
Operation mechanism of organic electrochemical transistors as redox chemical transducers†
Siew Ting Melissa
Tan
 a,
Scott
Keene
b,
Alexander
Giovannitti
a,
Armantas
Melianas
a,
Maximilian
Moser
c,
Iain
McCulloch
cd and
Alberto
Salleo
*a
a,
Scott
Keene
b,
Alexander
Giovannitti
a,
Armantas
Melianas
a,
Maximilian
Moser
c,
Iain
McCulloch
cd and
Alberto
Salleo
*a
aDepartment of Materials Science and Engineering, Stanford University, California 94305, USA. E-mail: asalleo@stanford.edu
bElectrical Engineering Division, Department of Engineering, University of Cambridge, Cambridge, UK
cDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Oxford, OX1 3TA, UK
dPhysical Science and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
First published on 2nd August 2021
Abstract
The ability to control the charge density of organic mixed ionic electronic conductors (OMIECs) via reactions with redox-active analytes has enabled applications as electrochemical redox sensors. Their charge density-dependent conductivity can additionally be tuned via charge injection from electrodes, for instance in organic electrochemical transistors (OECTs), where volumetric charging of the OMIEC channel enables excellent transconductance and amplification of low potentials. Recent efforts have combined the chemical detection with the transistor function of OECTs to achieve compact electrochemical sensors. However, these sensors often fall short of the expected amplification performance of OECTs. Here, we investigate the operation mechanism of various OECT architectures to deduce the design principles required to achieve reliable chemical detection and signal amplification. By utilizing a non-polarizable gate electrode and conducting the chemical reaction in a compartment separate from the OECT, the recently developed Reaction Cell OECT achieves reliable modulation of the OECT channel's charge density. This work demonstrates that systematic and rational design of OECT chemical sensors requires understanding the electrochemical processes that result in changes in the potential (charge density) of the channel, the underlying phenomenon behind amplification.
Introduction
Organic mixed ionic electronic conductors (OMIECs), commonly conjugated polymers, are promising materials for biosensors because they are well-suited for detecting chemical analytes, especially in aqueous media, due to their redox-activity as well as ionic and electronic conductivity.1 Charge transfer processes, via faradaic reactions with chemically reactive redox analytes or capacitive charge injection from conductive electrodes, change the charge density on their backbone and consequently the electrochemical potential of electrons. As with any semiconductor, charge density (related to the position of the Fermi level in the OMIEC) modulates electronic conductivity, which can be measured by applying a probing bias and measuring the ensuing current. Charge density also affects the OMIECs’ electronic charge mobility and volumetric capacitance, giving rise to second-order effects on the materials’ conductivity. Furthermore, side chain engineering enables the intercalation of charge compensating ions to permeate the bulk, facilitating operation in aqueous electrolytes and volumetric charging throughout the material.2–7 These multifunctional properties of OMIECs have enabled their applications in a variety of electrochemical devices such as sensor electrodes in 3-electrode electrochemical cells to detect redox-active analytes.8 Additionally, control of electronic conductivity with charge density (or, equivalently, potential) has enabled their usage as the channel material in organic electrochemical transistors (OECTs).9–15Operation of an OECT in the traditional sense is potentiometric – changes in potential difference between the gate electrode and the channel modulate the latter's charge density and hence conductivity. Conventionally, the source electrode is grounded and the gate potential is measured with respect to the source (VGS). The change in conductivity can be probed by application of a drain bias with respect to the source (VDS) across the channel and measuring changes in the drain current (ID). Transconductance  is the figure of merit that captures the sensitivity of channel conductivity in response to a change in its potential or, equivalently and perhaps more intuitively, its charge density. Due to the ability of the OMIEC channel to charge volumetrically, OECTs display exceptionally large transconductances, allowing deep modulations in channel conductivity to arise from small VGS modulations, thus amplifying minute potential perturbations.15–17
is the figure of merit that captures the sensitivity of channel conductivity in response to a change in its potential or, equivalently and perhaps more intuitively, its charge density. Due to the ability of the OMIEC channel to charge volumetrically, OECTs display exceptionally large transconductances, allowing deep modulations in channel conductivity to arise from small VGS modulations, thus amplifying minute potential perturbations.15–17
Recognizing that OMIECs can function as redox-active electrodes and as the channel material of OECT amplifiers, it is convenient to combine both functionalities within the same device to yield a compact amplifying sensor. Indeed, there have been multiple reports utilizing OECTs as chemical redox sensors, where the analyte is introduced directly into the OECT electrolyte while an external VGS is applied.18–21 However, this approach, as we22 and Macchia et al.23 have pointed out, merits further analysis. Firstly, the amplification of these reactions by the OECT is unclear. Indeed, all-in-one operation of an OECT sensor with a non-polarizable Ag/AgCl gate showed sub-unity current gain.22 Furthermore, conducting redox reactions within the electrolyte of the OECT in the presence of asymmetric electric fields (from both VGS and VDS) results in complex and site-specific side reactions on both the channel and gate as these parasitic redox reactions may be triggered only at specific voltages. These unknown parasitic reaction currents are superimposed onto the desired sensing current. Thus, while using the OECT as a sensor does provide a response to the analyte concentration, a deeper understanding of the device physics is needed in order to understand the complete nature of such response.
To fully leverage the excellent transconductance of the OECT and avoid parasitic side reactions, we proposed a different device architecture, a Reaction Cell OECT (RC-OECT),22 that first transduces the redox reaction into a potential change that subsequently gates the OECT. By conducting the redox reaction on a separate 2-electrode cell from the OECT, we avoid any side reactions within the OECT. This architecture enables the OECT to truly amplify the redox reaction in the RC, resulting in far larger modulations in OECT channel conductivity than the all-in-one approach.
We emphasize that our analysis pertains to sensing via faradaic redox chemical reactions, where sustained electron transfer occurs between species in the electrolyte and the device electrodes, and is hence distinct from capacitive potentiometric devices e.g. Ion-Selective FETs. Faradaic reactions in the RC enable the RC (and its corresponding gate circuit) to be self-powered, distinguishing it from other two-compartment device architectures that operate capacitively e.g. Floating Gate FETs.24,25
The RC-OECT may be one of many potential approaches to design redox chemical sensors that fully take advantage of the transconductance of the OECT. To enable further development of high-gain chemical transducers based on OECTs, it is useful to fundamentally understand the device physics of all OECT-based sensing approaches. To do so, we investigate the electrochemical mechanisms that enable changes in the potential (charge density) of the OECT channel, which is the underlying phenomena behind the OECT's amplification abilities.
Similar to our initial RC-OECT study, H2O2, which is reduced to H2O via the Hydrogen Peroxide Reduction Reaction (HPRR, H2O2 + 2H+ + 2e− → 2H2O), is utilized as the model redox-active species due to its ubiquity in electrochemical sensors, allowing it to be generalized to enzymatic sensors of common metabolites e.g. glucose that form H2O2 as by-products. We similarly utilize a homo-3,3′-dialkoxybithiophene polymer, p(g3T2),26 a p-type conjugated polymer with glycolated side-chains, as the model sensing OMIEC in the OECT channel and the RC anode since we found it to be readily oxidized by H2O2. This study delves into greater details of the electrochemical processes occurring at various electrodes while combining the operational understanding of transistors. We chose a model “analyte” and a model OMIEC, nevertheless our conclusions are general. We envision that this study will enable the systematic optimization of RC-OECT devices as well as spur further designs of potentiometric OECT-based chemical sensors.
Results
Traditionally, the most reliable sensing approach of faradaic redox reactions is operation in a 3-electrode electrochemical cell, where the OMIEC working electrode is deposited on a current collecting substrate and immersed in an electrolyte. The OMIEC's potential is measured against a standard reference electrode (e.g. Ag/AgCl in saturated KCl for aqueous electrolytes). While utilizing the true reference electrode results in reliable measurements, it is difficult to miniaturize and integrate into devices that are micro or nanofabricated. Hence, other electrochemical device architectures have been developed to enable the utilization of a “reference electrode” that is compatible with these fabrication techniques.The OECT is in fact one such alternative electrochemical device. Most commonly, an Ag/AgCl pellet is utilized as pseudo-reference electrode and simultaneously gate electrode for the OECT (Fig. 1(a)). To integrate the gate electrode with microfabricated devices, the Ag/AgCl pellet can be miniaturized by depositing Ag and partially reacting it with HCl to form Ag/AgCl. The pellet is not a true reference electrode because it is immersed in an OECT electrolyte where the Cl− concentration is different from that of the saturated electrode and in principle can vary, if for instance Cl− is absorbed in the p-type OMIEC to compensate for hole injection. Typically, one relies on maintaining a high Cl− concentration in the electrolyte to consider it approximately constant. Furthermore, the OECT is inherently a 2-electrode electrochemical cell comprising of the channel (working electrode) and the gate electrode (pseudo-reference), with the absence of a current-collecting counter electrode. Hence, gate currents flow through the gate electrode to compensate for the change in charge on the channel. In typical OECT operation, the device finds itself in the limit of small current flows (∼nA). Under these conditions the 3-phase equilibrium at the Ag/AgCl gate electrode is largely unperturbed, ensuring that the gate potential is fixed, and by virtue of applying a VGS, the OMIEC channel potential can be reliably determined as well. We note that channel potential is defined by the potentials on the source and drain contacts, with a gradient on the channel length defined by the applied VDS. Fig. 1(b) is a schematic of a typical transfer curve of such an OECT where the change in VGS (red rectangle) modulates the drain current (ΔID). Its characteristics can be approximately modelled by the following equation:13

 summarizes the channel geometry, where W, d, and L are the channel width, thickness, and length, respectively. VT is the threshold voltage of the OECT, related to the oxidation or reduction onset of the channel as well as the gate electrode's work function.
summarizes the channel geometry, where W, d, and L are the channel width, thickness, and length, respectively. VT is the threshold voltage of the OECT, related to the oxidation or reduction onset of the channel as well as the gate electrode's work function.
 | ||
| Fig. 1 Schematics of various electrochemical device architectures for redox analyte detection. (a) Standard organic electrochemical transistor with OMIEC as the channel between two source and drain contacts as well as an Ag/AgCl pellet pseudo-reference electrode as gate immersed in aqueous electrolyte. (b) Schematic of transfer curve of an OECT where a change in drain current (ΔID) is measured upon applying a change in gate voltage (ΔVGS). (c) Amperometric OECT with an Ag/AgCl pellet as gate electrode as well as addition of redox analyte to OECT electrolyte while applying a fixed VGS. (d) Transfer curve showing an increase in faradaic currents upon addition of redox analyte to OECT electrolyte (e) amperometric OECT with an OMIEC as gate electrode as well as addition of redox analyte to OECT electrolyte while applying a fixed VGS. (f) Schematic of shift in threshold voltage and change in transfer curve of A-OECT (OMIEC gate) after addition of redox analyte to OECT electrolyte. At the fixed VGS, a ΔID arises from the shift in threshold voltage. (g) Reaction Cell OECT where the redox analyte is added to an electrochemical cell separate from the OECT, resulting in a potential change (VRC) that is utilized to gate the OECT. (h) Transfer curve of OECT where ΔID is a result of the change in VGS = VRC. | ||
Instead of a pseudo-reference electrode, the gate electrode can be of the same material as the channel for ease in depositing and patterning the same material on both the gate and channel in a single deposition step.11,20 However, the lack of a reference electrode poses challenges in determining the channel's potential (and therefore charge density). The transistor's transfer characteristics – the value of VT in particular – are strongly dependent on the work function of the gate electrode.27 The Ag/AgCl gate electrode's work function is reasonably constant, the work function of an OMIEC gate electrode however may vary depending on its processing history and redox reactions with other species present in the electrolyte (e.g. molecular oxygen).28 Applying VGS only determines the potential difference between the gate and channel but does not control the potentials of either electrode (hence the position of the Fermi level) with respect to a reference. This leads to many challenges in operating an OECT with OMIEC gate electrodes.
In this study, we compare two commonly seen all-in-one designs in literature and the RC-OECT22 as redox chemical sensors. The first is an amperometric OECT with Ag/AgCl gate electrode (Fig. 1(c)). Due to the fixed potential of the gate and the fixed potential difference between gate and source (VGS), the potential on the channel is pinned, which prevents any permanent charge accumulation on the OMIEC channel backbone that otherwise would lead to amplification: every charge introduced in the channel must be swept out, otherwise its potential (and doping level) would change. Hence, changes to drain current equal the total reaction current due to current continuity within the device. It is noteworthy that these currents are potential-dependent (Fig. 1(d)). We note that this operation mode is fundamentally different from the traditional potentiometric operation of the device as an OECT. Alternatively, the gate electrode can be an OMIEC (Fig. 1(e)). As the gate electrode's work function (potential) is not fixed, the channel potential is no longer pinned and the energy of the electrons that could react in the channel is not controlled. Hence, charge transfer reactions on either the gate or channel lead to changes in their potentials, resulting in a shift in the OECT's threshold voltage, VT as the position of the Fermi level in the semiconductor is no longer fixed (Fig. 1(f)). Lastly, the reaction can instead be separated on a reaction cell where the change in potential across the 2-electrode cell, akin to a VGS, is used to gate the OECT using a non-polarizable gate electrode (Fig. 1(g)). The OECT is operated in a similar manner as Fig. 1(a and b) but the origin of VGS is now VRC, the reaction cell voltage (Fig. 1(h)). The following sections discuss the physical origins of the transfer curve behaviors and the respective merits of the various OECT architectures in Fig. 1(d, f and h).
Effect of gate material on OECT operation
We first investigate the operation mechanism of an OECT in the absence of any redox chemicals using different gate electrodes: a pseudo-reference electrode (Ag/AgCl) vs. an OMIEC (p(g3T2)). Utilizing Ag/AgCl as the gate electrode results in more stable OECT operation compared to an OMIEC because of the Ag(s)/AgCl(s)/Cl−(aq) 3-phase equilibrium. Hence, the potential of the Ag/AgCl gate electrode remains constant during regular OECT operation. By applying a fixed VGS, the channel potential and thus its charge density and conductivity can be measured reproducibly. This reproducible and deterministic OECT operation is readily verified experimentally (Fig. 2(b and c)).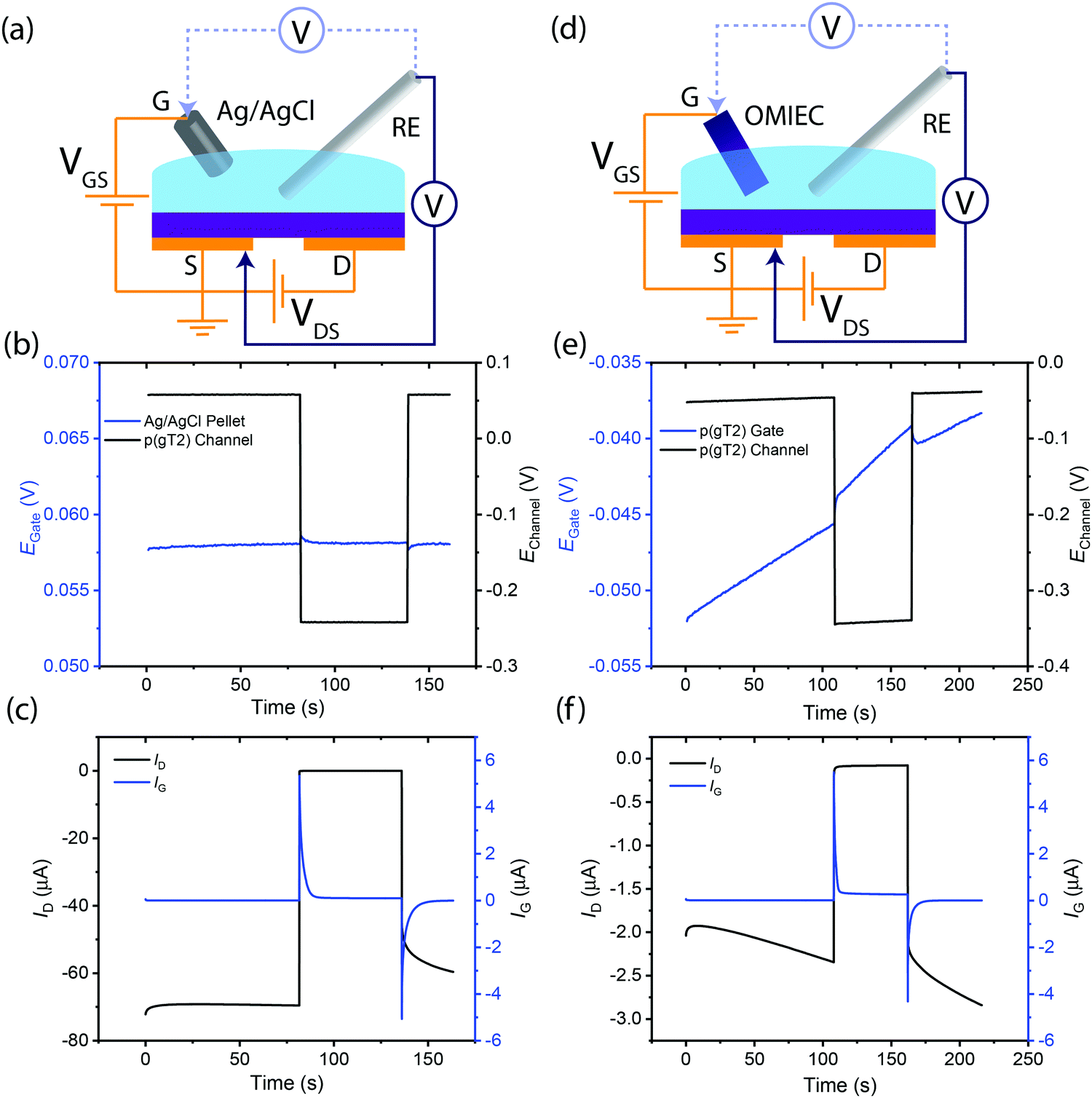 | ||
| Fig. 2 Potential and current stabilities of OECTs using Ag/AgCl vs. OMIEC gate electrodes upon pulsing VGS from 0 to +0.3 V. (a) Schematic of OECT with Ag/AgCl gate while conducting potential measurements of the Ag/AgCl gate and source of the channel. (b) Plot of gate (blue) and source (black) potentials in an OECT utilizing Ag/AgCl pellet as the gate electrode. (c) Plot of gate and drain current in an OECT utilizing Ag/AgCl pellet as the gate electrode. (d) Schematic of OECT with OMIEC gate while conducting potential measurements of the OMIEC gate and source of the channel. (e) Plot of gate (blue) and source (black) potentials in an OECT utilizing OMIEC as the gate electrode. (f) Plot of gate and drain current in an OECT utilizing OMIEC as the gate electrode. | ||
In contrast, utilizing an OMIEC gate electrode (Fig. 2(d)) of less well-defined potential may lead to challenges in fixing the potential of the channel. The potential of the OMIEC electrode can vary depending on the material's energy levels (which additionally determines its susceptibility to side reactions with ambient oxidants such as oxygen) as well as its processing history and microstructure. The observed drift in potential on the gate over time is likely due to possible side reactions with molecular oxygen dissolved in the electrolyte,28 which can be further exacerbated by the applied potentials in the OECT, resulting in the steady oxidation of the OMIEC gate. Hence, applying constant VGS = 0 V, the drift in gate potential is reflected in a concomitant drift in channel (source) potential (Fig. 2(e)). Due to the dependence of carrier mobility on charge density and hence potential, the drift in channel potential results in drain current drifts (Fig. 2(f)). We note that this instability may arise for any gate electrode material that is susceptible to parasitic redox side reactions, or any other electronic, chemical, or microstructural changes that may occur during device operation.
In summary, a gate electrode with a stable redox couple ensures that by applying a known VGS, the position of the Fermi level (i.e. the electrochemical potential of the electrons) in the channel can be reproducibly and definitively fixed.
Effect of faradaic chemical reactions in electrochemical cells
While utilizing a pseudo-reference gate electrode results in stable and reproducible performance, this all-in-one OECT architecture does not amplify chemical electron transfer reactions.22 After addition of H2O2 to the electrolyte, large gate currents occur at positive gate voltages VGS > +0.1 V in Fig. 3(a), corresponding to the accelerated rate of HPRR at negative channel potentials (ESI,† Fig. S1). However, ID (∼0.5 μA) is less than IG (∼1 μA) (Fig. 3(b)) due to current continuity illustrated in ESI,† Fig. S2(b). The sum of currents across the whole channel (from source to drain) must equal the gate current due to the lack of change in doping level of the channel, which prevents the formation of current sinks or sources. Hence, the change in drain current is a fraction of the total gate current, resulting in sub-unity current gains. | ||
| Fig. 3 Operation mechanism of Amperometric all-in-one OECTs comparing gate electrodes Ag/AgCl and OMIEC. (a–c) Ag/AgCl gate. (d–f) OMIEC gate (a) Transfer curve (black), gate currents (red) and transconductance (blue) of the amperometric OECT in its initial state (solid traces) and after addition of the redox analyte to the electrolyte (dashed). (b) Top: Plot of Ag/AgCl gate and source potentials vs. a saturated Ag/AgCl reference electrode with VGS = +0.3 V. Redox analyte (33 mM H2O2) is added to electrolyte at 60 s. Bottom: Corresponding plot of gate and drain currents. (c) Schematic of electron energy levels (defined as negative potentials, −E) in the Ag/AgCl pellet gate electrode (grey) and the valence band, Highest Occupied Molecular Orbital (HOMO), of the OMIEC channel (purple) at VGS = 0 V (left) and VGS = +0.3 V (right) as well as the energy level of electrons in the redox analyte (blue). Red arrows indicate the flow of electrons from the channel to the analyte in solution. (d) Transfer curve (black) and transconductance (blue) of the amperometric OECT with OMIEC gate in its initial state (solid traces) and after addition of the redox analyte to the electrolyte (dashed). (e) Top: Plot of OMIEC gate and source potentials vs. a saturated Ag/AgCl reference electrode with VGS = +0.3 V. Redox analyte (33 mM H2O2) is added to electrolyte at 60 s. Bottom: Corresponding plot of gate and drain currents. (f) Schematic of electron energy levels in the valence bands of the OMIEC gate electrode (left) and OMIEC channel (right) at VGS = +0.3 V as well as the energy level of electrons in the redox analyte. Before: upon addition of redox analyte to the electrolyte, red arrows indicate flow of electrons from OMIEC electrodes to redox analyte. After: the resulting oxidation of both the gate and channel results in the shift in potentials of both gate and channel by ΔVOxidation. | ||
Addition of H2O2 also results in small increase in potential ∼5 mV on both the gate and source (Fig. 3(b)). Ideally, the Ag/AgCl gate should not experience a potential shift due to its infinite capacitance. However, the large reaction current flowing through the gate (due to absence of a current collecting counter electrode) may have resulted in significant oxidation of Ag, changing the electrochemical potential of the gate electrode. The pinning of the channel potential to the gate potential by applying a fixed VGS results in the former mirroring the latter. This potential shift is small here, therefore its effect may not be very deleterious. Nevertheless, the shift will depend on the faradaic current, e.g. the analyte concentration, and can therefore in principle be much larger leading to the same problems outlined in the description of the OECT operated with an OMIEC gate electrode in the next section. Furthermore, the degradation of the pseudo-reference electrode calls into question the long-term stability of the device.
The magnitude of reaction currents may be controlled by the applied VGS. More positive VGS result in lower channel potentials and higher electron energies in the channel, increasing the driving force (kinetics) for electron transfer to the oxidant in the electrolyte (Fig. 3(c)). This presents a conundrum for the amperometric OECT. To ensure stable operation of the OECT, a pseudo-reference electrode is utilized as the gate. However, generating too large gate current from the redox reactions (∼1 μA) results in shifts of gate potential, which defeats the purpose of utilizing such an electrode. Hence, even if the OECT were to be optimized to maximize faradaic reaction currents from the analyte, e.g. by increasing VGS, it would result in deleterious effects on device stability. Furthermore, the faradaic current only emerges above the background current when the OECT is in its off-state as the on-current of an OECT is typically larger than a few μA, which restricts the VGS range of operation. Therefore, this OECT architecture not only does not amplify the reaction current, but it also performs more poorly than the simpler chronoamperometric setup where the absolute reaction current is measured.
While utilizing a non-polarizable Ag/AgCl electrode ensures operational stability, it fundamentally prevents changes in channel potential. Without the change in channel charge density (i.e. fixed hole concentration and doping level), there can be no change in channel conductivity and hence no amplification of the reaction current. This challenge may be resolved by utilizing a gate electrode with a tunable potential, such as an OMIEC. This material can be the same as that of the channel for fabrication convenience, but our analysis is independent of this choice. While this OECT design may exhibit less reliable performance due to the variability of the gate potential (Fig. 2(d–f)), it is worth investigating as this design has been widely explored as chemical sensors due to their ease in fabrication and their ability to amplify.
Identical measurements were conducted for this OECT with the only difference being the gate material – p(g3T2) instead of an Ag/AgCl pellet (ESI,† Fig. S2(c)). While our system involves both gate and channel participating in the oxidation reaction, the following arguments also apply to OECTs where the reaction is selectively limited to either electrode11 (e.g. biological catalysts selectively immobilized on one of the two electrodes). Due to the application of a fixed VGS, potential changes on the reactive electrode would result in similar potential changes on the other as well. Transfer curves obtained in the OECT's initial state and after addition of H2O2 (Fig. 3(d)) show a large difference after conducting redox reactions within the electrolyte. The shift in threshold voltage and maximum transconductance to more positive VGS provides clear evidence for oxidation of both the gate and channel. This observation is further supported by potential measurements on the gate and source electrodes (Fig. 3(e)), where potentials on both gate and source increase by ∼200 mV due to their redox-activity. Unlike the previous case where a non-polarizable gate was used, the channel potential, hence its charge state, is no longer pinned to a fixed value. Instead, it changes in concert with the gate electrode by applying a fixed potential difference, VGS. Fig. 3(f) summarizes the changes in electron energy levels on both the gate and channel before and after addition of a redox analyte. In addition to the change in drain current from changes in charge density, there exists additional potential-dependent reaction currents (red arrows) (see ESI,† Fig. S3 for gate currents). While the potential difference between gate and channel is fixed, the position of the Fermi level in the semiconductors and, consequently, the OECT I–V characteristics depend on the reaction currents. Hence, these redox reactions result in a shift in potential and corresponding change in charge density of the OECT channel, enabling the amplification of reaction currents.
However, the amplification is unreliable and this OECT architecture encounters several additional challenges. It is common practice to identify the OECT's maximum transconductance in its pristine state (i.e. VGS = −0.3 V) and operate the device at that predetermined gate voltage for analyte detection. However, the maximum transconductance point moves during operation and thus results in poorly reproducible performance, as seen from the drastic VT shift due to over-oxidation of the channel (Fig. 3(d) and ESI,† Fig. S4). Furthermore, over-oxidation of the channel (ESI,† Fig. S1) may lead to irreversible chemical degradation that changes the channel's electrochemical characteristics. Alternatively, from Fig. 3(d), it is tempting to conclude that operating the OECT anywhere between VGS = −0.1 V to +0.1 V can result in amplification. However, it is important to note that this ∼200 mV shift in VT corresponds to one particular H2O2 concentration i.e. 33 mM. Indeed, different concentrations of H2O2 result in varying shifts in VT (ESI,† Fig. S5), making the determination of optimal applied VGS a circular process of first having to know the concentration of an unknown analyte before measuring it. Furthermore, the oxidation of the gate is irreversible, requiring a new gate electrode for each measurement. The initial potential of the gate may be recovered by applying a potential difference with respect to a standard reference electrode. However, incorporation of this third electrode defeats the purpose of utilizing a simple 2-terminal OECT architecture. Alternatively, the potential on the gate may be reset to its original value by washing the analyte thoroughly off the electrode e.g. via continuous flow in a microfluidic system.29 The presence of ambient redox-active agents (such as molecular oxygen as an oxidizing agent for a reduced n-type electrode) may also enable the resetting of electrode potentials. However, we note that kinetics of resetting the electrode may be limited by the large volume and capacitance of the gate electrode (a prerequisite for effective gating of the OECT channel), hindering applications that require rapid continuous monitoring. Furthermore, reliance on ambient redox agents to reset the electrode's potential may result in competing reaction during the sensing measurements.
Due to the poor reproducibility of operating at gate voltages above VT (ESI,† Fig. S4), we resorted to operation of the OECT below threshold i.e. VGS = +0.3 V. Due to operation in its OFF state, the faradaic current results in negligible changes in channel charge density. Hence, the performance of the OECT then mimics that of its counterpart with Ag/AgCl gate (Fig. 3(b)), but with the additional challenge of drain current drifts due to drifts in the gate potential. Hence, it is challenging to obtain a reliable drain current vs. concentration calibration curve for the A-OECT with OMIEC gate. While a “calibration curve” (ESI,† Fig. S5) may obtained by extracting threshold voltages at different analyte concentrations, one must be careful as the gate potential sweep needed to measure transfer curves can affect the sensor response as faradaic reactions are potential dependent. The OECT with OMIEC gate experiences the same potential-dependent complexities while suffering additional challenges arising from irreversible changes in its transfer curve characteristics. As an amplifier, having variable transfer characteristics is undesirable device design as it fundamentally changes the amplifying characteristics irreversibly. Furthermore, we note that the peaking transconductance implies that linear shifts in VT induce non-linear changes in ID. This effect may be less pronounced for small VT shifts, but this would limit the detectable concentration range of analytes. Alternatively, these non-linear effects can be ameliorated by utilizing transistors with a constant transconductance. However, there has yet to be an OECT with such characteristics operated in aqueous electrolytes. These challenges in conjunction with the irreversible nature of the OMIEC oxidation makes measurement of an unknown concentration extremely challenging as the sensor cannot be calibrated and re-used.
The pitfalls encountered by the amperometric all-in-one OECTs offer important learning points for achieving reliable amplification in a faradaic chemical sensor. Firstly, the OECT as an amplifier should have stable and reproducible characteristics, implying the need for a non-polarizable gate electrode such as Ag/AgCl. Secondly, to achieve amplification, the transduction mechanism should involve steady-state changes to the charge density of the channel, i.e. its potential. These design principles are inherently contradictory for OECTs that combine both chemical detection and transistor function in the same compartment. To resolve these apparent contrasting principles, the chemical reaction should first be transduced into a potential change that subsequently gates the OECT. Furthermore, the separation of chemical reactions from the OECT compartment prevents complex reactions in the OECT due to the interplay of gate and drain voltages. These insights support the design of the RC-OECT described in our previous work, which amplifies reaction currents by 103 with high reproducibility.22 The RC-OECT's ability to transduce and amplify reaction currents on the RC enables it to reliably detect low analyte concentrations (sub millimolar) as the drain current modulations are orders of magnitude larger than the noise floor of our instrument, unlike A-OECTs. It can further reliably detect higher analyte concentrations compared to the all-in-one OECTs as the absence of applied electric fields prevent accelerated chemical degradation commonly seen in A-OECTs.
To understand the performance of the RC-OECT, similar potential measurements were conducted on the Ag/AgCl gate and source electrodes of the OECT as well as the ITO cathode and p(g3T2) anode of the RC while H2O2 was added to the RC's electrolyte (Fig. 4(a)). The electrode potentials are plotted in Fig. 4(d). The difference in potential between the anode and cathode is defined as VRC while the difference between the gate and source is defined as VGS (Fig. 4(e)). After addition of H2O2 to the RC at 60 s, the potentials on both the RC anode and cathode increase, with the anode increasing by a factor of two more than the cathode. The potential on the Ag/AgCl gate pellet remains constant, as expected, hence all the VRC potential is transferred to the OECT channel. To further verify the RC's effectiveness at gating the OECT, VRC and VGS are plotted in Fig. 4(e). VRC and VGS are almost identical, indicating that the total change in potential across the RC is dropped across the gate–source of the OECT and since a non-polarizable gate is used, the channel experiences all the potential change. As VRC is akin to applying an external VGS, the OECT's ability to transduce potential to drain current modulation was further verified by calculating the expected drain current (Fig. 4(e)) based on the OECT's transfer characteristics (ESI,† Fig. S6). The calculated drain current agrees closely with the experimental current, indicative that the RC indeed acts as a potentiometric modulator for the OECT. Furthermore, as no chemical reactions are conducted within the OECT and a stable gate electrode is used, the OECT retains its transfer characteristics (ESI,† Fig. S6). This feature allows the OECT to be reused for multiple measurements, while retaining consistent amplification characteristics. Lastly, by designing the RC-OECT to be modular, different RCs can be multiplexed to the same OECT for monitoring different reactions.
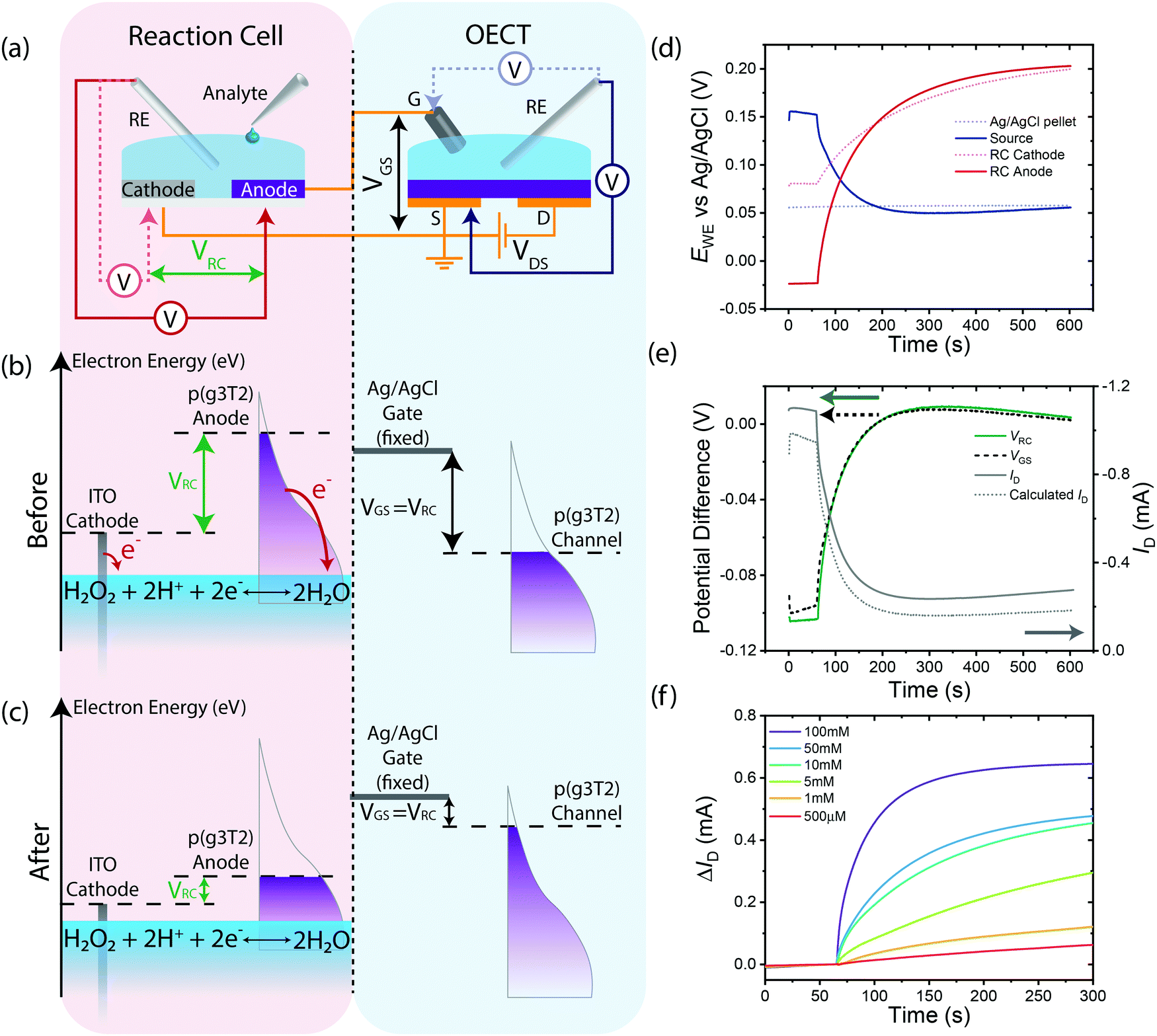 | ||
| Fig. 4 Operation mechanism of a reaction cell OECT using an Ag/AgCl gate electrode. (a) Schematic of RC and OECT where the RC anode is in electrical contact with the OECT's Ag/AgCl gate electrode and the RC cathode is in electrical contact with the source. Redox analyte (33 mM H2O2) is added to RC's electrolyte while conducting potential measurements on the RC's anode, cathode as well as the OECT's gate and source. (b) Schematic of electron energy levels in the RC ITO cathode (grey), p(g3T2) anode (purple, left), OECT's Ag/AgCl gate electrode (solid line), and OECT p(g3T2) channel (purple, right) in the system's initial state. Electron transfer from RC electrodes to the analyte in the RC electrolyte is shown in red arrows. Difference in energy levels between RC cathode and anode is indicated by VRC. Due to the fixed potential on the Ag/AgCl gate, VGS = VRC is imposed on the channel. (c) Change in electron energy levels in the electrode after system has reached a new equilibrium in the presence of the oxidative analyte in the RC. Drop in VGS = VRC results in filling of the channel valence band, dedoping the OECT channel. (d) Plot of potentials of Ag/AgCl gate electrode (blue dashed), OECT source (blue solid), RC anode (red solid) and RC cathode (red dashed). (e) Plot of potential differences across RC (green) and OECT's gate–source (black dashed) as well as the drain current (grey) and the calculated drain current (grey dashed). (f) RC-OECT calibration characteristics. Magnitude of change in RC-OECT drain current over time upon addition of 200 μL H2O2 of different concentrations to 400 μL RC PBS electrolyte. | ||
These findings are summarized in two schematics: the flow of charges within the RC-OECT (ESI,† Fig. S2(f)) and changes in electron energy levels of the electrodes (Fig. 4(b and c)). The loss of electrons from the RC anode to H2O2 is compensated by the flow of electrons from the Ag/AgCl pellet, via oxidation reactions on the gate electrode (Ag + Cl− → AgCl + e−). The resultant drop in [Cl−] in the electrolyte is compensated by the injection of anions from the OECT channel due to the reduction of the channel via electrons injected from the source. This change in charge density on the channel enables large changes in conductivity, thereby amplifying the small reaction currents on the RC anode into large drain current modulations. In Fig. 4(b and c). The cathode and anode have different energy levels initially. Upon addition of the oxidant, electrons are transferred to the analyte, changing potentials on both electrodes, and the overall potential difference between them (VRC). This potential difference is then utilized to gate the OECT (VRC = VGS). Due to the virtually infinite reservoir of electrons on the Ag/AgCl pellet, its electron energy level remains mostly constant. Hence, all changes occur on the channel where its reduction results in increase in its electron energy levels.
From our earlier analyses of OECTs with different gate electrodes, utilizing a non-polarizable gate electrode is crucial in maintaining reproducible performance. To further test this hypothesis on the RC-OECT architecture, we substituted the Ag/AgCl pellet with a p(g3T2) gate electrode (ESI,† Fig. S7). While the RC-OECT is still able to exhibit large drain current modulations and amplification, it suffers from poorer stability and reproducibility. Despite disconnecting the RC from the OECT after measurements, the OECT did not recover its initial characteristics, where the threshold voltage shifted by ∼100 mV, corresponding to the change in VRC = VGS. This indicates that using a polarizable or reactive OMIEC gate is not desirable in either potentiometric or amperometric OECT-based sensors.
The separation of chemical transduction and amplification into two compartments enables individual optimization of the RC and OECT. Firstly, ΔVRC can be maximized by selecting anode materials that are most sensitive to the redox analyte of interest while the cathode is passive to the analyte. To demonstrate this concept, we utilized an Ag/AgCl pellet instead of ITO as the cathode to minimize the cathode's oxidation (Fig. 5(a)). In selecting an anode material, PEDOT:PSS, a common air-sensitive conductive polymer, p(gPyDPP-MeOT2),28 a recently developed air-stable polymer, and p(g3T2) were investigated. p(g3T2) was selected for the study as it undergoes the largest degree of oxidation among the polymer candidates (Fig. 5(b)). See ESI,† Fig. S8 for more details on designing an optimal RC anode material.
 | ||
| Fig. 5 Optimization of RC-OECT. ΔVRC is maximized by minimizing oxidation on RC cathode and maximizing oxidation on RC anode. ΔID is maximized by optimizing OECT to exhibit maximum current modulation in complementary voltage range to ΔVRC. (a) RC cathode: potentials of ITO (grey dashed) and Ag/AgCl pellet (black solid) vs. saturated Ag/AgCl reference electrode with addition of H2O2 at 60 s. (b) RC anode: potentials of PEDOT:PSS (lightest blue, dotted), p(gPyDPP-MeOT2) (medium blue, dash), and p(g3T2) (dark blue, solid) vs. saturated Ag/AgCl reference electrode with addition of H2O2 at 60 s. (c) OECT transfer characteristics of p(g3T2) (solid lines) and PEDOT:PSS (dashed lines). (d) VRC (black) and ID (grey) of optimized RC-OECT using Ag/AgCl pellet as RC cathode, p(g3T2) as RC anode, and p(g3T2) as OECT channel. | ||
Taking the difference in potentials between the RC anode and cathode gives a VRC modulation range of −0.05 V to +0.125 V. Next, to maximize ΔID, this range of VRC should overlap with the range of VGS where the OECT exhibits maximum transconductance and/or encompass VT. The transfer characteristics of OECTs with p(g3T2) or PEDOT:PSS as the channel material were compared (Fig. 5(c)). While the PEDOT:PSS channel is highly conductive in this VGS range, its conductivity modulation is a small fraction of its baseline, rendering the measurement of a small ΔID over a large ID baseline difficult. In contrast, the p(g3T2) OECT undergoes ΔID ∼ 103 in the same VGS range (Fig. 5(d)). See ESI,† Fig. S9 for more details on optimizing channel aspect ratio for maximum transconductance and ESI,† Fig. S10 for full characterization of the RC-OECT. With the RC's potential range close to or centered around 0 V, OECT channel materials should be designed with turn on voltages close to 0 V vs. Ag/AgCl gate (i.e. ionization potentials ∼4.5 eV). Furthermore, as the OECT does not need to be interfaced with the sensing medium, a larger design space is open for OECT channel materials and electrolytes such as organic solvents,30 solid state electrolytes (ion gels)31 and ionic liquids32 that could enable large OECT transconductance and sensitivity.
As the RC and OECT perform distinct roles as redox to potential transducer and amplifier respectively, RC electrode and OECT channel materials can be selected to fulfill their specific metrics rather than attempting to find compromises between competing design metrics as needed in the all-in-one OECTs. Furthermore, the RC-OECT's transduction mechanism provides greater flexibility in OECT choice as long as the region of maximum transconductance is within the range of RC potentials. This condition is significantly less restrictive than the operation of an A-OECT where a discrete and fixed VGS must be identified prior to measurements. To improve the reaction kinetics, catalysts can be added to the RC anode (e.g. Pt to catalyze HPRR), thereby improving RC-OECT performance. However, this optimization would not benefit an A-OECT with OMIEC gate as it may exacerbate the shift in transfer characteristics and OMIEC degradation.
Discussion
Unlike solid-state field-effect transistors (FETs), potential differences (VGS and VDS) during OECT operation do not provide sufficient information nor control of the phenomena occurring at the OECT's electrodes. Hence, fully utilizing the amplification properties of OECTs for reliable faradaic chemical detection requires more detailed understanding of the potentials and charge density of its electrodes.Our analysis of electrochemical processes in various OECT architectures shows fundamentally that a non-polarizable gate electrode such as Ag/AgCl is imperative to preserve the stability and reproducibility of the OECT's I–V characteristics. However, conducting faradaic reactions within the electrolyte of this OECT does not amplify reaction currents. Conversely, an OMIEC gate, in principle leads to amplification however the resulting shift of the OECT I–V characteristics, which is irreversible in a 2-electrode geometry, makes this geometry fundamentally challenging to use as a sensor. We note that this shift is inevitable as it is inherent to the sensing process. Moreover, the non-constant transconductance leads to difficulties in sensor calibration, which will be sensitive to the specifics of how transconductance depends on VGS. Finally, the presence of the potentials needed to drive the OECT in the sensing environment leads to parasitic reactions, with molecular oxygen for instance. Any parasitic side reaction on the OECT gate and channel are extremely complex to model due to the asymmetric electric fields within the OECT, exacerbated by the potential-dependence of reaction currents (see ESI†). These additionally lead to further irreversible changes in the OECT characteristics, exacerbating the irreproducibility of the sensing measurements.
To achieve both operational stability and amplification, the reactive OMIEC material is physically separated from the OECT in a Reaction Cell which is utilized to gate an OECT with an Ag/AgCl gate electrode. By modulating the OECT channel potentiometrically, the RC fully leverages the excellent transconductance of the OECT, enabling the amplification of minute reaction currents into easily detected drain current modulations. The modularity of the RC-OECT architecture enables designing of the RC and OECT separately, where each component can be optimized for specific metrics of potential change and transconductance, respectively as demonstrated in the optimal materials choice to match sensing range with peak OECT transconductance. This avoids constraints faced by all-in-one OECTs where one material (e.g. channel) has to fulfill multiple functions simultaneously while ultimately not being optimized for any. Improving amplification of the RC-OECT requires further maximization of the OECT's transconductance by changing the channel W/L aspect ratio, enabling large improvements for small changes to device structure. This, in addition to the ability to select different materials and device geometries for the RC electrodes and OECT, enables great flexibility during RC-OECT device optimization, opening myriad opportunities for device development.
Following the general engineering design principles of minimizing complexity while maximizing performance, the RC-OECT architecture presents an effective approach to OECT-based redox chemical sensors. By demonstrating the importance of understanding the underlying electrochemical phenomena in OECTs to rationalize device design, we envision that these insights allow further systematic development of electrochemical device architectures for chemical sensing.
Materials and methods
OECT fabrication
Photolithographically patterned chips with gold contacts and patterned Parylene to expose regions for the channel were used as OECT chips. p(g3T2) was dissolved in chloroform at 10 mg mL−1 and spin coated at 1000 rpm, annealed at 60 °C for 10 minutes, followed by parylene lift-off to form the channel. The OECT device dimension are L = 10 μm, W = 2 mm, d = 120 nm and operated at VD = −0.1 V for both the amperometric OECT and RC[p(g3T2)/ITO]–OECT [p(g3T2)].RC fabrication
p(3gT2) was dissolved in chloroform at 10 mg mL−1 and spin coated on ITO/glass at 1000 rpm, and annealed at 60 °C for 10 minutes in ambient conditions to yield film thickness of 120 nm.OECT measurements
Transfer and output characteristics of the OECTs were obtained using a custom LabVIEW program with a K2612 Keithley. Phosphate buffered saline solution was used as the electrolyte and Ag/AgCl pellet as gate.Potential and current measurements
Open circuit measurements were conducted using a Biologic SP-300 with a saturated Ag/AgCl reference electrode (BASi). A-OECT: PDMS wells were adhered to the OECT substrate to confine the electrolyte (phosphate buffered saline solution) with either an Ag/AgCl pellet or dropcast p(g3T2) on conductive carbon paper electrode as gate. The reference electrode was immersed in the same electrolyte and the working electrode connected to the pellet while the counter electrode was connected to the source. At the same time, the OECT was operated using a custom LabVIEW program with a K2612 Keithley to impose VGS and VDS and to measurement drain and gate currents. A constant drain voltage (VD = −0.1 V) and gate voltage (VG ranging from −0.3 V to +0.3 V) was applied. The drain and gate currents were measured simultaneously after addition of 100 μL 100 mM H2O2 to 200 μL of PBS electrolyte. RC-OECT: the VD across the OECT was supplied by a K2612 Keithley and ID measured using a custom LabVIEW program. The reference electrode was immersed in the RC or OECT electrolytes and the working electrode was connected to the RC anode or Ag/AgCl pellet as well as the RC cathode and OECT source. 400 μL of Phosphate Buffered Saline solution was drop cast on the surface of the reaction cell and 200 μL of 100 mM H2O2 added after 1 minute.Chronoamperometry measurements
Potential of the working electrode was held at +0.3 V, 0 V, −0.1 V, and −0.3 V vs. a saturated Ag/AgCl reference electrode and the current flow into the film measured after 200 μL of 100 mM H2O2 to 400 μL PBS solution.Cyclic voltammetry
The scan rate for the cyclic-voltammogram measurements was 50 mV s−1 and conducted in phosphate buffered saline solution in ambient conditions unless stated otherwise.Conflicts of interest
There are no conflicts to declare.References
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Nat. Mater., 2020, 19, 13 CrossRef CAS PubMed.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, Chem. Mater., 2018, 30, 2945 CrossRef CAS PubMed.
- A. Giovannitti, D. T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12017 CrossRef CAS.
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Adv. Mater., 2020, 2002748, 1 Search PubMed.
- J. Gladisch, E. Stavrinidou, S. Ghosh, A. Giovannitti, M. Moser, I. Zozoulenko, I. Mcculloch and M. Berggren, Adv. Sci., 2019, 1901144 Search PubMed.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604 CrossRef CAS.
- M. Moser, L. R. Savagian, A. Savva, M. Matta, J. F. Ponder, T. C. Hidalgo, D. Ohayon, R. Hallani, M. Reisjalali, A. Troisi, A. Wadsworth, J. R. Reynolds, S. Inal and I. McCulloch, Chem. Mater., 2020, 32(15), 6618–6628 CrossRef CAS.
- D. S. Bindra and G. S. Wilson, Anal. Chem., 1989, 61, 2566 CrossRef CAS.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, DOI:10.1038/natrevmats.2017.86.
- A. M. Pappa, V. F. Curto, M. Braendlein, X. Strakosas, M. J. Donahue, M. Fiocchi, G. G. Malliaras and R. M. Owens, Adv. Healthcare Mater., 2016, 5, 2295 CrossRef CAS PubMed.
- G. Méhes, A. Roy, X. Strakosas, M. Berggren, E. Stavrinidou and D. T. Simon, Adv. Sci., 2020, 2000641, 1 Search PubMed.
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1 CrossRef CAS.
- D. A. Bernards and G. G. Malliaras, Adv. Funct. Mater., 2007, 17, 3538 CrossRef CAS.
- J. T. Friedlein, R. R. McLeod and J. Rivnay, Org. Electron., 2018, 63, 398 CrossRef CAS.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J. M. Badier, C. Bernard and G. G. Malliaras, Sci. Adv., 2015, 1, 1 Search PubMed.
- D. Khodagholy, J. Rivnay, M. Sessolo, M. Gurfinkel, P. Leleux, L. H. Jimison, E. Stavrinidou, T. Herve, S. Sanaur, R. M. Owens and G. G. Malliaras, Nat. Commun., 2013, 4, 1 Search PubMed.
- J. Rivnay, P. Leleux, M. Sessolo, D. Khodagholy, T. Hervé, M. Fiocchi and G. G. Malliaras, Adv. Mater., 2013, 25, 7010 CrossRef CAS.
- D. A. Bernards, D. J. MacAya, M. Nikolou, J. A. Defranco, S. Takamatsu and G. G. Malliaras, J. Mater. Chem., 2008, 18, 116 RSC.
- D. Ohayon, G. Nikiforidis, A. Savva, A. Giugni, S. Wustoni, T. Palanisamy, X. Chen, I. P. Maria, E. Di Fabrizio, P. M. F. J. Costa, I. Mcculloch and S. Inal, Nat. Mater., 2020, 19, 456–463 CrossRef CAS.
- A. M. Pappa, D. Ohayon, A. Giovannitti, I. P. Maria, A. Savva, I. Uguz, J. Rivnay, I. McCulloch, R. M. Owens and S. Inal, Sci. Adv., 2018, 4(6), eaat0911 CrossRef PubMed.
- M. Braendlein, A.-M. Pappa, M. Ferro, A. Lopresti, C. Acquaviva, E. Mamessier, G. G. Malliaras and R. M. Owens, Adv. Mater., 2017, 29, 1605744 CrossRef.
- S. T. M. Tan, A. Giovannitti, A. Melianas, M. Moser, B. L. Cotts, D. Singh, I. McCulloch and A. Salleo, Adv. Funct. Mater., 2021, 2010868, 1 Search PubMed.
- E. Macchia, R. A. Picca, K. Manoli, C. Di Franco, D. Blasi, L. Sarcina, N. Ditaranto, N. Cioffi, R. Österbacka, G. Scamarcio, F. Torricelli and L. Torsi, Mater. Horiz., 2020, 7, 999–1013 RSC.
- S. Lai, F. A. Viola, P. Cosseddu and A. Bonfiglio, Sensors, 2018, 18, 1 Search PubMed.
- S. P. White, K. D. Dorfman and C. D. Frisbie, Anal. Chem., 2015, 87, 1861 CrossRef CAS.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 1349 RSC.
- S. E. Doris, A. Pierre and R. A. Street, Adv. Mater., 2018, 30, 1 CrossRef.
- A. Giovannitti, R. B. Rashid, Q. Thiburce, B. D. Paulsen, C. Cendra, K. Thorley, D. Moia, J. T. Mefford, D. Hanifi, D. Weiyuan, M. Moser, A. Salleo, J. Nelson, I. McCulloch and J. Rivnay, Adv. Mater., 2020, 1908047, 1 Search PubMed.
- A. Koklu, D. Ohayon, S. Wustoni, A. Hama, X. Chen, I. McCulloch and S. Inal, Sens. Actuators, B, 2021, 329, 129251 CrossRef CAS.
- D. T. Duong, Y. Tuchman, P. Chakthranont, P. Cavassin, R. Colucci, T. F. Jaramillo, A. Salleo and G. C. Faria, Adv. Electron. Mater., 2018, 4, 1 Search PubMed.
- A. Melianas, T. J. Quill, G. LeCroy, Y. Tuchman, H. V. Loo, S. T. Keene, A. Giovannitti, H. R. Lee, I. P. Maria, I. McCulloch and A. Salleo, Sci. Adv., 2020, 6(27), eabb2958 CrossRef CAS PubMed.
- X. Wu, A. Surendran, J. Ko, O. Filonik, E. M. Herzig, P. Müller-Buschbaum and W. L. Leong, Adv. Mater., 2019, 31, 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc02224e |
| This journal is © The Royal Society of Chemistry 2021 |