
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new alternative tool to analyse glycosylation in pharmaceutical proteins based on infrared spectroscopy combined with nonlinear support vector regression†
Sabrina
Hamla
 *a,
Pierre-Yves
Sacré
a,
Allison
Derenne
b,
Kheiro-Mouna
Derfoufi
b,
Ben
Cowper
c,
Claire I.
Butré
d,
Arnaud
Delobel
d,
Erik
Goormaghtigh
b,
Philippe
Hubert
a and
Eric
Ziemons
a
*a,
Pierre-Yves
Sacré
a,
Allison
Derenne
b,
Kheiro-Mouna
Derfoufi
b,
Ben
Cowper
c,
Claire I.
Butré
d,
Arnaud
Delobel
d,
Erik
Goormaghtigh
b,
Philippe
Hubert
a and
Eric
Ziemons
a
aUniversity of Liege (ULiege), CIRM, Vibra-Sante Hub, Department of Pharmacy, Laboratory of Pharmaceutical Analytical Chemistry, Liege, Belgium. E-mail: sabrina.hamla@uliege.be; Tel: +3243664316
bCenter for Structural Biology and Bioinformatics, Laboratory for the Structure and Function of Biological Membranes, ULB, Campus Plaine CP206/02, 1050 Brussels, Belgium
cNational Institute for Biological Standards and Control, Blanche Lane, South Mimms, Potters Bar, Hertfordshire EN6 3QG, UK
dQuality Assistance, Techno Parc de Thudinie 2, 6536 Thuin, Belgium
First published on 17th February 2022
Abstract
Almost 60% of commercialized pharmaceutical proteins are glycosylated. Glycosylation is considered a critical quality attribute, as it affects the stability, bioactivity and safety of proteins. Hence, the development of analytical methods to characterise the composition and structure of glycoproteins is crucial. Currently, existing methods are time-consuming, expensive, and require significant sample preparation steps, which can alter the robustness of the analyses. In this work, we suggest the use of a fast, direct, and simple Fourier transform infrared spectroscopy (FT-IR) combined with a chemometric strategy to address this challenge. In this context, a database of FT-IR spectra of glycoproteins was built, and the glycoproteins were characterised by reference methods (MALDI-TOF, LC-ESI-QTOF and LC-FLR-MS) to estimate the mass ratio between carbohydrates and proteins and determine the composition in monosaccharides. The FT-IR spectra were processed first by Partial Least Squares Regression (PLSR), one of the most used regression algorithms in spectroscopy and secondly by Support Vector Regression (SVR). SVR has emerged in recent years and is now considered a powerful alternative to PLSR, thanks to its ability to flexibly model nonlinear relationships. The results provide clear evidence of the efficiency of the combination of FT-IR spectroscopy, and SVR modelling to characterise glycosylation in therapeutic proteins. The SVR models showed better predictive performances than the PLSR models in terms of RMSECV, RMSEP, R2CV, R2Pred and RPD. This tool offers several potential applications, such as comparing the glycosylation of a biosimilar and the original molecule, monitoring batch-to-batch homogeneity, and in-process control.
Introduction
Glycosylation is considered the most common post-translational modification (PTM) of proteins and affects more than 60% of therapeutic proteins, including monoclonal antibodies (mAbs) and fusion proteins.1 This PTM results in the binding of carbohydrates to specific sites of the amino acid chain of proteins.2 These carbohydrates are linked to an oxygen or a nitrogen atom, and they are called O-glycans or N-glycans, respectively. All N-glycans (Fig. S3†) have a common main structure of five monosaccharides, composed of a linear chain of two N-acetylglucosamines (GlcNAc), mannose and two further branching mannoses. This core structure is named M3 (Fig. S3†). In addition, mannose can be added to this elementary structure to form M4, M5, M6… and form a high-mannose glycan. Furthermore, the addition of fucose, GlcNAc, galactose (Gal), and N-acetylneuraminic acid (Neu5Ac; sialic acid) monosaccharides form a complex N-glycan. The most abundant N-glycan in mAbs is the complex-type FA2, characterised by the M3 core structure with fucose linked to the first GlcNAc and extension of the branching mannoses with additional GlcNAc monosaccharides. Furthermore, the addition of β-linked galactose to this complex-type FA2 forms FA2G1 and FA2G2 glycans, which can be terminated with sialic acids (N-glycolylneuraminic acid, Neu5Gc) and/or α-linked galactose.3–5Glycosylation is considered a critical quality attribute because it impacts the stability, pharmacokinetics, pharmacodynamics (PK/PD), and the safety (immunogenicity) of the product.5 It is a complex process involving hundreds of enzymes specific to each cell. This PTM is influenced by many parameters during the manufacturing process, including host system type (mammalian cells, yeast strains, plant cells, insect cells or genetically modified animals) and environmental conditions of culture (bioreactor type, culture media and process parameters).6–8 Even though these parameters are controlled during the production, the process generally generates micro- and macro-heterogeneity in the glycosylation of proteins. Micro-heterogeneity corresponds to heterogeneity in the structures of glycans occupying a particular glycosylation site, at the same time macro-heterogeneity can be defined as the difference in frequencies of occupation of glycosylation sites in a glycoprotein batch.1,8 These differences significantly affect the protein glycosylation profile, and therefore the quality and safety of the final therapeutic product. Hence, the regulatory authorities require the systematic characterisation of the composition and structure of glycoproteins throughout the drug development and manufacturing processes to ensure the quality and consistency of the final drug product.9,10
Current glycosylation analysis methods are complex and time-consuming processes, consisting of long sample preparation protocols requiring a minimum of three steps: glycan release, labelling and glycan purification. Each step can suffer from several potential sources of error.11 Therefore, the efficiency is not always consistent and can lead to variations in the results from one laboratory to another.7 To overcome these limitations, it is proposed to use a vibrational spectroscopy technique – FT-IR spectroscopy. Firstly, this technique has already been used to analyse biomolecules for a wide range of applications. For example, it is a tool of choice for the structural characterisation of proteins.12–15 It has also been used to monitor the carbohydrate content, notably in algae cultures.16 The study by Khajehpour et al. demonstrated the correlation between the intensity of the spectral bands in 1200 and 1000 cm−1 and the amount of carbohydrates in glycoproteins. These absorption bands are mainly due to the stretching of the C–C and C–O bonds of the carbohydrate skeleton.17 Moreover, a recent study by Derenne et al.5 showed that the use of FT-IR for the analysis of glycosylation profiles presents several advantages. Firstly, it is a simple, fast, and non-destructive tool. Secondly, FT-IR spectra of glycoproteins generate an average but the accurate fingerprint of the glycosylation profile. This signature reflects not only important differences such as the presence or absence of certain monosaccharides but also small modifications of the global content of glycans or monosaccharides.
The objective of the present work is twofold. First, the study aims to demonstrate the use of FT-IR spectra to analyse the total quantity of carbohydrates present in various types of glycoproteins. Second, the study intends to extract from FT-IR spectra the relative amount of each major monosaccharide (mannose, N-acetylglucosamine, galactose, fucose and sialic acid). Therefore, several regression models were calibrated to assess the global rate of glycosylation and determine each monosaccharide's relative quantity. In this work, the global rate of glycosylation is defined as the ratio between the mass of glycans and the total mass of the protein. To build such predictive models, infrared spectra of reference samples were measured. Two databases of FT-IR spectra were constituted: the first one included 18 proteins and was used to calibrate the model for the global rate of glycosylation. The second one gathered 32 proteins and was exploited to calibrate the models used to predict the relative quantity of each monosaccharide. The global rate of glycosylation and the composition in monosaccharides were determined using reference MALDI and LC-FLR-MS methods respectively.
Two regression algorithms were considered and compared to correlate the spectral information and the quantitative reference values. These methods differ in their ability to model complex, potentially nonlinear, relationships. The Partial Least Squares Regression model (PLSR), one of the most used regression models for analysing spectroscopic data, was applied. PLSR, whose main strengths are its simplicity and interpretability,18 is often used to process high dimensional data.19 It deals with a linear relationship between the parameter to be predicted and the intensity of the spectral absorption bands. Therefore, it is appropriate for the analysis of chemical processes that follow the Beer–Lambert law.19–21 PLSR regression was applied to manage some highly correlated and possibly noisy predictor variables. It is based on a dimension reduction process in order to deal with the strong collinearities of the spectral data.22 In most cases, PLSR methods are able to cope with low degrees of non-linearity, including additional latent variables. However, spectra can contain highly nonlinear effects for various reasons, such as differences in viscosity, temperature, or chemical biological composition of the sample matrix.19 Thereby, PLSR may not predict the parameter of interest well enough. Support Vector Regression (SVR) method has emerged as a powerful alternative to PLSR thanks to its many attractive features. First, it can find nonlinear global solutions and thus properly model complex nonlinear relationships and select only the samples representative of the problem, which are then called support vectors. Second, this method gives high prediction accuracy18,19 and is less sensitive to spectral noise. Moreover, SVR models are robust models that can handle possible spectral variations due to nonlinear interference.23
Materials and methods
Chemicals, reagents, and proteins for the analysis of the global rate of glycosylation
Eighteen therapeutic proteins were used in this study to build a database. The glycoproteins were provided by the Saint-Pierre hospital (Brussels, Belgium). They include three monoclonal antibodies (mAbs) and one therapeutic Fc-fusion protein. These proteins and their producers are as follows:- Infliximab (Remicade® 100 mg, Janssen Biologics)
- Cetuximab (Erbitux® 5 mg mL−1, Merck KGaA)
- Nivolumab (Opdivo® 10 mg mL−1, Bristol Myer Squib)
- Aflibercept (Zaltrap® 25 mg mL−1, Sanofi-Aventis)
The following 13 other glycoproteins were purchased from Sigma-Aldrich (Merck):
- Alpha1-acid glycoprotein
- Alpha-crystallin
- Apo-transferrin
- Carboxypeptidase Y
- Conalbumin
- Avidin
- Fetuin
- Lactoferrin
- Lectin from Glycine max
- Lectin from Phaesolus vulgaris
- Lectin from Maackia amurensis
- Peroxidase
- Ribonuclease B
Finally, the last protein called PPp was provided by Xpress Biologics (Liege, Belgium). It was produced in Pichia pastoris and was received at a concentration of 25.97 mg mL−1 in PBS buffer (pH 7.2). The proteins were dissolved in 0.9% NaCl at 10 mg mL−1.
Size exclusion spin columns were used to remove residual salts and excipients present in the formulations of the therapeutic proteins. These excipients (mannitol, Tween 80, Tween 20, trehalose, sodium citrate and sodium acetate, etc.…), can indeed interfere with FT-IR measurements in the spectral region between 1200 and 950 cm−1 due to the vibrational frequencies of their chemical bonds, which are close to those of carbohydrates shown in Fig. S1-A and S1-B.† Therefore, as explained in the work of Derenne et al.,5 for FT-IR measurements a buffer exchange step with 0.9% NaCl was performed using Micro Bio-SpinTM P-6 Gel columns (Tris buffer, sample volume 10–75 μL, 6000 Da MW limit). Also, the samples were prepared using the same method with (NH4)2CO3 for buffer exchange for the MALDI measurements.
Chemicals, reagents, and proteins for the analysis of the composition in monosaccharides
Thirty-two therapeutic proteins were used to build a second database, and for some proteins*, two batches were analysed. These glycoproteins were provided by the Saint-Pierre hospital (Brussels, Belgium) and by the pharmacy of the University Hospital Center of Liège (CHU Liège, Belgium). They include twenty-three monoclonal antibodies (mAbs), one therapeutic Fc-fusion protein and three s mAbs biosimilars.The 27 proteins and their producers are as follows:
- Adalimumab* (Humira® 40 mg, Abbvie)
- Aflibercept* (Zaltrap® 25 mg mL−1, Sanofi-Aventis)
- Avelumab* (Bavencio® 20 mg mL−1, Merck)
- Bevacizumab* (Avastin® 25 mg mL−1, Roche Pharma)
- Cetuximab* (Erbitux® 5 mg mL−1, Merck KGaA)
- Daratumumab (Darzalex® 20 mg mL−1, Janssen Biologics)
- Durvalumab (Imfinzi® 50 mg mL−1, AstraZeneca)
- Golimumab (Simponi®100 mg, Janssen Biologics)
- Infliximab (Remicade® 100 mg, Janssen Biologics)
- Ipilimumab* (Yervoy® 5 mg mL−1, Bristol Myer Squibb)
- Natalizumab (Tysabri® 300 mg, Biogen)
- Nivolumab* (Opdivo® 10 mg mL−1, Bristol Myer Squibb)
- Ocrelizumab (Ocrevus® 300 mg, Roche)
- Omalizumab (Xolair® 150 mg mL−1, Novartis)
- Panitumumab* (Vectibix® 25 mg mL−1, Amgen)
- Pembrolizumab* (Keytruda® 50 mg, Merck)
- Pertuzumab* (Perjeta® 30mg mL−1, Roche Pharma)
- Ramucirumab* (Cyramza® 10mg mL−1, Eli-Lilly)
- Rituximab* (Mabthera® 10 mg mL−1, Roche Pharma)
- Secukinumab (Cosentyx® 150 mg mL−1, Novartis)
- Tocilizumab* (Roactemra® 20 mg mL−1, Roche)
- Trastuzumab* (Herceptin® 150 mg, Roche Pharma)
- Trastuzumab-emtasine (Kadycla® 160 mg, Roche Pharma)
- Vedolizumab (Entyvio® 60 mg mL−1, Takeda)
- Biosimilar of Rituximab* (Truxima® 10 mg mL−1, Celltrion)
- Biosimilar of Infliximab (Remisma® 100 mg, Celltrion)
- Biosimilar of Infliximab (Inflectra® 100 mg, Hospira)
The following glycoproteins were purchased from Sigma-Aldrich (Merck):
- Alpha1-acid glycoprotein
- Etanercept (European Pharmacopoeia Reference Standard)
- Avidin
- IgG1 Kappa from Human Myeloma
- Ribonuclease B
Salts and excipients were removed from all samples using size exclusion spin columns, following the method used previously for the analysis of the global rate of glycosylation.
Chemicals, reagents, and proteins used to investigate the cause of non-linearity
Monosaccharides were purchased from Sigma-Aldrich (Merck) and from Dextra* at 10 mg ml−1:- Sialic acid
- Galactose
- Fucose
- N-Acetylglucosamine
- D-Mannose*
Glycans (Fig. S3†) were purchased from Dextra at 1 mg ml−1:
- FA2, FA2G1, FA2G2
- M5, M6, M7
Reference analysis of the global rate of glycosylation
Alpha crystallin, PPp and the three lectins were diluted to 0.1 mg mL−1 in 0.1% formic acid. All the other proteins were diluted to 0.2 mg mL−1 with 0.1% formic acid. The samples were loaded on the target using the dried droplet method with the addition of 0.5 μL sample followed by 0.5 μL of the matrix (10 mg mL−1 sinapinic acid in H2O/ACN/FA 70/30/0.1). The target was dried under vacuum.
Eluent A was 0.1% formic acid (FA) in H2O, and eluent B was 0.1% FA in acetonitrile. The elution profile was as follows: 0–2 min, isocratic on 5% B; 2–3 min, linear gradient to 20% B and 3–13 min, linear gradient from 20% B to 90% B, 13–15 min, isocratic on 90% B, 15–17 min, linear gradient to 10% B and from 17 to 19 min, linear gradient to 90% B, and 19–21 min; linear gradient to 5% B and isocratic for 2 min and 5% B. The flow rate was 0.3 mL min−1. Lactoferrin and Ribonuclease B were diluted to 0.5 mg mL−1 in eluent A. 2 μL of each sample were injected into the column, which was thermostated at 80 °C. The samples were kept at 5 °C and the detection was performed at a wavelength of 280 nm using a UV detector.
The mass spectra were acquired on the m/z range of 400 to 5000 in positive ion mode. The capillary voltage was set at 2.5 kV, the sample cone at 120 V and the source operated at 100 °C. Nitrogen was used as desolvation gas (500 °C, 800 L h−1) and cone gas at 100 L h−1. An on-line mass correction was applied using Leucine Enkephalin. Molecular mass was calculated by deconvoluting the mass spectra using MaxEnt1 algorithm.
Reference analysis of the composition of monosaccharides
Glycoworks RapiFluor-MS N-glycan. The Glycoworks RapiFluor-MS N-glycan 24 samples kit (#176003713) was purchased from Waters Corporation (MA, USA). This analytical method allows quick deglycosylation followed by rapid labelling and cleaning of the labelled glycans (N-glycans release).
UPLC-FLR-MS analysis. Labelled N-glycans were analysed via HILIC separation combined with fluorescence (FLR) and mass spectrometry (MS) detection using a UPLC-MS system equipped with an ACQUITY UPLC BEH Amide (2.1 × 150 mm, 1.7 μm particle diameter and 130 Å pore size) column (Waters Milford, MA). The details of this method were described in the article by Derenne et al.5 MS data were obtained using a Single Quadrupole Detector 2, SQD2 (Waters Milford, MA) in ESI positive mode and the data were acquired using Empower 3.1 software.
FT-IR measurements
The Bruker Tensor 27 FT-IR spectrometer (Bruker Optics GmbH, Ettlingen, Germany) with Opus 6.5 software (Bruker Optics GmbH, Ettlingen, Germany) equipped with a mercury-cadmium-telluride detector was used for spectra acquisition. The recordings were performed in ATR mode using a Golden GateTM ATR accessory (Specac, Orpington, United Kingdom) with an integrated total reflection element composed of a single reflection diamond, with an angle of incidence of 45°. The spectra were acquired over the spectral range between 4000 and 600 cm−1 at a resolution of 2 cm−1 and with 128 scans. 0.5 μL were deposited on the diamond crystal and dried quickly with a constant and gentle nitrogen flow for 5 minutes.5 After acquiring each spectrum, the crystal was cleaned with water and a cleaning check was performed spectroscopically.Furthermore, a background was recorded before the start of the measurements and prior to each new sample. Regarding the global rate of glycosylation analysis, for each glycoprotein, 6 spectra were recorded with 6 distinct deposits. As for the analysis of the composition of monosaccharides, three independent samples were prepared for each glycoprotein on three distinct days to obtain triplicate measurements. In total, 18 deposits were made, resulting in the measurement of 18 spectra. Therefore, for the analysis of the global rate of glycosylation, a total of 108 spectra were acquired (18 glycoproteins × 6 spectra), and for the analysis of the composition of monosaccharides, a total of 846 spectra were acquired (one batch for 17 glycoproteins and two batches for 15 glycoproteins: 47 samples × 18 spectra). Finally, for the investigation of the cause of non-linearity, the glycans (FA2, FA2G1, and the high mannose: Man-1, Man-3, Man-5, Man-6, Man-8) were studied. In this context, 6 spectra (6 distinct deposits) were recorded for each sample.
FT-IR data analysis
The reference water vapour spectrum was obtained, in the absence of sample, as the difference between a spectrum recorded before and after purging the sample room with dry air. Therefore, the reference water vapour spectrum was recorded in the same conditions of samples acquisition and was subtracted from all FT-IR spectra, with 1956–1935 cm−1 as reference peak.24,25
The preprocessing retained for the model of the global rate of glycosylation is the Savitzky–Golay 1st derivative (polynomial order: 2, window size: 15) to improve the signal/noise ratio and the standard normal variate (SNV) to reduce the effect of the variation of the signal linked to the quantity of samples deposited on the crystal and the impact of sample drying on the crystal.
The FT-IR spectra used to build the monosaccharide model were preprocessed by the Savitzky–Golay 2nd derivative (polynomial order: 3, window size: 15) and SNV.
All instrumental techniques, including FT-IR spectroscopy, are affected by noise. The latter is considered in our case as an additional spectral perturbation, which is not related to the chemical nature of the sample26 but rather to the parameters of the spectrophotometer, such as the number of scans and the spectral resolution.27 It should be noted that a low sample concentration generates a weak signal and has a high noise impact. In this study, the noise was defined as the standard deviation in the spectral region from 2000 to 1900 cm−1 (since there is no biological related absorption band). The signal was defined as the maximum intensity between 1180 and 965 cm−1 in the spectrum after subtraction of a baseline drawn through these two points. Consequently, the signal-to-noise ratio was calculated for each IR spectrum to assess its spectral quality. All IR spectra recorded with a signal-to-noise ratio less than 60 were eliminated. In addition, spectral smoothing of the remaining IR spectra was applied to further reduce the noise level. It was carried out by apodizing the Fourier transform using a Gaussian function with a final resolution of 4 cm−1.
Principal component analysis (PCA) was performed on the spectra of each glycoprotein to detect outliers. The first principal component (PC) was selected to minimize the total distance between the data and its projection on the PC. Also, the variance of the projected points was maximized. An 85% Hotelling's T2 confidence ellipse was built around the mean position of each glycoprotein in the score plot, based on a PC1-PC2 space. For each glycoprotein, the FT-IR spectra outside the confidence ellipse were considered outliers. The PCA was performed on both the training set and the test set (Table S4†). The future samples will be projected onto the defined PCA to ensure that they have the same variance as the calibration data set. This verification is performed by looking at the orthogonal and score distances of the new samples to the pre-defined PCA space. A new spectrum exhibiting orthogonal and score distances higher than the 85% confidence limit will be considered as an outlier.
SVR method. The SVR models were evaluated using the Gaussian RBF kernel (Radial Basis Function). It is a Gaussian kernel that expresses sample-to-sample similarities by the equation K(xi,xj) = exp(−γ∥xi − xj∥2). With γ > 0 and xi, xj are input features values for i and j samples and γ is the kernel parameter.28–30 The nonlinear SVR model requires the determination of three meta-parameters: the cost C, and the variables ε and γ.29,31C represents the error regularization parameter, ε corresponds to the size of the margin, and is correlated to the number of support vectors selected. Only samples with prediction errors larger than ±ε are considered support vectors and then contribute to the final prediction of the model.18,32 The γ parameter represents the variance of the RBF kernel. It determines the nonlinear mapping of the input data and characterises the degree of non-linearity of the model.33 The excellent generalization performance of SVR models requires the simultaneous optimization of its three meta-parameters (C, ε and γ). This optimization is performed through a grid search using 2-step cross-validation.30,34 First, a coarse grid search is performed to select approximately the best region, followed by a finer grid search to obtain optimal values. Regarding the PLSR model, it only requires the determination of a single meta-parameter, namely the number of latent variables (LVs).22
Performance evaluation of the PLSR and SVR models. The calibration and validation sets were the same for each PLSR and SVR model. The samples were split into a training set (66%) and a test set (33%). For the study of the global rate of glycosylation, the Kennard – Stone algorithm was used to perform the split because it enables a uniform selection of samples covering most of the sources of variation in the data set.35 Different spectral ranges were considered to build the models, and based on low value in terms of cross-validation performance (RMSECV) and of external validation performance (RMSEP), the spectral range between 1179 and 965 cm−1 was retained. Regarding the study of the composition of monosaccharides, the data were split manually into a training and test set. Therefore, the influence of the splitting on the performance of the models was evaluated. Two different models with different samples splits were built. To calibrate and optimize the models, Venetian blinds was used as a cross-validation strategy while keeping the replicates together. Venetian blinds was tuned by certain parameters to build the models with a data split of 10 and one sample per blind (thickness). Consequently, the calibration models were evaluated using the Root Mean Square Error of cross-validation (RMSECV). Furthermore, the optimal number of LVs in PLSR models was selected based on a minimum RMSECV.
where ŷCV,i is the value predicted by the cross-validated model for sample i. yi is the measured value obtained for sample i and n is the number of samples.
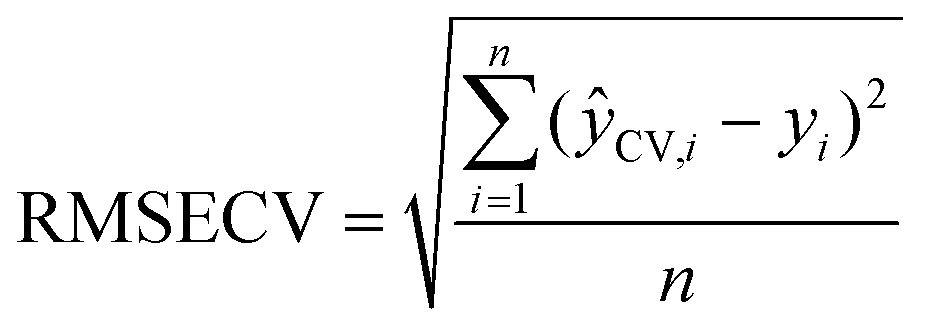
In the last step, the models were evaluated by external validation, by projecting an independent test set. These models were evaluated using the Root Mean Square Error of Prediction (RMSEP) and R2 of prediction.

Accordingly, the performances of the PLSR and SVR models were evaluated by comparing the results obtained by the calibration performances: (RMSECV) and R2CV and by the external validation performances: (RMSEP) and R2 of prediction. Low values of RMSECV and RMSEP are expected to indicate high accuracy, and a high value of R2 indicates that the model correctly handles the spectral variability and is, therefore, able to accurately estimate the concentration. Additionally, the ratio of performance to deviation (RPD) was evaluated by dividing the standard deviation (SD) of the reference values of the samples in the validation set by the standard error of prediction (SEP).36,37
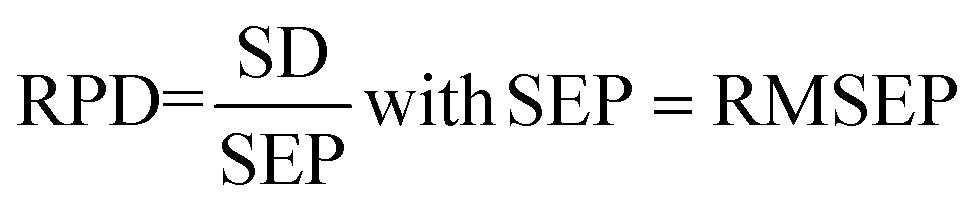
where yi is the measured value for predicting sample i. ȳ is the mean of the reference data for the samples in the validation set. n is the number of observations in the prediction set.
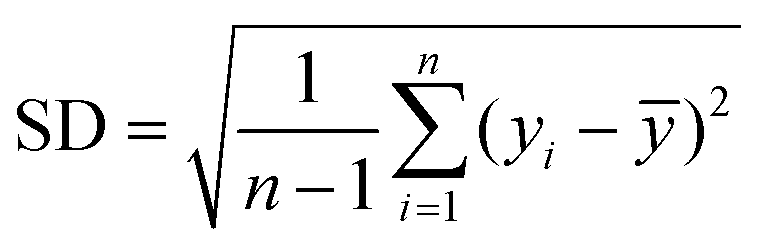
The Ratio of Performance to Deviation (RPD) corresponds to the factor by which the prediction accuracy increases related to the mean composition. Ideally, in the case of good model calibration, the ratio is greater than two.37,38 Three categories of the RPD ratio were identified to interpret the model's reliability: RPD > 2: excellent model, 1,4 < RPD < 2: fair model, and RPD < 1,4: non-reliable model.
Results and discussion
Comparison of the performances of PLSR and SVR to determine the global rate of glycosylation
The first objective of this study was to build a model to predict the global rate of glycosylation. This study focused on three monoclonal antibodies (mAbs), one fusion protein (Fc), fourteen other glycoproteins, among which some have a very distinctive glycosylation profile. For example, alpha1-acid glycoprotein only contains sialylated glycans; O- and N-glycans of fetuin have a high content of sialic acid; ribonuclease B and avidin have a high content of high-mannose glycans.5 In this study, only three antibodies were investigated since most antibodies will have a very similar global rate of glycosylation. If all antibodies were to be considered in the model of the global rate of glycosylation, we would have faced a lack of variance in the model. The total mass of each protein was expressed in Da and was obtained based on the MALDI-TOF data, except for lactoferrin and Ribonuclease B, whose masses were obtained based on ESI-QTOF measurements. These data are illustrated in Table 1. The mass of the glycans is obtained by subtracting the theoretical mass of the amino acid sequence from the total mass. The assumption made during this study was that there is no post-translational modification other than glycosylation. The model of the global rate of glycosylation was established between 0% and 41% (w/w).| Proteins | Theoretical mass of the sequence (Da) | Intact mass (Da) | Global rate of glycosylation (% (w/w)) | Measurement method |
|---|---|---|---|---|
| Aflibercept | 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 918 918 |
114606 |
15.43 | MALDI-TOF |
| Alpha1-acid glycoprotein | 21560 |
36725 |
41.29 | |
| Alpha-crystalline | 19790 |
19915 |
0.63 | |
| Apo-transferrin | 75195 |
80039 |
6.05 | |
| Avidin | 14343 |
15955 |
10.10 | |
| Carboxypeptidase Y | 47319 |
57553 |
17.78 | |
| Cetuximab | 145440 |
153811 |
5.44 | |
| Conalbumin | 75828 |
77837 |
2.58 | |
| PPp | 7700 | 7686 | 0 | |
| Fetuin | 36353.24 |
46434 |
21.71 | |
| Infliximab | 145889.86 |
150598 |
3.13 | |
| Lactoferrin | 76165.29 |
81584 |
6.64 | ESI-QTOF |
| Lectin (Glycine max) | 27571 |
29445 |
6.36 | MALDI-TOF |
| Lectin (Maackia amurensis) | 27044.85 |
31982 |
15.44 | |
| Lectin (Phaseolus vulgaris) | 27347 |
29904 |
8.55 | |
| Nivolumab | 143616 |
147008 |
2.31 | |
| Peroxidase | 33918 |
43799 |
22.56 | |
| Ribonuclease B | 13690.3 |
15037 |
8.96 | ESI-QTOF |
Fig. 1 illustrates all the recorded spectra and, Fig. 2 shows the results of data modelling for the prediction of the global rate of glycosylation and Table 2 illustrates the performance criteria of both PLSR and SVR models. This result confirms the possibility to measure the global rate of glycosylation by FT-IR. The SVR model shows better results compared to PLSR in terms of R2CV of cross-validation (0.99 vs. 0.97) and RMSECV (0.38 vs. 2.20), R2Pred of external validation performance (0.99 vs. 0.89) and RMSEP (0.64 vs. 4.05) over a calibration range from 2 to 41% (w/w).
 | ||
| Fig. 1 FT-IR spectra recorded after the removal of residual salts and excipients present in the formulations of therapeutic proteins, using size exclusion spin columns for the analysis of the global rate of glycosylation. FT-IR spectra are measured over the 4000 and 600 cm−1 spectral range and are coloured according to the global rate of glycosylation. | ||
 | ||
| Fig. 2 Regression models with all glycoproteins in the spectral range between 1179 and 965 cm−1 for the analysed global rate of glycosylation. (A) PLS regression model. (B) SVR regression model. ◆ test set ● training set. | ||
| Model | PLSR | SVR | |
|---|---|---|---|
| Calibration | Number of latent variables | 7 | |
| Number of support vectors | 113 | ||
| R 2Cal | 0.98 | 1.00 | |
| R 2CV | 0.96 | 0.99 | |
| RMSEC (% (w/w)) | 1.70 | 0.17 | |
| RMSECV (% (w/w)) | 2.20 | 0.38 | |
| Validation | R 2Pred | 0.84 | 0.99 |
| RMSEP (% (w/w) | 4.05 | 0.64 | |
The optimal SVR parameters selected by cross-validation were set as: C = 10, ε = 0.0001 and γ = 0.01. In our case, a small ε leads to a narrow margin, which means a large number of support vectors were selected in the model. In this study, the SVR model was considered an adequate method for the prediction of the global rate of glycosylation. Indeed, in this case, the SVR method is better at handling the complex relationship between FT-IR spectra and the global rate of glycosylation than PLSR. This is possible since SVR adjusts the error within a particular threshold (±ε) with a maximum number of calibration samples. Thereby, the possibly nonlinear problem is transformed into a linear problem based on the mapping kernel function, particularly the Gaussian kernel with radial basis function (RBF). RBF has the particularity of being applied in the case of a strong nonlinear regularization of a complex system or when there is no prior knowledge of the data set. Finally, with a RPD ratio of 10.26 for the SVR model compared to 1.62 for the PLSR model (SD = 6.57), this confirms that the model of the global rate of glycosylation built with the SVR model presents good accuracy.
Comparison of the performances of PLSR and SVR to determine the composition of monosaccharides
The reference data were obtained via UPLC-FLR-MS with the mass spectrometry data to identify N-glycans and fluorescence data used for the glycan quantification. The Glycoworks RapiFluor MS method used in this study can only analyse N-glycans. As a result, in this study, all proteins containing O-glycans were excluded from the database used to build monosaccharide prediction models. This study was carried out on thirty-two therapeutic proteins among which: twenty-three monoclonal antibodies (mAbs), three mAbs biosimilars, one fusion protein (Fc) and five other glycoproteins of which three specific glycoproteins (alpha1-acid glycoprotein, avidin, ribonuclease B). Table S1† groups the composition of the main N-glycans for each glycoprotein. The composition is expressed in mass percentages and was obtained by relative peak areas – %RPA. Table S2† presents the overall mass percentage of the 5 monosaccharides present in each glycoprotein. Overall mass percentage of the monosaccharides was calculated from the mass percentages of glycans based on the structural combination between glycans and monosaccharides (Fig. S3†). We can deduce, as presented in Tables S3 and S4,† that ribonuclease B and avidin have elevated proportions of mannose monosaccharide, thus corresponding to high-mannose glycans. In contrast, alpha1-acid glycoprotein and aflibercept have elevated ratios of sialic acid, which corresponds to sialylated glycans. Cetuximab has the highest overall galactose content, which corresponds to N-glycans containing alpha-linked galactose. It also has the lowest mannose content. As for aflibercept, it contains the highest sialic acid and fucose levels.18Fig. 3 illustrates the measured spectra. The quantitative models predicting the amount of the monosaccharides (mannose, N-acetylglucosamine, galactose, fucose, sialic acid) were built based on the FT-IR spectra of these compounds. In addition, a signal-to-noise filter, as described previously, was applied to improve the performance of the models. Therefore, the methodology followed allowed us to overcome a possible spectral variability (Fig. S5†). As a result, 233 spectra were removed, and 613 spectra were retained to build the regression models in the spectral range between 1200 and 900 cm−1. These spectra are shown in Fig. S4.† 354 spectra (58%) were used as calibration set and 259 spectra (42%) were used as test set. The distribution of the samples for calibration and validation of this model is illustrated in Table S4.†Table 3 demonstrate the performances of both PLSR and SVR models in predicting the amount of each monosaccharide. Fig. 4 and 5 show the results of data modelling by the PLSR model and the SVR model, respectively. These results show that SVR models have a low value in terms of calibration (RMSEC), cross-validation performance (RMSECV) and of external validation performance (RMSEP). Also, these results show that SVR models have high R2CV and R2Pred values indicating that the models captured most of the correlation between the spectral data and the quantitative values.
| Mannose | N-Acetylglucosamine | Galactose | Fucose | Sialic acid | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Models | PLSR | SVR | PLSR | SVR | PLSR | SVR | PLSR | SVR | PLSR | SVR | |
| Calibration | Number of latent variables | 6 | 5 | 7 | 5 | 3 | |||||
| Number of support vectors | 262 | 263 | 308 | 247 | 191 | ||||||
| R 2Cal | 0.98 | 1.00 | 0.98 | 1.00 | 0.94 | 1.00 | 0.98 | 1.00 | 0.98 | 1.00 | |
| R 2CV | 0.98 | 0.99 | 0.98 | 0.99 | 0.94 | 0.99 | 0.98 | 0.99 | 0.98 | 1.00 | |
| RMSEC (% (w/w)) | 0.94 | 0.13 | 0.81 | 0.08 | 1.11 | 0.08 | 0.31 | 0.03 | 0.90 | 0.05 | |
| RMSECV (% (w/w)) | 0.99 | 0.40 | 0.83 | 0.27 | 1.17 | 0.46 | 0.33 | 0.09 | 0.91 | 0.14 | |
| Validation | R 2Pred | 0.85 | 0.84 | 0.91 | 0.93 | 0.80 | 0.90 | 0.91 | 0.97 | 0.98 | 0.99 |
| RMSEP (% (w/w)) | 1.91 | 0.84 | 1.34 | 1.09 | 2.31 | 1.27 | 0.47 | 0.29 | 0.99 | 0.55 | |
| SD | 2.10 | 3.87 | 3.47 | 1.64 | 4.29 | ||||||
| RPD | 1.10 | 2.51 | 2.88 | 3.56 | 1.50 | 2.73 | 3.47 | 5.58 | 4.33 | 7.77 | |
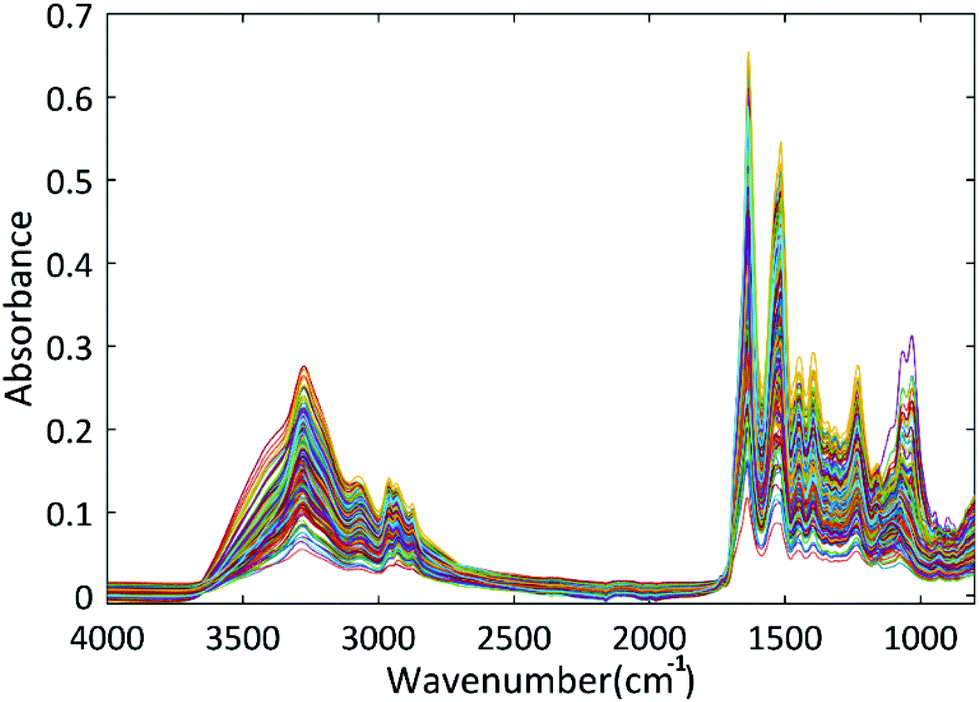 | ||
| Fig. 3 FT-IR spectra recorded after removal of residual salts and excipients present in the formulations of therapeutic proteins using size exclusion spin columns for analysis of the composition of monosaccharides. FT-IR spectra were recorded over the 4000 and 600 cm−1 spectral range. | ||
 | ||
| Fig. 4 Measured versus predicted amounts of monosaccharides obtained by PLS regression between 1200 and 950 cm−1 for the analysis of the amount of each monosaccharide. ◆ Test set ● Training set. | ||
 | ||
| Fig. 5 Measured versus predicted amounts of monosaccharides obtained by SVR regression between 1200 and 950 cm−1 to analyse the quantity of each monosaccharide. ◆ Test set ● Training set. | ||
Optimized SVR parameters are presented in Table S5.† Also, the Ratio of Performance to Deviation (RPD) of each model PLSR and SVR was calculated and presented in Table 3. As expected, it appears that the good values of RPD (ratio greater than two) were obtained by the SVR model indicating the good performances of this model. Moreover, for both PLSR and SVR models, the influence of the test and training set on the models was evaluated through the construction of two different models. These models were built by varying the distribution of proteins between the test and training set as shown in Tables S6 and S7.† The results of the analytical performances of each respective model are shown in Tables S8 and S9.† It emerged that the SVR models have a low value in terms of calibration (RMSEC), cross-validation performance (RMSECV) and external validation performance (RMSEP). In addition, the SVR models have high R2CV and R2Pred values.
Table S10† shows the results of the calculation of Ratio Performance to Deviation RPD1 for a first distribution of samples in the test and training set, and RPD2 for a second distribution. As expected, it appears in both variations, the models exhibit good values of RPD (ratio greater than 2) and these were obtained by the SVR model, which confirms a good robustness of these models. It emerges from this study that it is possible to measure the relative amounts of monosaccharides in protein glycosylation by FT-IR using SVR.
Hypothesis of the cause of non-linearity
During this study, the SVR models systematically outperformed PLSR models to capture the relationship between the amount of monosaccharides in glycoproteins calculated based on reference UPLC-FLR-MS analysis and the FT-IR data. This result seems to indicate that a complex relationship exists between these two parameters and that PLSR cannot model correctly.The theory underlying the application of PLSR assumes that spectra follow the Beer–Lambert law = εcl, where the absorbance A follows a linear model depending on the molar extinction coefficient ε of the analyte of concentration c, and on the optical path traveled by the optical beam l. Thereby, a system is considered linear if a simple dependency ratio is established. In the case where we have K constituents, the equation becomes: D = c1s1 + c2s2 + … + cisi. Where ci represent the concentration for sample i and si the spectra for sample i.
Therefore, it has been attempted to show that a more complex relationship exists by comparing theoretical (weighted sum of the spectra of each monosaccharide) and experimentally measured glycans spectra. In this context, the spectra of glycans (FA2, FA2G1, as well as the high mannose: Man-1, Man-3, Man-5, Man-6, Man-8) were studied.
For example, the theoretical spectrum of glycan FA2 is computed as:
| DFA2 = (4sN-acetylglucosamine + 3smannose + 1sfucose) |
Since it is known that FA2 is a combination of 4 N-acetylglucosamine, 3 mannose and 1 fucose. By comparing this DFA2 weighted spectrum with the reference spectrum of glycans FA2. Fig. 6 clearly shows that the calculated spectrum of glycans FA2 differs from the reference spectrum FA2 in terms of intensity and positions of the IR bands. In addition, similar results are observed for other glycans as illustrated in Fig. S6–S9.†
 | ||
| Fig. 6 (A) Illustrates the reference FT-IR spectrum of glycans FA2 recorded in ATR at 1 g L−1. (B) Illustrates the calculated theoretical FT-IR spectrum of glycans FA2 recorded at 10 g L−1 with DFA2 = (4sN-acetylglucosamine + 3smannose + 1sfucose)/10. | ||
As expected, the measured FT-IR spectrum of a glycan is not the simple sum of the different monosaccharide spectra. This reinforces the fact that a more complex relationship exists between the spectral data and the amount of monosaccharides. This might be explained by the existence of different environments surrounding the chemical bonds (different protein sequences implying different conformations) implying minor differences in the vibrational. Glycans contain the same monosaccharides but with different complex structures (Fig. S3†). Thus, the modes of vibration of the monosaccharide molecule can be influenced by the non-covalent interaction effects that may occur; also the vibration of molecules affects the vibration of other molecules.
Conclusions
Glycosylation is one of the critical attributes of biopharmaceuticals to be monitored from development to production. However, the conventional liquid chromatography and mass spectrometry analysis are complex with long sample preparation protocols. To overcome these limitations, we suggest using FT-IR spectroscopy in ATR mode to monitor the global glycosylation rate and the composition in monosaccharides of proteins. This approach has many advantages: reduced sample preparation since the analysis is carried out on the whole protein (no cleavage, labelling or separation step) and concise measurement time (approximately 5 minutes).First, the global rate of glycosylation on the intact proteins was modelled. To increase the range of application of the model, a wide variety of proteins was included in the calibration. To build the model, SVR regression was used in the spectral range between 1179 and 965 cm−1. The model presents good predictive performances in terms of RMSEP with 0.64% (w/w) and of RMSECV with 0.38% (w/w).
The second part of the study is specifically related to antibodies and their biosimilars, representing a large part of the biotherapeutic market. This study demonstrated the capacity of FT-IR spectroscopy to quantify the relative amount of each monosaccharide. In this context, the regression models were established on the spectral region of glycans, between 1200 and 950 cm−1. It was shown that the SVR models outperformed the PLSR ones exhibiting good performances in terms of RMSEP, RMSECV, underlining high robustness and high predictive accuracy of the models. Finally, the Ratio of Performance to Deviation (RPD) was evaluated for the different models. The RPD for SVR models value were more than doubled compared to PLS. Thus it confirms the accuracy of the SVR models.
This approach based on FT-IR spectroscopy combined with the SVR models, paves the way to three potential applications: comparing the glycosylation of a biosimilar and the original molecule, monitoring batch-to-batch homogeneity, and for in-process control.
Author contributions
Sabrina Hamla: Conceptualization, methodology, investigation, writing – original draft, writing – review & editing, software, formal analysis, visualization. Pierre-Yves Sacré: Conceptualization, writing – review & editing. Allison Derenne: Conceptualization, methodology, supervision, formal analysis, visualization, funding acquisition, project administration, writing – review & editing. Kheiro-Mouna Derfoufi: Investigation, formal analysis. Ben Cowper: Investigation, formal analysis, data curation, validation. Claire I. Butré: Investigation, formal analysis. Arnaud Delobel: Investigation, formal analysis. Erik Goormaghtigh: Funding acquisition, writing – review & editing. Philippe Hubert: Supervision, funding acquisition, project administration. Eric Ziemons: Conceptualization, supervision, funding acquisition, project administration, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This project was supported by the “Service Public de Wallonie-DGO6” (Walinnov 2017/2, convention # 1710032). We are grateful to the Saint-Pierre Hospital (Brussels, Belgium) and to the pharmacy of the University Hospital Center of Liège (CHU Liège, Belgium) for providing us all the therapeutic proteins. A special thanks to Hermane Avohou, Alice Kasemiire and Priyanka Kumari for proofreading the article. We acknowledge FNRS grant no. 001518F (EOS-convention # 30467715). E. G. is Research Director with the National Fund for Scientific Research (Belgium).References
- A. Planinc, J. Bones, B. Dejaegher, P. Van Antwerpen and C. Delporte, Glycan characterization of biopharmaceuticals: Updates and perspectives, Anal. Chim. Acta, 2016, 921, 13–27, DOI:10.1016/j.aca.2016.03.049.
- L. Zhang, S. Luo and B. Zhang, Glycan analysis of therapeutic glycoproteins, mAbs, 2016, 8(2), 205–215, DOI:10.1080/19420862.2015.1117719.
- M. Berger, M. Kaup and V. Blanchard, Protein Glycosylation and Its Impact on Biotechnology, in Genomics Syst. Biol. Mamm. Cell Cult, ed. W. S. Hu and A.-P. Zeng, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 165–185. DOI:10.1007/10_2011_101.
- J. C. Egrie, E. Dwyer, J. K. Browne, A. Hitz and M. A. Lykos, Darbepoetin alfa has a longer circulating half-life and greater in vivo potency than recombinant human erythropoietin, Exp. Hematol., 2003, 31, 290–299, DOI:10.1016/S0301-472X(03)00006-7.
- A. Derenne, K.-M. Derfoufi, B. Cowper, C. Delporte and E. Goormaghtigh, FTIR spectroscopy as an analytical tool to compare glycosylation in therapeutic monoclonal antibodies, Anal. Chim. Acta, 2020, 1112, 62–71, DOI:10.1016/j.aca.2020.03.038.
- V. Padler-Karavani, H. Yu, H. Cao, H. Chokhawala, F. Karp, N. Varki, X. Chen and A. Varki, Diversity in specificity, abundance, and composition of anti-Neu5Gc antibodies in normal humans: potential implications for disease, Glycobiology, 2008, 18, 818–830, DOI:10.1093/glycob/cwn072.
- M.-E. Lalonde and Y. Durocher, Therapeutic glycoprotein production in mammalian cells, J. Biotechnol., 2017, 251, 128–140, DOI:10.1016/j.jbiotec.2017.04.028.
- P. Hossler, Protein Glycosylation Control in Mammalian Cell Culture: Past Precedents and Contemporary Prospects, BT – Genomics and Systems Biology of Mammalian Cell Culture, ed. W. S. Hu and A.-P. Zeng, Springer Berlin Heidelberg, Berlin, Heidelberg, 2016, vol. 127, pp. 187–219 Search PubMed.
- P. Hossler, S. F. Khattak and Z. J. Li, Optimal and consistent protein glycosylation in mammalian cell culture, Glycobiology, 2009, 19(9), 936–949, DOI:10.1093/glycob/cwp079.
- L. Hajba, E. Csanky and A. Guttman, Liquid phase separation methods for N-glycosylation analysis of glycoproteins of biomedical and biopharmaceutical interest. A critical review, Anal. Chim. Acta, 2016, 943(November), 8–16, DOI:10.1016/j.aca.2016.08.035.
- M. Hilliard, et al., Glycan characterization of the NIST RM monoclonal antibody using a total analytical solution: From sample preparation to data analysis, mAbs, 2017, 9(8), 1349–1359, DOI:10.1080/19420862.2017.1377381.
- A. Barth, Infrared spectroscopy of proteins, Biochim. Biophys. Acta, Bioenerg., 2007, 1767(9), 1073–1101, DOI:10.1016/j.bbabio.2007.06.004.
- J. Kong and S. Yu, Fourier transform infrared spectroscopic analysis of protein secondary structures, Acta Biochim. Biophys. Sin., 2007, 39(8), 549–559, DOI:10.1111/j.1745-7270.2007.00320.x.
- E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, “Evaluation of the information content in infrared spectra for protein secondary structure determination, Biophys. J., 2006, 90, 2946–2957 CrossRef CAS PubMed.
- J. De Meutter and E. Goormaghtigh, FTIR Imaging of Protein Microarrays for High Throughput Secondary Structure Determination, Anal. Chem., 2021, 93(8), 3733–3741, DOI:10.1021/acs.analchem.0c03677.
- J. M. Girard, J. S. Deschênes, R. Tremblay and J. Gagnon, FT-IR/ATR univariate and multivariate calibration models for in situ monitoring of sugars in complex microalgal culture media, Bioresour. Technol., 2013, 144, 664–668, DOI:10.1016/j.biortech.2013.06.094.
- M. Khajehpour, J. L. Dashnau and J. M. Vanderkooi, Infrared spectroscopy used to evaluate glycosylation of proteins, Anal. Biochem., 2006, 348(1), 40–48, DOI:10.1016/j.ab.2005.10.009.
- U. Thissen, M. Pepers, B. Üstün, W. J. Melssen and L. M. C. Buydens, Comparing support vector machines to PLS for spectral regression applications, Chemom. Intell. Lab. Syst., 2004, 73(2), 169–179, DOI:10.1016/j.chemolab.2004.01.002.
- R. Tange, M. Rasmussen, E. Taira and R. Bro, Application of support vector regression for simultaneous modelling of near infrared spectra from multiple process steps, J. Near Infrared Spectrosc., 2015, 23, 75, DOI:10.1255/jnirs.1149.
- J. Workman, M. Koch and D. Veltkamp, Process analytical chemistry, Anal. Chem., 2007, 79(12), 4345–4363, DOI:10.1021/ac070765q.
- S. Challa and R. Potumarthi, Chemometrics-based process analytical technology (PAT) tools: Applications and adaptation in pharmaceutical and biopharmaceutical industries, Appl. Biochem. Biotechnol., 2013, 169(1), 66–76, DOI:10.1007/s12010-012-9950-y.
- S. Wold, M. Sjöström and L. Eriksson, PLS-regression: A basic tool of chemometrics, Chemom. Intell. Lab. Syst., 2001, 58(2), 109–130, DOI:10.1016/S0169-7439(01)00155-1.
- R. M. Balabin and E. I. Lomakina, Support vector machine regression (SVR/LS-SVM) - An alternative to neural networks (ANN) for analytical chemistry? Comparison of nonlinear methods on near infrared (NIR) spectroscopy data, Analyst, 2011, 136(8), 1703–1712, 10.1039/c0an00387e.
- E. Goormaghtigh, FTIR Data Processing and Analysis Tools, in Biol. Biomed. Infrared Spectrosc, ed. A. Barth and P. I. Haris, IOS Press, 2009, pp. 104–128 Search PubMed.
- E. Goormaghtigh and J. Ruysschaert, Subtraction of atmospheric water contribution in Fourier transform infrared spectroscopy of biological membranes and proteins, Spectrochim. Acta, 1994, 50, 2137–2144 CrossRef.
- L. Blanchet, J. Réhault, C. Ruckebusch, J. P. Huvenne, R. Tauler and A. de Juan, Chemometrics description of measurement error structure: Study of an ultrafast absorption spectroscopy experiment, Anal. Chim. Acta, 2009, 642(1–2), 19–26, DOI:10.1016/j.aca.2008.11.039.
- A. Gaigneaux, J. M. Ruysschaert and E. Goormaghtigh, Cell discrimination by attenuated total reflection-fourier transform infrared spectroscopy: The impact of preprocessing of spectra, Appl. Spectrosc., 2006, 60(9), 1022–1028, DOI:10.1366/000370206778397416.
- S. Paudel, P. H. Nguyen, W. L. Kling, M. Elmitri, B. Lacarrière and O. Le Corre, Support vector machine in prediction of building energy demand using pseudo dynamic approach, ECOS 2015 – 28th Int. Conf. Effic. Cost, Optim. Simul. Environ. Impact Energy Syst., no. July, 2015 Search PubMed.
- B. Schölkopf, Learning with kernels, Proc. 2002 Int. Conf. Mach. Learn. Cybern., 2002, vol. 1, pp. 11–48 Search PubMed.
- R. I. Tange, M. A. Rasmussen, E. Taira and R. Bro, Benchmarking support vector regression against partial least squares regression and artificial neural network: Effect of sample size on model performance, J. Near Infrared Spectrosc., 2017, 25(6), 381–390, DOI:10.1177/0967033517734945.
- D. A. Zavala-Ortiz, et al., Support Vector and Locally Weighted regressions to monitor monoclonal antibody glycosylation during CHO cell culture processes, an enhanced alternative to Partial Least Squares regression, Biochem. Eng. J., 2020, 154(November 2019), 107457, DOI:10.1016/j.bej.2019.107457.
- N. Long, D. Gianola, G. J. M. Rosa and K. A. Weigel, Application of support vector regression to genome-assisted prediction of quantitative traits, Theor. Appl. Genet., 2011, 123(7), 1065–1074, DOI:10.1007/s00122-011-1648-y.
- M. Ghorbani, G. Zargar and H. Jazayeri-Rad, Prediction of Asphaltene Precipitation using Support Vector Regression tuned with Genetic Algorithms, Petroleum, 2016, 2(3), 301–306, DOI:10.1016/j.petlm.2016.05.006.
- R. G. Brereton and G. R. Lloyd, Support Vector Machines for classification and regression, Analyst, 2010, 135(2), 230–267, 10.1039/b918972f.
- W. Ng, B. Minasny, B. Malone and P. Filippi, In search of an optimum sampling algorithm for prediction of soil properties from infrared spectra, PeerJ, 2018, 6, e5722, DOI:10.7717/peerj.5722.
- T. R. Viegas, A. L. M. L. Mata, M. M. L. Duarte and K. M. G. Lima, Determination of quality attributes in wax jambu fruit using NIRS and PLS, Food Chem., 2016, 190, 1–4, DOI:10.1016/j.foodchem.2015.05.063.
- B. Minasny and A. B. McBratney, Why you don ‘ t need to use RPD By Budiman Minasny & Alex. McBratney University of Sydney Why you don ‘ t need to use RPD, Pedometron, 2013,(33), 14–15 Search PubMed.
- V. Bellon-Maurel, E. Fernandez-Ahumada, B. Palagos, J. M. Roger and A. McBratney, Critical review of chemometric indicators commonly used for assessing the quality of the prediction of soil attributes by NIR spectroscopy, TrAC, Trends Anal. Chem., 2010, 29(9), 1073–1081, DOI:10.1016/j.trac.2010.05.006.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an00697e |
| This journal is © The Royal Society of Chemistry 2022 |