reduction step with Δν(CN) of 7 cm−1 in 3TQ and 12 cm−1 in B-3TQ and B3-3TQ. From the point of view of the Δν(CN) values, the way to conciliate these findings in the thienoquinoidal compounds is assuming a transition from class III full delocalized to class II localized mixed valence species. A class II localized situation would confine the charge around one of the dicyanomethylene terminals by which the Δν(CN) of the first reduction step would be similar in all thienoquinoidal cases irrespective of the size/nature of the bridge. In addition, from such localized state post-reduction to the dianion would impart smaller variations. Hence, according to the infrared spectra, the radical anions of B-3TQ and B3-3TQ are closer to the class II species with confinement of the charge on the dicyano groups. In the case of 3TQ, the infrared data discussion places this as a class II compound whereas the interpretation of the UV-Vis-NIR absorption features revealed this as class III. As a result, 3TQ could behave more in the transition region between classes III and II.
A tentative reason for the stabilization of the class II form at the expense of the class III structure in the anions of B-3TQ and B3-3TQ can be related with the significant reduction of the bond length alternation (i.e., BLA) pattern between the consecutive CC bonds of the π-conjugated core on passing from the neutral to the radical anions. The proximity of the structure to a vanishing BLA prepares this to undergo pseudo Jahn–Teller (p-JT) distortion towards an asymmetric structure with increasing BLA. This p-JT effect is often acting together with solvation effects meaning the formation of a structure with the solvated charge mainly around one of the two dicyanomethylene parts or localized class II form, as described in Scheme 2. Also in agreement with this vision is the two-electron transfer process observed in the cyclic voltammetry experiment of the thienoquinoidal trimers which reveals the introduction of two electrons at potentials below the electrochemical threshold in line with each one somehow entering in independent moieties.
 | ||
| Scheme 2 Representation of the evolution of the 3TQ structure upon reduction/oxidation. In the radical anion, specific solvation (solvent molecules are denoted as shaded discs) of the charge and p-JT might justify the existence of an equilibrium of the full delocalized and localized forms. | ||
 | ||
| Scheme 3 Comparison between delocalized/localized cyanines and delocalized/localized forms of the radical anion of 3TQ. | ||
The data discussed in the radical anions of the tetracyano thienoquinoidal trimers establish a parallelism with the behavior of cyanines. The similitude between the radical anions of the tetracyano quinoidal compounds and the cyanines are (i) both disclose an odd number of π-electrons in conjugation; (ii) whereas the amine/imine pair of the nitrogen functionalization produces the transmission of the cation along the polymethine path, in the quinoidal compounds, the cyano/imine character of the CN groups imparts the conjugation effect of the negative charge in the radical anions; and (iii) the absorption spectra evolve from intense well resolved NIR bands in the smaller bridge compounds (see the spectra of the radical anion of 2TQ in Fig. S5, ESI† and 3TQ in Fig. 3) towards broader less intense bands in the radical anions of the benzoannulated quinoid trimers. This comparison agrees with the existence of a transition between the delocalized and localized states in the radical anions of the quinoidal trimers, an evolution that will be completed in the case of the longer oligomer, such as the case of the tetracyano thienoquinoidal hexamer that will be discussed in Section III.2.5.
 | ||
| Scheme 4 Chemical structure of the quinoidal 3TQ in comparison with that of its aromatic counterpart, 3TA (Bu: n-butyl), a prototypical oligothiophene able to form π-dimeric dications. | ||
One way to explore the presence and formation of π-dimers is by addressing the evolution of the electronic absorption spectra in solution on cooling as the aggregation reaction is thermodynamically favored by reduction of the entropic term by lowering the temperature. In this situation, the spectroscopic fingerprint of the π-dimer formation should then appear, which, in the case of the terthiophene dication π-dimer of 3TA,47 is characterized by new bands at 1012 nm and 590 nm that grow up progressively on cooling at the expense of the 1106 nm and 654 nm features of the individual radical cations of 3TA at room temperature.
The radical anion of 3TQ has been obtained by chemical reduction and low temperature spectroscopic measurements of its solution carried out such as shown in Fig. 7. At room temperature, the spectrum of the radical anion of 3TQ features 675 and 1570 nm bands. In the initial steps of the cooling, these bands of the individual molecule increase their intensity up to 210 K upon which a new pair of bands at 680 nm and more notably at 1001 nm rises isosbestically. This behavior is very similar to that described for the radical cation of 3TA47 and the similitude of the wavelength position of the NIR bands of low temperature species of both the radical anion of 3TQ (1001 nm) and the radical cation of 3TA (1012 nm) is certainly indicative that similar species have been generated on cooling. This situation suggests the formation of a π-dimer dianion from two radical anions of 3TQ which, for the first time, is described for the cathodic species of thienoquinoidal molecules. The same experiment has been carried out for the radical anion of B-3TQ in Fig. 7; upon cooling, its electronic absorption spectra evolve with some similarity to the case of 3TQ with the intensification of a new band at 820 nm which emerges isosbestically from the band at 938 nm of the radical anion at room temperature. This might reveal that B-3TQ also shows propensity to form π-dimer dianions.
 | ||
| Fig. 7 UV-Vis-NIR absorption spectra of the radical anions of B3-3TQ (left) and of 3TQ (right) from 298 K to 210 K (blue shaded spectrum) in CH2Cl2. | ||
The formation of π-dimer dianions from the radical anions of the thienoquinoidal compounds is indicating that the charge in the individual ions is distributed over the whole π-conjugated skeleton which is the preferred form to accomplish efficient multi-bonding center coupling to stabilize the aggregated structure. This description is more consistent with a full delocalized class III mixed valence system rather than a class II in which the confinement of the charge would make the intermolecular interaction less favorable. This discussion for 3TQ is in agreement with the description that its radical anion behaves in the class II/III mixed valence frontier and corroborates how external stimuli could move the charge in the direction of delocalization or confinement depending on the perturbation.
III.3. Electronic structure of tetracyanoquinoidal hexathiophene
Extension of the π-conjugated structure from thienoquinoidal trimers to a thienoquinoidal hexamer (i.e., B2-6TQ in Fig. 1) introduces the stabilization of an open-shell diradical ground electronic state as previously reported for a similar quinoidal hexamer.8,16In Fig. 8, DFT/UB3LYP/6-31G** calculations of the complete series of neutral tetracyano thienoquinoidal compounds are shown calculated either as closed or open-shell singlets. Two series consisting of unsubstituted quinoidal thiophenes (nTQ) and all benzothienoquinoidals (Bn-nTQ) from a monomer to heptamer (n = 1–7) have been calculated. We find that for the nTQ series starting from the pentamer the open-shell singlet diradical is the most stable situation for the ground electronic state with a very reduced singlet–triplet gap of <2 kcal mol−1. Conversely, substitution of the thiophenes with benzene groups in benzothienoquinoidal units destabilizes the open-shell form and all the calculated systems become singlet closed-shell ground electronic states. Within these two extremes, B2-6TQ should display a situation between the two hexamers. In particular, calculations for B2-6TQ disclose a theoretical singlet–triplet gap of 4.2 kcal mol−1 and a diradical index52y0 = 0.142 (i.e., y0 varies from y0 = 0 for closed-shell molecules to y0 = 1 for completely formed diradicals) revealing an incipient diradical character for B2-6TQ. On the other hand, it is clear that all the above studied thienoquinoidal trimers disclose a closed-shell configuration in their ground electronic state.
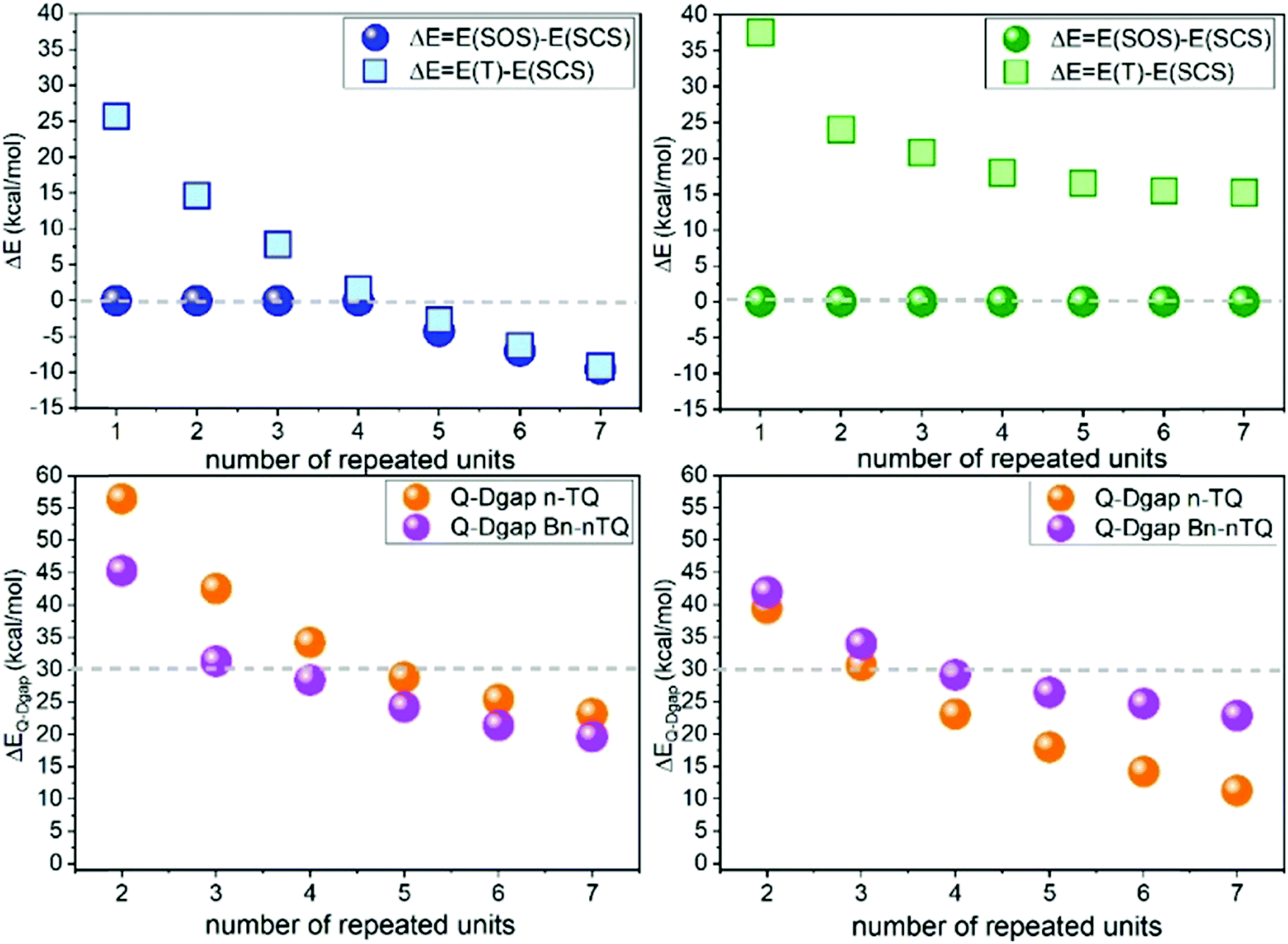 | ||
| Fig. 8 Top: DFT/(U)B3LYP calculations of the energy differences (ΔE) between the singlet closed (SCS) or open-shell (SOS) ground electronic states and the first excited state triplets (T) in the nTQ (left) and Bn-nTQ (right) series. Bottom: DFT/UB3LYP calculations of the energy differences between the ground electronic state doublet (D) and first quarter excited state (Q) of the radical anions (left) and cations (right) of the same series. | ||
The electronic absorption spectrum of B2-6TQ in Fig. 9 is constituted by a broad band with the peak maximum at 978 nm which is due to the electric dipole allowed S0 → S1 excitation (as in the trimer compounds). This band displays a vibronic structure with shoulders at high and lower energies. However, by deconvolution (Table S1, ESI†), we obtain four components (Fig. 9) which compared with the pattern of three vibronic peaks on the trimers suggests that the deconvoluted peaks at 890, 974 and 1037 nm might belong to the S0 → S1 band, whereas the additional peak red-shifted at 1207 nm might arise from the S0 → S2 excitation described as a dark state in the previous sections. Contrarily to the other quinoidal compounds, this S0 → S2 band emerges in the electronic absorption spectrum of B2-6TQ as a result of the diradical character imprinted in its ground electronic state. The appearance of this two-photon absorption band in the one photon optical spectrum as a weak feature is the confirmation of its open-shell diradical character.
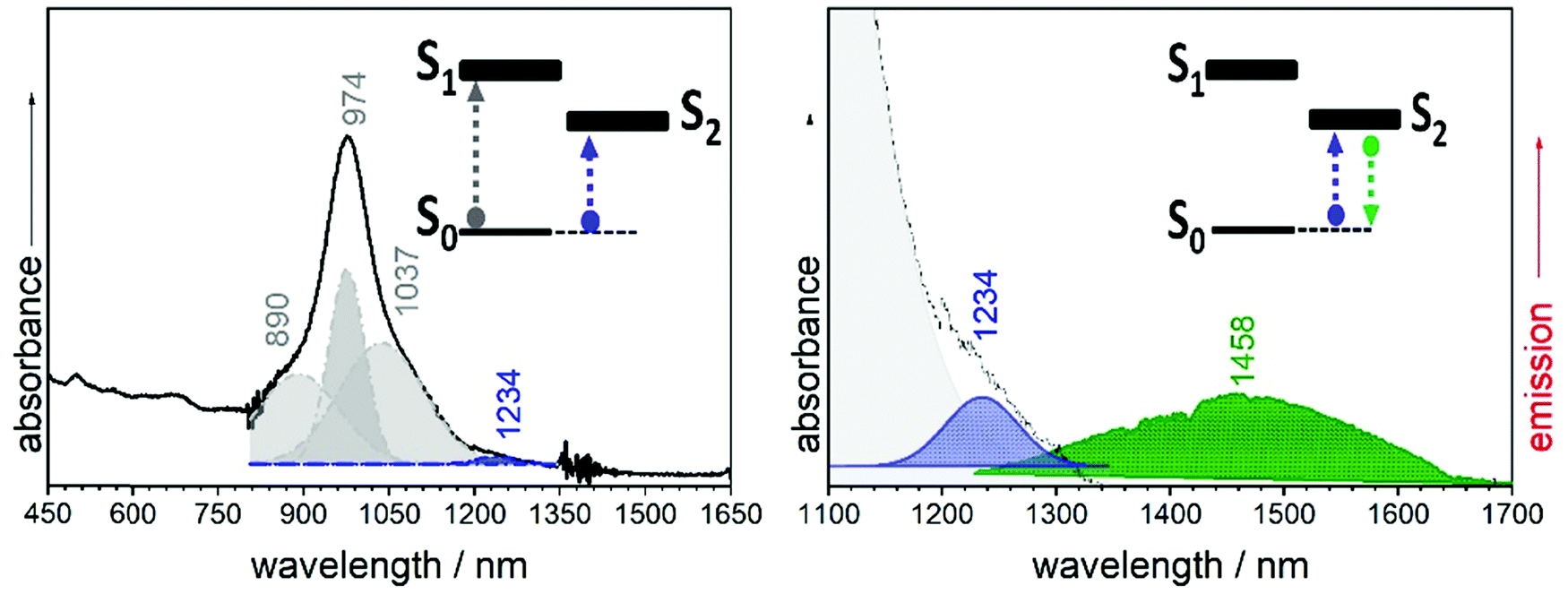 | ||
| Fig. 9 Left: UV-Vis-NIR absorption spectrum (black line) of B2-6TQ with its deconvoluted components (grey and blue shaded). Right: 1064 nm emission spectrum (green shaded) of B2-6TQ at room temperature. | ||
In Fig. 9 the emission spectrum of B2-6TQ is also shown. By excitation at 785 nm of the strongest absorption band, no emission spectrum is recorded, a situation that is reversed by excitation at longer wavelengths in the S0 → S2 band (blue shaded band in Fig. 9) which gives rise to a weak NIR emission with a maximum at 1458 nm assigned as the S2 → S0 emission from the dark excited state. Therefore, a difference with the quinoidal trimers is that in B2-6TQ we detect the S0 → S2/S2 → S0 absorption/emission pair. As shown in Fig. 2, the larger energy difference between the S2/S1 excited states in B2-6TQ reduces internal conversion between the two; hence, the low energy emission only appears upon excitation on its own absorption band.
III.4. Electronic structure of the charged species of B2-6TQ
Similar to the quinoidal trimers, B2-6TQ displays a two-electron reduction wave. However, contrarily to the case of the smaller size homologues, in the spectroelectrochemical experiment, the spectra of radical anions could not be resolved neither in the UV-Vis-NIR nor in the infrared scanning, and only one species due to the dianion appears in the whole cathodic potential range. The electronic absorption spectrum of the formed dianion species in Fig. 10 displays a band with a maximum at 571 nm (no NIR bands) in line with the 583 nm band in the dianion of B-3TQ. In the infrared spectroelectrochemical experiment, the ν(CN) band of the dianion is recorded at 2168 cm−1 which directly evolves from the ν(CN) band in the neutral at 2202 cm−1 meaning a change, Δν(CN), upon incorporation of two charges of 34 cm−1, a variation that is similar to that found in the conversion from the neutral to the anion radical of the thienoquinoidal trimers. In agreement with the above discussion, this comparison corroborates the localization of the charge in the environment of one dicyanomethylene group of the molecule in the monocharged trimers being a localized structure with one charge on one terminal group similar to that of the dianion of B2-6TQ which features one charge at one terminal group.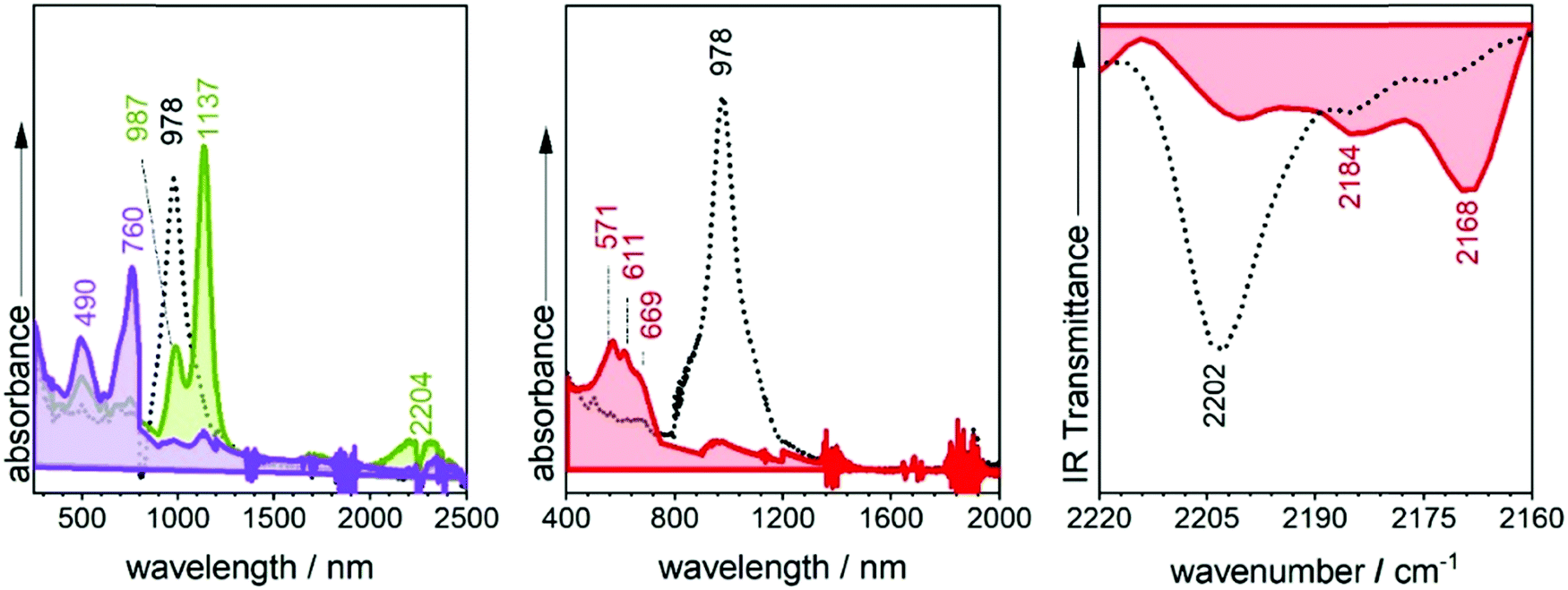 | ||
| Fig. 10 UV-Vis-NIR electronic absorption spectra of B2-6TQ taken during the spectroelectrochemical oxidation (left, radical cation green shaded and dication purple shaded) and reduction (middle, dianion red shaded) in 0.1 M TBPF6 in CH2Cl2 at room temperature, together with the infrared spectra of B2-6TQ (right, dianion, red shaded) taken under the same spectroelectrochemical reduction conditions. | ||
Finally, B2-6TQ can be oxidized to the radical cation and dication in two reversible one-electron oxidation processes. The formation of these two species has been followed by UV-Vis-NIR spectroelectrochemistry in Fig. 10 as well. The formation of the radical cation gives way to the appearance of a new strong and narrow band at 1137 nm together with a new weak component at 2204 nm. Contrarily to the formation of a radical cation in the homologue thienoquinoidal trimers (i.e., formed by oxidation of a closed shell structure), oxidation of B2-6TQ proceeds through electron extraction in a pseudo-aromatic thienyl core (aromatization generated by diradical stabilization). This radical cation can be stabilized in two different forms, either as a quartet ground electronic state (three unpaired electrons, two unpaired electrons already in the neutral plus one more upon oxidation) or as a ground state doublet (one unpaired electron, the other two being paired), as described in Scheme 5. The relative stability of these configurations for the radical cation of B2-6TQ has been explored by quantum chemical calculations in Fig. 8.
 | ||
| Scheme 5 Representation of the evolution of the B2-6TQ structure upon reduction/oxidation and upon formation of the radical cation (transiting between a triradical or a radical form). | ||
It is observed that the radical cations of the smaller tetracyanoquinoidal compounds with closed-shell structures in the neutral state disclose a doublet ground electronic state generated upon electron extraction from the doubly occupied HOMO orbital (i.e., data are analyzed up to heptamers) with a first quartet high spin state always at very high energies. The energy difference between the doublet and the quartet excited state is progressively decreasing displaying a saturation effect already for the longer oligomers. The objective of this analysis was to explore the possibility to find a situation of HOMO–SOMO energy level inversion in the neutral compounds. The existence of neutral radical molecules showing HOMO–SOMO energy inversion53,54 is now of relevance as it has been shown that these electronic configurations produce a series of benefits in the application in organic electronic devices. In the case of the neutral radical molecules, a manifestation of the SOMO–HOMO energy inversion is that oxidation of the neutral radical produces a triplet ground electronic state. In the attempt to investigate this behavior in neutral diradicals (such as B2-6TQ), we inspect the stabilization of the quartet ground electronic state in the oxidized radical cations as a way to confirm the SOMO–HOMO inversion effect in the starting neutral diradicals. As shown in Fig. 8, the doublet configuration is always the preferred configuration of the radical cations of the whole quinoidal series; hence, the newly generated radical, despite being stabilized in the central part of the molecule, ferromagnetically interacts with the radicals at both sides. In principle, the enlargement of the central thiophenic spine does not produce the doublet → quartet reversal (i.e., such as it would be expected for SOMO–HOMO inversion in the neutral) and, as stated, the behavior tends towards a saturation effect. The same study, as a reference, has been done for the radical anions, and Fig. 8 also discloses the energy differences between the doublet and quartet configurations. In the two series of radical anions analyzed, the stabilization of the doublet state regarding the quartet one is larger than for the same states in the series of radical cations.
III.5. Comparison with Veciana and Rovira contributions
These π-conjugated molecules refer to several systems reported by Veciana and Rovira's group based on para-substituted thiophene-vinylene cores functionalized with perchlorinated triphenylene methanes (i.e., PTM), as displayed in Scheme 6.55–57 | ||
| Scheme 6 Chemical structures of the Veciana and Rovira compounds with diradical character (PTM-nTV-PTM, with n = 1–6) and the PTM tetrathiafulvalene neutral radical, PTM-TTF. | ||
The analogies with our studied compounds rely on their thiophenic nature, on the diradical character and on the stabilization of the radical centers by surrounding them with electron acceptor groups. PTM-nTV-PTM compounds display reversible reductions to the radical anion and dianion. In the case of the monovalent species, these are formally mixed valence systems which are reported to be class III delocalized systems in the case of PTM-1TV-PTM and PTM-2TV-PTM whereas for PTM-4TV-PTM and longer, all are class II/I localized mixed valence systems. There is a correlation between the behavior of B2-6TQ and that of the longer PTM derivatives: (i) both disclose diradical characters in the neutral state and give rise to localized radical anion systems. And (ii) the longer PTM-nTV-PTM compounds also display reversible electron oxidations, such as for example B2-6TQ, and give rise to doublet ground electronic states due to coupling of the thiophene-centered radical cation with those unpaired electrons in the PTM terminals. Contrarily to these cases, the PTM-TTF neutral radical does display SOMO–HOMO inversion (yielding triplet species upon one electron oxidation), a situation that is evaded in the medium-large size elements of the quinoidal diradicals and in the PTM-nTV-PTM compounds.
IV. Conclusions
In this paper, tetracyano thienoquinoidal compounds of different sizes and chemical compositions have been studied, and their features have been understood in the context of the properties of the well-known oligoenes (in the case of the neutral quinoidal compounds) and of the behavior of cyanines (in the case of the charged anionic states). The optical properties of these compounds in the neutral state are controlled by the energy proximity and relative energy disposition of the bright (electric dipole–dipole allowed) and dark (electric dipole–dipole forbidden) singlet excited states. Interestingly, the emission features of the quinoidal oligothiophenes fall in the near infrared spectral region which is an uncommon finding in organic molecules for NIR emitters. In the anionic state, the trimeric compounds disclose behaviors compatible with being either charge localized or delocalized mixed valence systems and can be catalogued as systems in the transition region between these two regimes of charge distribution.The hexamer compound discloses incipient diradical character which permits the detection of the NIR emission at energies well below 1 eV. While benzoannulation of the thiophene modulates the diradical character, in parallel, it forces the confinement of the charge in the vicinity of the terminal dicyanomethylenes in the radical anions and dianions having these charge localized character. The oxidized species of the hexamer correspond with systems with charge localization in the molecular center. In the radical cation, electron extraction generates another radical in the molecular core which together with the flanking radicals (i.e., already present in the neutral species) composes a triradical state which is proven to have a doublet ground electronic state. We show that by increasing the diradical character of the starting neutrals, the doublet–quartet energy gap in the radical cations is reduced but this gap is not reversed among the studied compounds.
This paper represents a research study based on spectroscopic data oriented to the elucidation of the electronic structure of organic quinoidal π-conjugated molecules of interest in organic electronic applications and in materials chemistry. The aspects of the electronic structure of the reduced species of quinoidal thienoquinoidal compounds revealed in this article might be of relevance to direct future chemical designs for n-channel organic field effect transistors and related devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Spanish Ministry of Science, Innovation and Universities MCIU (PGC2018-098533-B-100 and RED2018-102815-T), and the Junta de Andalucía, Spain (UMA18FEDERJA057). We also thank the Research Central Services (SCAI) of the University of Málaga.Notes and references
- (a) A. Mishra, C. Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141 Search PubMed; (b) I. F. Perepichka and D. F. Perepichka, Handbook of Thiophene-based Materials, Wiley VCH, Weinheim, 2009 Search PubMed.
- N. S. Zhang, B. P. Colella, S. C. Cherniawski, B. Mannsfeld and A. L. Briseno, ACS Appl. Mater. Interfaces, 2014, 6, 5327 Search PubMed.
- H. E. Katz, L. Torsi and A. Dodabalapur, Chem. Mater., 1995, 7, 2235 Search PubMed; A. Dodabalapur, L. Torsi and H. E. Katz, Science, 1995, 268, 270 Search PubMed.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99 Search PubMed; H. Sirringhaus, Adv. Mater., 2014, 26, 1319 Search PubMed.
- K. Schulten and M. Karplus, Chem. Phys. Lett., 1972, 14, 305 Search PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891 Search PubMed.
- D. Casanova, Chem. Rev., 2018, 118, 7164 Search PubMed.
- T. Takahashi, K. Matsuoka, K. Takimiya, T. Otsubo and Y. Aso, J. Am. Chem. Soc., 2005, 127, 8928 Search PubMed.
- S. Di Motta, F. Negri, D. Fazzi, C. Castiglioni and E. V. Canesi, J. Phys. Chem. Lett., 2011, 1, 3334 Search PubMed.
- L. Ren, F. Liu, X. Shen, C. Zhang, Y. Yi and X. Zhu, J. Am. Chem. Soc., 2015, 137, 11294 Search PubMed.
- C. Zhang and X. Zhu, Adv. Funct. Mater., 2020, 30, 765 Search PubMed.
- J. E. Raymond, J. Casado, J. T. Lopez Navarrete, K. Takimiya and T. Goodson III, J. Phys. Chem. Lett., 2011, 2, 2179 Search PubMed.
- O. Varnavski, N. Abeyasinghe, J. Arago, J. J. Serrano-Perez, E. Orti, J. T. Lopez-Navarrete, K. Takimiya, D. Casanova, J. Casado and T. Goodson III, High Yield Ultrafast Intramolecular Singlet Exciton Fission in a Quinoidal Bithiophene, J. Phys. Chem. Lett., 2015, 6, 1375–1384 Search PubMed.
- K. H. Kim, B. Keller, R. Ho-Wu, N. Abeyasinghe, R. J. Vázquez, T. Goodson III and P. M. Zimmerman, J. Am. Chem. Soc., 2018, 140, 7760 Search PubMed.
- J. Casado, L. L. Miller, K. T. R. Mann, T. M. Pappenfus, H. Higuchi, E. Ortí, B. Milián, R. Pou-Amérigo, V. Hernández and J. T. López Navarrete, J. Am. Chem. Soc., 2002, 124, 12380 Search PubMed.
- R. P. Ortiz, J. Casado, S. R. González, V. Holín, J. T. López-Navarrete, M. Viruela, E. Ortí, K. Takimiya and T. Otsubo, Chem. – Eur. J., 2010, 16, 470 Search PubMed.
- Y. Suzuki, E. Miyazaki and K. Takimiya, J. Am. Chem. Soc., 2010, 132, 10453 Search PubMed.
- J. C. Ribierre, S. Watanabe, M. Matsumoto, T. Muto, D. Hashizume and T. Aoyama, J. Phys. Chem. C, 2011, 115, 20703 Search PubMed.
- D. Yuan, D. Huang, S. Medina Rivero, A. Carreras, C. Zhang, Y. Zou, X. Jiao, C. R. McNeill, X. Zhu, C. Di, D. Zhu, D. Casanova and J. Casado, Chem, 2019, 5, 964–976 Search PubMed.
- T. Izumi, S. Kobashi, K. Takimiya, Y. Aso and T. Otsubo, J. Am. Chem. Soc., 2003, 125, 5286 Search PubMed.
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336 Search PubMed.
- Y. Zheng, M. Miao, G. Dantelle, N. D. Eisenmenger, G. Wu, I. Yavuz, M. L. Chabinyc, K. N. Houk and F. Wudl, Adv. Mater., 2015, 27, 1718 Search PubMed.
- R. Kishi, Y. Murata, M. Saito, K. Morita, M. Abe and M. Nakano, J. Phys. Chem. A, 2014, 118, 10837 Search PubMed.
- R. Kishi, M. Dennis, K. Fukuda, Y. Murata, K. Morita, H. Uenaka and M. Nakano, J. Phys. Chem. C, 2013, 117, 21498 Search PubMed.
- K. Yamamoto, Y. Ie, M. Nitani, N. Tohnai, F. Kakiuchi, K. Zhang, W. Pisula, K. Asadi, P. W. M. Blom and Y. Aso, J. Mater. Chem. C, 2018, 6, 7493 Search PubMed.
- K. Yamamoto, S. Jinnai, T. Takehara, T. Suzuki and Y. Ie, Org. Lett., 2020, 22, 547 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 Search PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 Search PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997 Search PubMed.
- H. H. Heinze, A. Gorling and N. Rosch, J. Chem. Phys., 2000, 113, 2088 Search PubMed.
- M. B. Robin and P. Day, Mixed Valence Chemistry. A survey and classification, Academic Press, 1968, vol. 10, p. 247 Search PubMed.
- N. S. Hush, Coord. Chem. Rev., 1985, 64, 135 Search PubMed.
- A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326 Search PubMed.
- J. S. Miller, Angew. Chem., Int. Ed., 2006, 45, 2508 Search PubMed.
- E. E. Havinga, J. L. van Donger, R. A. J. Janssen, J. Cornil and J.-L. Brédas, Chem. – Eur. J., 1998, 4, 1509 Search PubMed.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18(10), 309 Search PubMed.
- K. Yui, H. Ishida, Y. Aso, T. Otsubo, F. Ogura, A. Kawamoto and J. Tanaka, Bull. Chem. Soc. Jpn., 1989, 62, 1547 Search PubMed.
- J. S. Chappell, A. N. Bloch, W. A. Bryden, M. Maxfield, T. O. Poehler and D. O. Cowan, J. Am. Chem. Soc., 1981, 103, 2442 Search PubMed.
- S. Ohira, J. M. Hales, K. J. Thorley, H. L. Anderson-Joseph, W. Perry and J. L. Brédas, J. Am. Chem. Soc., 2009, 131, 6099 Search PubMed.
- P. A. Bouit, C. Aronica, L. C. Toupet, B. Le Guennic, C. Andraud and O. Maury, J. Am. Chem. Soc., 2010, 132, 4328 Search PubMed.
- L. M. Tolbert and X. Zhao, J. Am. Chem. Soc., 1997, 119, 3253 Search PubMed.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192 Search PubMed.
- J. L. Zafra, L. Qiu, N. Yanai, T. Mori, M. Nakano, M. P. Alvarez, J. T. López Navarrete, C. J. Gómez-García, M. Kertesz, K. Takimiya and J. Casado, Angew. Chem., Int. Ed., 2016, 55, 14563 Search PubMed.
- L. L. Miller and K. R. Mann, Acc. Chem. Res., 1996, 29, 417 Search PubMed.
- J. C. Evans, A. G. Evans, N. H. Nouri-Sorkhabi, A. Y. Obaid and C. C. Rowlands, J. Chem. Soc., Perkin Trans. 2, 1985, 315–318 Search PubMed.
- D. D. Graf, J. P. Campbell, L. L. Miller and K. R. Mann, J. Am. Chem. Soc., 1996, 118, 5480 Search PubMed.
- A. Harbuzaru, I. Arrechea-Marcos, A. D. Scaccabarozzi, Y. Wang, X. Guo, M. Caironi, J. T. López-Navarrete, M. C. R. Delgado and R. P. Ortiz, J. Mater. Chem. C, 2020, 8, 15759 Search PubMed.
- J. S. Miller and J. J. Novoa, Acc. Chem. Res., 2007, 40, 189 Search PubMed.
- J. J. Novoa, P. Lafuente, R. E. D. Sesto and J. S. Miller, Angew. Chem., Int. Ed., 2001, 113, 2608 Search PubMed.
- J. Casado, P. Mayorga Burrezo, F. J. Ramírez, J. T. López Navarrete, S. H. Lapidus, P. W. Stephens, H. L. Vo, J. S. Miller, F. Mota and J. J. Novoa, Angew. Chem., Int. Ed., 2013, 52, 6421 Search PubMed.
- K. Yamaguchi, Chem. Phys. Lett., 1975, 33, 330 Search PubMed.
- Y. Wang, H. Zhang, M. Pink, A. Olankitwanit, S. Rajca and A. Rajca, J. Am. Chem. Soc., 2016, 138, 7298 Search PubMed.
- A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Nat. Mater., 2020, 19, 1224 Search PubMed.
- C. Franco, P. M. Burrezo, V. Lloveras, R. Caballero, I. Alcón, S. T. Bromley, M. Mas-Torrent, F. Langa, J. T. López-Navarrete, C. Rovira, J. Casado and J. Veciana, J. Am. Chem. Soc., 2017, 139(2), 686 Search PubMed.
- J. Guasch, L. Grisanti, V. Lloveras, J. Vidal-Gancedo, M. Souto, D. C. Morales, M. Vilaseca, C. Sissa, A. Painelli, I. Ratera, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2012, 51, 11024 Search PubMed.
- M. Souto, J. Guasch, V. Lloveras, P. Mayorga, J. T. Lopez-Navarrete, J. Casado, I. Ratera, C. Rovira, A. Painelli and J. Veciana, J. Phys. Chem. Lett., 2013, 4, 2721 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc01436f |
| This journal is © The Royal Society of Chemistry 2021 |