
Separation, identification, and confirmation of cyclic and tadpole macromolecules via UPLC-MS/MS†
Jason M.
O'Neill
a,
Jialin
Mao
a,
Farihah M.
Haque
b,
Fabienne
Barroso-Bujans
 cde,
Scott M.
Grayson
b and
Chrys
Wesdemiotis
*a
cde,
Scott M.
Grayson
b and
Chrys
Wesdemiotis
*a
aDepartment of Chemistry, The University of Akron, Akron, OH 44325, USA. E-mail: wesdemiotis@uakron.edu
bDepartment of Chemistry, Tulane University, New Orleans, LA 70118, USA
cDonostia International Physics Center (DIPC), Paseo Manuel Lardizábal 4, Donostia - San Sebastián 20018, Spain
dMaterials Physics Center, CSIC-UPV/EHU, Paseo Manuel Lardizábal 5, Donostia - San Sebastián 20018, Spain
eIKERBASQUE - Basque Foundation for Science, María Díaz de Haro 3, E-48013 Bilbao, Spain
First published on 27th April 2022
Abstract
Macrocyclic poly(glycidyl phenyl ether) (pGPE) synthesized via zwitterionic ring opening polymerization is typically contaminated by chains with linear and tadpole architecture. Although mass spectrometry (MS) analysis can readily confirm the presence of the linear byproduct, due to its unique mass, it is unable to differentiate between the cyclic and tadpole structures, which are constitutional isomers produced by backbiting reactions in monomeric or dimeric chains, respectively. To overcome this problem, ultraperformance reversed-phase liquid chromatography interfaced with electrospray ionization tandem mass spectrometry (UPLC-ESI-MS/MS) was employed. The separation achieved by UPLC revealed that the tadpole isomer elutes before the cyclic structure because of the increased polarity afforded by its distinctive substituents. The ratio of tadpole to cyclic species increased with the degree of polymerization, in agreement with the synthetic method used, as the potential for forming tadpole structures by backbiting is entropically favored in longer polymer chains. Once separated, the two isomers could be independently characterized by tandem mass spectrometry. The macrocyclic and tadpole species exhibit unique fragmentation patterns, including structurally diagnostic fragments for each structure.
Introduction
Cyclic polymers, often referred to as macrocycles or rings, are a class of polymers that exhibit unique chemical and physical properties, endowed by their lack of free chain ends (Fig. 1). When compared to linear polymers of the same molecular weight, cyclic polymers exhibit higher density, lower intrinsic viscosity, lower translational friction coefficients, higher glass transition temperatures, higher critical coelution temperatures, increased rates of crystallization, and higher refractive indexes.1–3 Unfortunately, the synthesis of purely cyclic polymeric material has proven to be challenging.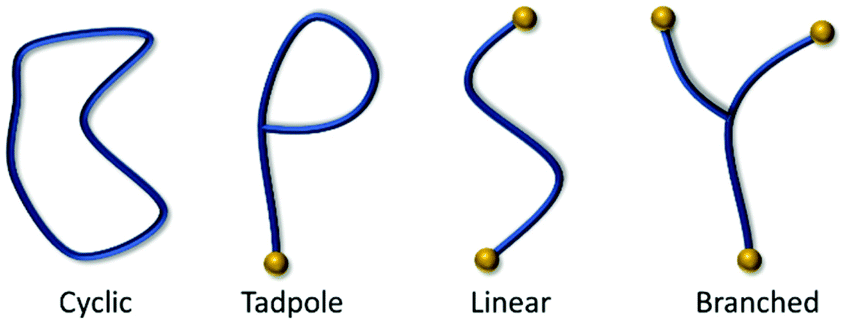 | ||
| Fig. 1 Examples of polymer topology ordered by increasing number of free end groups. | ||
Although the first observation of a cyclic molecule was reported over 100 years ago,4 interest in the cyclic topology only peaked after several cyclic biomacromolecules were discovered in the 1960s.3 Twenty years later, advances in the preparation of synthetic ring polymers began to progress and have recently reached a consistent flow of literature regarding strategies for the controlled synthesis of cyclic macromolecules.1–10 Most synthetic strategies for macrocycles can be divided into two categories, ring closure (RC) and ring expansion (RE). In RC approaches, the free chain ends of a linear polymer are reacted together to produce the macrocycle. Such ring closure can happen between identical end groups within the same polymer (homodifunctional unimolecular RC), different end groups within the same polymer (heterodifunctional unimolecular RC), or via the joining of two different linear polymers (homodifunctional bimolecular RC). Recent advances in RC reactions have significantly increased yields through the use of highly specific “click” chemistry to achieve ring closure; despite this progress, RC pathways still suffer from two serious drawbacks.9 First, ring formation has an associated entropic penalty that must be overcome. This entropic penalty increases with the degree of polymerization, limiting RC to the production of lower molecular weight (<20 kDa) species. Second, RC reactions are optimized at low concentrations, making them difficult to scale-up.3,8,10 To combat these challenges, synthetic chemists turned to the use of RE techniques, in which polymerization occurs via the addition of monomers into a weak bond of a pseudo cyclic chain. Because the polymerization takes place within a cyclic species the entire time, the entropic penalty associated with RC is reduced.7 This effect makes RE more suitable for the synthesis of high molecular weight macrocycles. Unfortunately, the mechanistic requirements of RE restrict the number of monomers that can undergo such reactions, thus limiting the diversity of cyclic polymers that can be synthesized by RE as compared to RC.3
Regardless of the synthetic pathway chosen, the formation of linear impurities and unwanted ring-containing byproducts in RE polymerizations, like tadpole oligomers, is inevitable. A presence of small amounts of non-cyclic impurity can cause a significant change in the chemical and physical properties of the polymeric material. As a result, the ability to detect, identify, and quantify these impurities is essential. Cyclic and non-cyclic species differ in their number of end groups. This makes analytical methods that can sense the presence of end groups the preferred approach for detecting non-cyclic impurities. Initial attempts involved the use of NMR spectroscopy and size exclusion chromatography (SEC), however, these tools are not ideal. NMR suffers heavily from bulk suppression; although the end groups may present unique chemical shifts, they only make up a small fraction of the total mass, depending on the degree of polymerization.3 SEC, on the other hand, does not have the resolving power to fully separate the cyclic and linear species, often sampling them as coeluting peaks.11 Moreover, neither of these techniques can distinguish between impurities with one or two end groups (i.e., linear vs. tadpole). As a result, mass spectrometry (MS) strategies are becoming the primary means for determining cyclic purity.12,13 A major advantage of MS is its ability to identify non-cyclic impurities of unique mass (literally unique mass-to-charge ratio, m/z). The existence of end groups in linear and tadpole species often results in their masses being different from those of the cyclic species, making them easily distinguishable by simple, one-dimensional MS. Nevertheless, depending on the type of synthesis employed, cyclic and non-cyclic species may be isomeric, making them indistinguishable by MS alone. In these cases, the use of separation techniques like ion mobility (IM) spectrometry or liquid chromatography (LC) prior to MS is often required. Liénard et al. recently demonstrated the successful separation of linear and cyclic species by ion mobility mass spectrometry (IM-MS), taking advantage of the more compact nature of cyclic ions.13 Unfortunately, due to differences in ionization efficiency between the linear and cyclic chains, cyclic purity could only be roughly estimated. In an alternative approach, Josse et al. reported the detection of linear contaminants in poly(lactic acid) (PLA) using survival yield (SY) data acquired by tandem mass spectrometry (MS/MS).12 In this technique, ions undergo fragmentation at gradually increasing collision energy, until they are totally depleted. Because cyclic structures require the scission of two bonds (rather than one in linear) to produce fragments, they can withstand higher collision energies than their linear counterparts before complete fragmentation. Using this logic, SY MS/MS was able to quantify as little as 2% linear contaminant. This approach requires the generation of a calibration curve from a series of standards with known cyclic to linear compositions. Such well controlled standards are difficult to find for many polymer types. Considering the shortcomings of even the most recent techniques, the characterization of polymer topology is an area of analytical chemistry in need of further development. The study reported here addresses this issue by putting forth an alternative methodology using LC-MS and LC-MS/MS for the relative quantitation of non-cyclic impurities. By targeting differences in polarity caused by polar end groups present in non-cyclic entities, LC-MS can facilitate the separation of these two species, so that they can be independently analyzed and quantified.
The macromolecular system investigated was poly(glycidyl phenyl ether), pGPE, formed by electrophilic zwitterionic ring expansion polymerization (ZREP) of glycidyl phenyl ether, GPE, using tris-(pentafluorophenyl)borane, B(C6F5)3, as Lewis acid initiator.14,15 This polymerization process provides a one-step route to prepare large quantities of macrocyclic polyethers (cf. Scheme S1†), but it cogenerates non-cyclic byproducts with linear and tadpole architecture (cf. Schemes S2–S5†).14,15 Linear contaminants (Schemes S2, S4, and S5†) and tadpole admixtures with a modified OH end group (Scheme S4†) can easily be detected by simple MS analysis, as their end group substituents shift them to a higher mass than those of the cyclic product, cf. Fig. S1.†15 On the other hand, the isomeric nature of the unmodified tadpole (TP-OH) and cyclic (C) species makes these two products indistinguishable via one-dimensional MS. This problem can be solved if the free hydroxy group of the tadpole (cf. Scheme S3†) is derivatized, for example by acetylation or propargylation, so that the tadpole mass is increased and does not overlap with that of the cyclic polyether. Such derivatization can also be used to remove the tadpole component and obtain pure macrocyclic polymer.15 However, this methodology is time and resource consuming and, more importantly, it depends upon the efficiency of the derivatization reaction; unreacted TP-OH would still remain undetected. In an alternative approach, the present study employs LC-MS with ultraperformance liquid chromatography (UPLC) for the separation and relative quantitation of the isomeric pGPE products. With UPLC, the TP-OH and C species can be separated for confident and conclusive identification by MS and MS/MS analysis, thus minimizing the use for end group derivatization.
Experimental section
Sample preparation
The pGPE polymer was prepared by ZREP, using solvents that were dried over calcium hydride to inhibit the formation of linear impurities; the detailed synthetic procedure has been reported elsewhere.15 Water (H2O) and acetonitrile (ACN) solvents used to make sample solutions and as UPLC mobile phase were purchased from Fischer Scientific (Fairlawn, NJ). Sodium acetate used as a UPLC mobile phase additive was purchased from Sigma Aldrich (St Louis, MO).A stock solution of pGPE was prepared in ACN at a concentration of 1 mg mL−1. This stock solution was diluted with H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ACN (1:1, v/v) to give a working concentration of 250 μg mL−1. Prior to preparation, the pGPE samples were kept at ∼3 °C.
:ACN (1:1, v/v) to give a working concentration of 250 μg mL−1. Prior to preparation, the pGPE samples were kept at ∼3 °C.
LC-MS analysis
All analyses were performed on a Waters Synapt G1 HDMS quadrupole/time-of-flight (Q/ToF) mass spectrometer equipped with electrospray ionization (ESI) and an inline Waters Acquity UPLC column (Waters Corp., Milford, MA). Separation was achieved via gradient elution on an Acquity UPLC Protein Bridged Ethylene Hybrid (BEH) C4 1.7 μm, 2.1 × 150 mm column; a C4 column led to superior separation efficiency as compared to columns of C8 or C18 functionality. The solvents were (A) H2O with 10 mM sodium acetate and (B) ACN with 10 mM sodium acetate; the added salt facilitated ionization by Na+ adduction. Gradient elution was performed as follows: 50% B to 60% B in 1 min; 60% B to 95% B in 12 min; hold at 95% B for 1 min (the rest was A).The eluates were ionized by ESI to positive ions under the following optimized conditions: ESI capillary voltage, 3.0 kV; sample cone voltage, 30 V; extraction cone voltage, 3.0 V; source temperature, 120 °C; desolvation temperature, 250 °C; desolvation gas (N2) flow, 500 L h−1. LC-MS spectra were acquired using ToF detection and Q in rf-only mode. LC-MS/MS spectra were acquired by setting Q in ion-selective mode to isolate the m/z of interest, applying a collision voltage of 80 V to the trap cell between the Q and ToF analyzers to cause collisionally activated dissociation (CAD), and detecting the CAD fragments in the ToF segment.
Results and discussion
LC-MS analysis of pGPE
Separation of the crude pGPE product was successfully performed via reversed-phase liquid chromatography (RPLC).16 The total ion chromatogram (TIC) acquired by ESI-MS of the eluates, and the corresponding combined ESI-MS spectrum of all eluates can be seen in Fig. 2. Separation was achieved based on polarity and the degree of polymerization, with the smaller and more polar oligomers eluting first (cf.Fig. 2a and S2†). The MS spectrum (Fig. 2b) contains a series of ions separated by a repeat unit of 150.07 Da, corresponding to the mass of the GPE monomer (C9H10O2). Accurate mass analysis of these ions identified them as sodiated cyclic/tadpole n-mers, [Cn/TPn-OH + Na]+, comprising n C9H10O2 repeat units (n = 4–13) and no additional substituents (no nominal end groups). The measured and theoretical m/z values of the identified oligomers are summarized in Table 1 and agree very well with each other (within <10 ppm), corroborating the composition [(C9H10O2)n + Na]+ for the observed cyclic/tadpole distribution.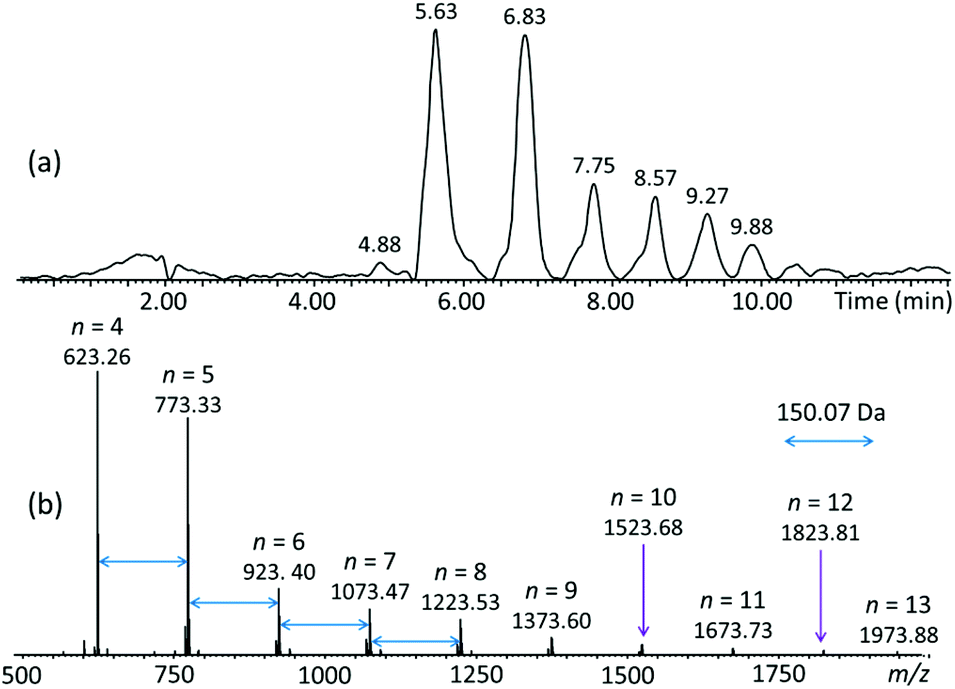 | ||
| Fig. 2 (a) LC-MS total ion chromatogram (TIC) of pGPE; the LC-MS spectra acquired at 4.88 and 5.63 min are depicted in Fig. S2.† (b) Summed ESI-MS spectrum of the TIC. The observed ions correspond to sodiated (C9H10O2)n oligomers with n = 4–13, this composition is common to the cyclic (C) as well as the tadpole (TP-OH) architecture. | ||
| Cn or TPn-OH | Chemical formula of M | Measured m/z | Theoretical m/z | Error in ppm |
|---|---|---|---|---|
| n = | ||||
| 4 | C36H40O8 | 623.2609 | 623.2615 | −0.96 |
| 5 | C45H50O10 | 773.3315 | 773.3296 | 2.46 |
| 6 | C54H60O12 | 923.3973 | 923.3977 | −0.43 |
| 7 | C63H70O14 | 1073.4728 | 1073.4658 | 6.52 |
| 8 | C72H80O16 | 1223.5347 | 1223.5339 | 0.65 |
| 9 | C81H90O18 | 1373.6038 | 1373.6019 | 1.38 |
| 10 | C90H100O20 | 1523.6799 | 1523.6700 | 6.50 |
| 11 | C99H110O22 | 1673.7291 | 1673.7381 | −5.38 |
| 12 | C108H120O24 | 1823.8090 | 1823.8062 | 1.54 |
| 13 | C117H130O26 | 1973.8795 | 1973.8743 | 2.63 |
Although the TIC in Fig. 2 shows separated LC peaks, these have unresolved shoulders, affirming the compositional complexity of the analyzed polyether sample. Noteworthily, oligomers of the same mass are eluted within distinct LC peaks, as illustrated in Fig. S2† for the pGPE 4-mers (m/z 623.26); this result strongly suggests that both the cyclic as well as the tadpole architectures are present in the product. To confirm this expectation, extracted ion chromatograms (XICs) were generated. An XIC only shows the elution times of a select m/z value, providing an isolated view of oligomers detected at this m/z. The XIC of m/z 623.26 (Fig. 3), which corresponds to the cyclic/tadpole 4-mer, shows two baseline resolved peaks, one peaking at the retention time of 4.91 min and one, with significantly higher abundance, slightly later at 5.63 min. Because this XIC only displays components with the chosen mass (m/z 623.26), the observation of two unique, separated peaks indicates successful separation of the two isomeric architectures. The tadpole was assigned to the earlier eluting peak in the chromatogram for two reasons: (i) since the product was formed via ZREP, which favors the formation of cyclic species,14,15 the cyclic architecture should dominate over the tadpole, especially at lower degrees of polymerization; (ii) the primary difference between the C4 and TP4-OH architectures is the inclusion of a polar hydroxy end group in the tadpole isomer, which should cause earlier elution for TP4-OH under RPLC conditions (see below).16
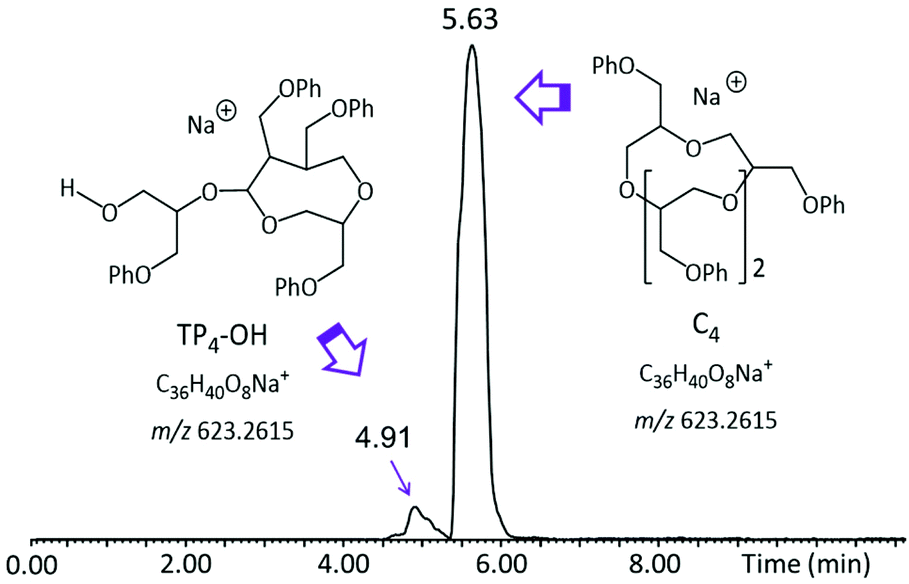 | ||
| Fig. 3 Extracted ion chromatogram (XIC) of the [M + Na]+ ion of the pGPE 4-mer (m/z 623.26), depicting RPLC separation of the tadpole and macrocyclic components of this oligomer. | ||
According to RPLC theory, the increased polarity of the TP-OH species decreases its interaction with the nonpolar stationary phase (C4), thereby shortening its retention time as compared to the cyclic counterpart.16 This relationship between polarity and retention time allows for the logical assignment of tadpole and macrocyclic structures to the tetramers eluting at 4.91 and 5.63 min, respectively. Such separation of the isomers by LC enables estimation of their relative amounts as well as their individual characterization by MS/MS (vide infra). To examine if the separation observed for the two 4-mers also happens for the other pGPE n-mers, XICs for the chain lengths from n = 5 to n = 13 were generated (Fig. 4). In most cases, two distinct peaks, corresponding to the tadpole and cyclic isomers, are observed. At shorter chain lengths (n = 4–8), the cyclic architecture dominates. However, as the degree of polymerization increases, the fraction of the tadpole architecture also increases, until eventually becoming more prevalent than the cyclic structure at n = 9. These trends agree with ZREP synthetic predictions, as the potential to form a tadpole via backbiting (Scheme S3†) should increase with increasing polymer chain length.14,15 Once the degree of polymerization exceeds 11, the tadpole species is the only structure observed, cf. Fig. 4 and Table S1.†
 | ||
| Fig. 4 Stacked XICs for the pGPE oligomers with n = 4–13 repeat units. Red and blue peaks represent tadpole and cyclic species, respectively. The ratio of the cyclic to tadpole peak areas is given in the column at the right side. See Table S1† for the corresponding retention times and absolute peak areas. | ||
LC-MS/MS analysis of pGPE
In order to confirm the architectures deduced by LC-MS and polarity considerations, the putative tadpole and cyclic isomers were subjected to LC-MS/MS via collisionally activated dissociation (CAD).16–20 The MS/MS spectra of the corresponding 7-mers (n = 7; m/z 1073.47) can be seen in Fig. 5.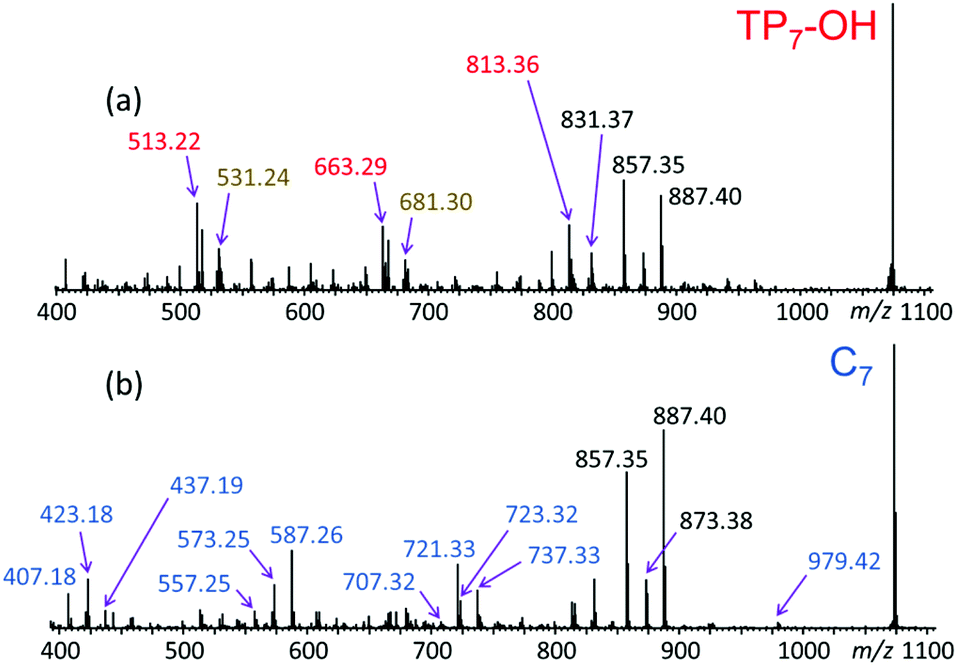 | ||
| Fig. 5 LC-MS/MS spectra of the sodiated 7-mers (m/z 1073.47) eluting at (a) 7.84 and (b) 8.57 min (cf.Fig. 4 and Table S1†). Based on their elution order in RPLC, these eluates correspond to (a) TP7-OH and (b) C7, respectively. | ||
The fragmentation patterns in Fig. 5a and b clearly attest that tadpole and cyclic pGPE architectures produce similar fragment ions in the upper m/z range, but uniquely different fragments in the middle and lower m/z ranges of the spectra. The upper fragment mass ranges of both MS/MS spectra include abundant fragment ions at m/z 887.40 and 857.35, corresponding to [M − 2PhO˙ + Na]+ and [M − 2PhOCH3 + Na]+, respectively (M = TP7OH or C7). Since the losses of PhO˙ and PhOCH3 occur within the side chain and not the backbone of pGPE, they do not help to distinguish between the cyclic and tadpole structures. It is noteworthy, however, that fragment ion [M − 2PhO˙ + Na]+ (m/z 887.40) is more abundant from the cyclic than the tadpole precursor, pointing out that the loss of two phenoxy radicals proceeds more efficiently from the cyclic architecture.
The middle and low mass regions of the tadpole LC-MS/MS spectrum (Fig. 5a) are dominated by fragments of m/z 813.36, 663.29, and 513.22 which all belong to the fragment ion series [150n + 40 + Na]+. These fragments are accounted for by a charge-remote fragmentation pathway initiated by the loss of a PhO˙ radical to form a primary –CH2˙ radical within the pGPE side chain, cf. Scheme 1a. The latter radical can isomerize to a more stable tertiary radical through a 1,5-H rearrangement (backbiting), denoted by 1,5-rH˙ in Scheme 1a. If this radical is formed along the tadpole tail, it can induce β scission of an adjacent O–C bond, resulting in the loss of a tail piece and a fragment ion with a terminal alkene as the new tail end. Because this pathway is dependent upon cleavage within the tail, it is not observed in the LC-MS/MS spectrum of the cyclic species (Fig. 5b). As a result, these structurally diagnostic fragment ions (Scheme 1b) can be used to positively identify the presence of tadpole species.
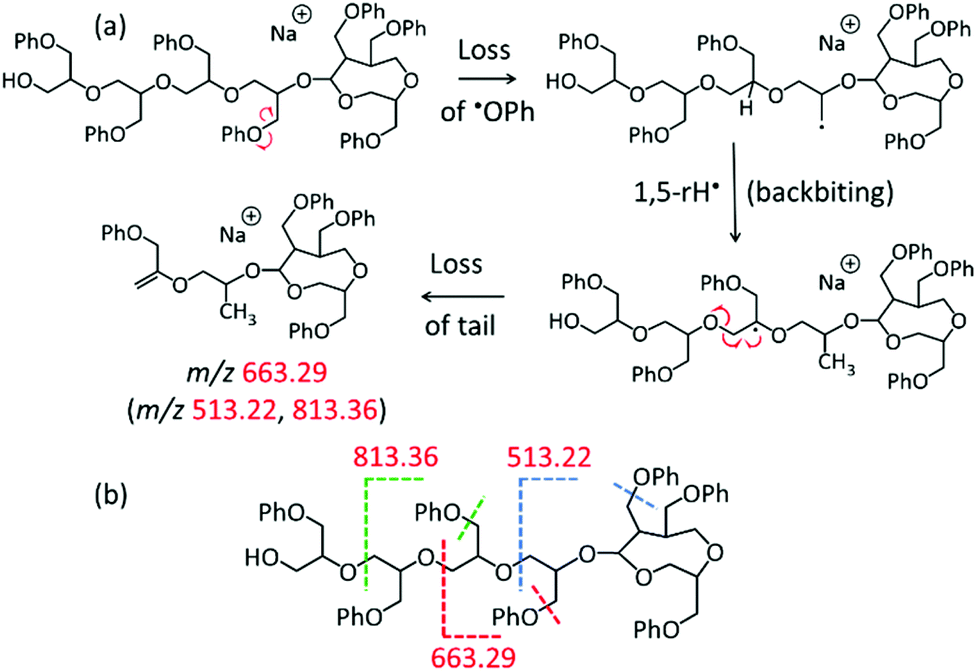 | ||
| Scheme 1 (a) Fragmentation pathway to the fragment ion series [150n + 40 + Na]+, illustrated for the formation of the fragment at m/z 663.29; (b) similar fragmentation pathways lead to the fragments at m/z 513.22 and 813.36. | ||
All three fragments in the [150n + 40 + Na]+ series of Fig. 5a (marked by red m/z) can arise from a TP7-OH tadpole consisting of a tail with four repeat units tethered to a head with three repeat units, cf. Scheme 1. The same fragments can also be formed from an isomeric tadpole with a smaller head and longer tail (2 and 5 repeat units, respectively), cf. Fig. S3.† Tadpoles with larger heads and shorter tails than that in Scheme 1 can only contribute to the heavier members of the [150n + 40 + Na]+ series (m/z 663.29 and/or 813.36, based on head/tail sizes). Larger head sizes would contribute fragments characteristic for the macrocyclic architecture, such as m/z 723.32 and 737.33 (cf.Fig. 5b); however, these are minuscule in the LC-MS/MS spectrum of the tadpole (Fig. 5a), suggesting that smaller head sizes (2–3 repeat units for the 7-mer) predominate.
A second set of unique fragments for the tadpole appear at m/z 681.30, 531.24, and 381.17 (Fig. 5a and S3†) and belong to the fragment ion series [150n + 58 + Na]+ (marked by brown m/z). All can be rationalized by β C–O bond cleavage (no prior backbiting), induced by the –CH2˙ radical created on a nearby side chain via PhO loss, cf. Scheme S6.† This reaction detaches the head and a segment of the tail, giving rise to a fragment ion containing the residual tail and a new propenyl end group (Scheme S6†).
The LC-MS/MS spectrum of the cyclic pGPE isomer C7 (Fig. 5b) includes several fragment ions that are significantly more abundant than in the spectrum of the tadpole and, hence, constitute signatures of the macrocyclic architecture (marked by blue m/z). These ions can arise after ring opening, initiated again by the loss of a PhO˙ radical to form a –CH2˙ terminated radical on one of the macrocycle's side chains. Subsequent β C–O bond scission and H˙ loss produce a linear chain with aldehyde and vinyl end groups (m/z 979.42 in Scheme 2, top). Alternatively, isomerization of the primary to a more stable tertiary radical may take place before ring opening. In this case, consecutive β C–O bond cleavage and H˙ loss give rise to an isomeric linear chain with keto and vinyl end groups (m/z 979.42 in Scheme 2, bottom). The tertiary radical can also undergo a competitive β C–O bond cleavage to release a second PhO˙ radical, which yields the most abundant fragment ion at m/z 887.40.
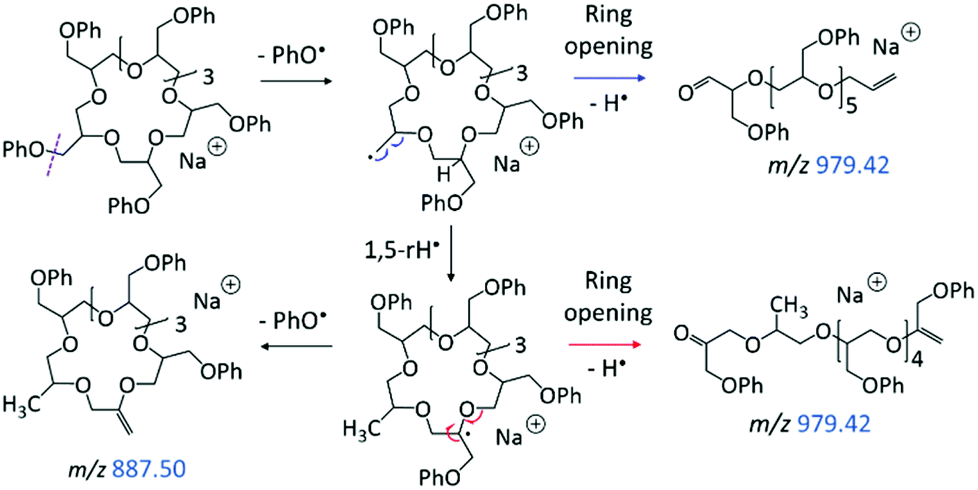 | ||
| Scheme 2 Homolytic ring-opening of sodiated C7 (m/z 1073.47), initiated by PhO˙ loss. Smaller fragment ions are attributed to the ring-opened structures (Schemes 3–4). | ||
The linear fragments emerging after PhO˙ + H˙ loss (m/z 979.42 in Scheme 2) are not observed in high intensity, as they can readily undergo further fragmentation to form smaller ions, such as m/z 423.18, 587.26, and 721.33 (cf.Fig. 5b); plausible dissociation pathways are presented in Schemes 3 and 4. Homolytic ether bond cleavages along the linear backbone result in the formation of radical ions, as illustrated in Scheme 3 for both ring-opened intermediates of m/z 979.42. Depending on the location of the unpaired electron, these radical ions can undergo H˙ rearrangement followed by the loss of PhO˙ to generate the fragment ions outlined in Scheme 3, top; or they can fragment via PhOCH2˙ elimination to produce the fragments outlined in Scheme 3, bottom. These pathways are exemplified for the formation of m/z 587.26 and 423.18, respectively, but also account for analogous fragments containing more or less repeat units (Scheme 3).
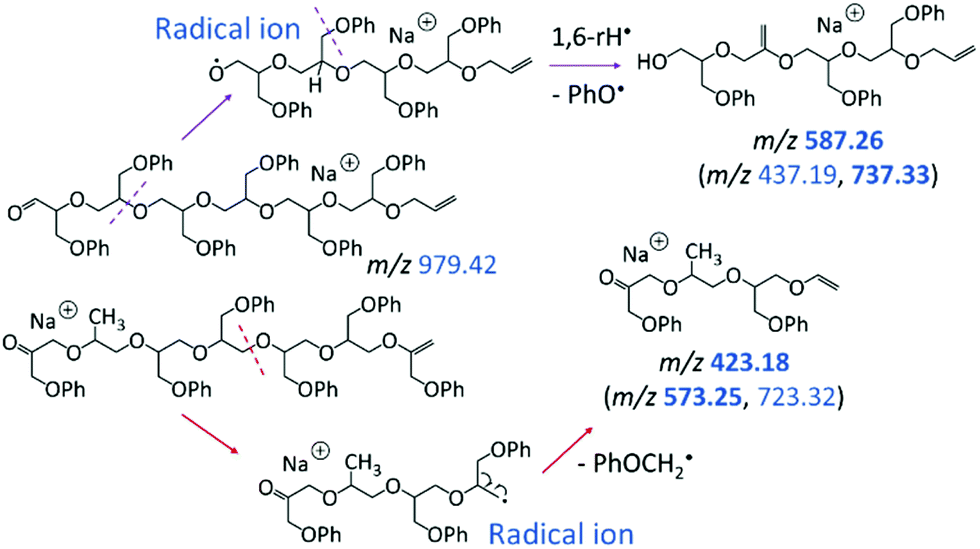 | ||
| Scheme 3 Dissociation of the ring-opened intermediates with aldehyde and vinyl or keto and vinyl end groups (both at m/z 979.42), leading to the fragments at m/z 587.26 and 423.18, respectively. Fragments with the same substituents, i.e. series [150n + 114 + Na]+ and [150n + 100 + Na]+, respectively, arise through analogous bond cleavages; their m/z values are given in parenthesis, with intense ions indicated in boldface. | ||
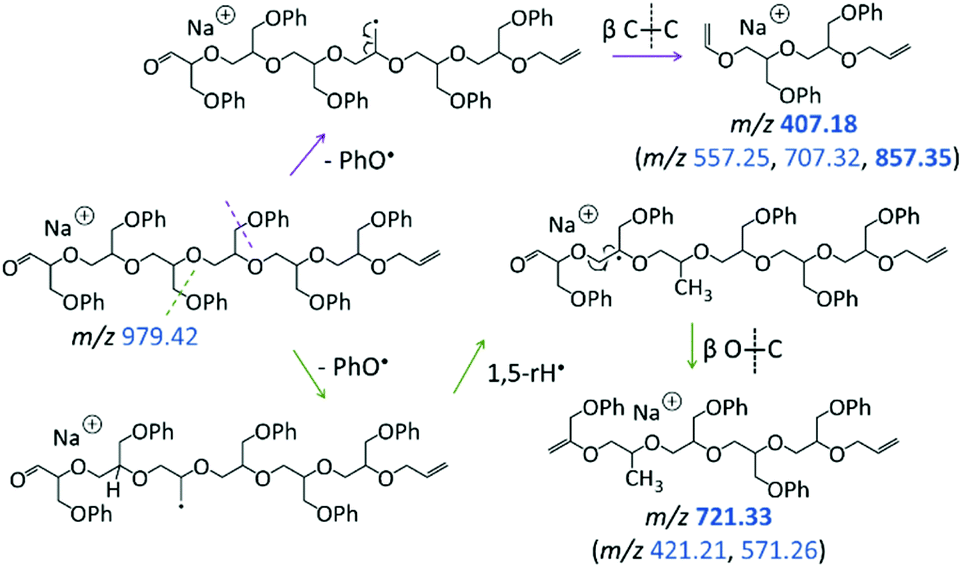 | ||
| Scheme 4 Dissociation of the ring-opened intermediate with aldehyde and vinyl end groups (m/z 979.42), leading to m/z 407.18, 721.33, and analogous fragments with the 150 Da repeat unit (i.e., series [150n + 84 + Na]+ and [150n + 98 + Na]+, respectively). The m/z values of intense ions are indicated in boldface. | ||
The other abundant fragment ions in the LC-MS/MS spectrum of the macrocycle are accounted for by initial PhO˙ loss from the ring-opened intermediate with aldehyde and vinyl end groups (Scheme 4). Successive β C–C bond scission gives rise to fragments with vinyl and propenyl end groups (Scheme 4, top); whereas radical migration followed by β O–C bond scission gives rise to fragments with phenoxy propenyl and propenyl end groups (Scheme 4, bottom). Collectively, due to their dominance from the cyclic structure, the fragment ions in Schemes 3 and 4 can be used to confidently determine the presence of pGPE species with cyclic architecture.
The LC-MS/MS characteristics of cyclic and tadpole isomers were discussed in detail for the pGPE 7-mers, but other oligomer sizes give rise to completely analogous fragmentation patterns, as affirmed by the corresponding 8-mers in Fig. S4.† Consistently, the relative abundances of the fragments generated via the pathways in Schemes 1 and S6† are higher in the MS/MS spectrum of the tadpole, whereas the fragments arising via the pathways in Schemes 2–4 show higher relative abundances in the MS/MS spectrum of the macrocyclic isomer.
It is worth noting that the chromatographic peaks for the tadpole and macrocycle species in Fig. 4 exhibit a shoulder as the oligomer size increases. This shape is attributed to different tail lengths within the tadpole structures and to different conformers of the sodiated macrocycles, respectively.
Conclusions
Cyclic polymers exhibit unique physical and chemical properties attributed to their cyclic topology. Non-cyclic impurities like linear and tadpole architectures, even in low concentration, can result in significant changes in the observed properties of the cyclic polymers. Unfortunately, the synthesis of highly pure cyclic polymers is challenging and requires either specific synthetic restraints or the use of complex modification and purification techniques for the removal of impurities. As a result, the ability to characterize the products of macrocycle syntheses is vital to the development of cyclic polymers. In this work, UPLC-MS/MS was used to facilitate the separation and characterization of pGPE prepared by ZREP. Oligomers with a degree of polymerization (DP, n) ranging from 4 to 13 were observed as sodiated ions. Separation of isomeric tadpole and cyclic architectures was achieved through RPLC. The presence of an additional free hydroxy end group in the tadpole structure resulted in faster elution as compared to the cyclic counterpart. Once separated, the relative amount of the cyclic and tadpole species was evaluated as a function of the DP (i.e., chain length). It was observed that the tadpole proportion increased with the DP. This observation coincides with synthetic predictions, as backbiting reactions that form the tadpole become increasingly more likely with further polymerization. Cyclic species were shown to dominate over tadpoles until n = 8, where the ratio shifted in favor of the tadpole species. Eventually by n = 12, cyclic species were no longer observed.The cyclic and tadpole structures assigned via UPLC-MS were confirmed for the n = 7 oligomer by MS/MS. The resulting spectra showed uniquely different fragment ions for the tadpole and cyclic isomers. Two series of ions corresponding to fragmentation events within the tadpole tail were identified (m/z 513.22, 663.29, and 813.36; as well as 381.17, 531.24, and 681.30). These ions are negligible in the LC-MS/MS spectrum of the macrocycle, which makes them ideal for identifying the presence of tadpole species. Conversely, several ions unique to the cyclic species were also observed (for example, m/z 423.18, 587.26, and 721.33). Analogous fragmentation characteristics were observed for the n = 8 oligomer. Hence, LC-MS/MS fragment ions can be used to establish a ratio of tadpole to cyclic architectures and provide information about the success of synthetic parameters designed to favor cyclic entities.
The described LC-MS (MS/MS) approach provides the ability to separate and quantitate non-cyclic impurities formed during the synthesis of cyclic polymers. This is an important task, as even small amounts of the non-cyclic admixtures can compromise product performance.
Author contributions
J. M. O. and C. W. conceptualized the separation and characterization strategy; F. M. H., F. B. B, and S. M. G designed the synthetic strategy and prepared pGPE; J. M. O and J. M. performed the LC-MS and MS/MS experiments; J. M. O. and C. W. analyzed and interpreted the experimental results; J. M. O. and C. W. composed the original draft; all authors reviewed and edited the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Science Foundation (CHE-1808115 to C. W.) is gratefully acknowledged.Notes and references
- K. Endo, Synthesis and Properties of Cyclic Polymers, Adv. Polym. Sci., 2008, 217, 121–183, DOI:10.1007/12_2008_157.
- Z. Jia and M. J. Monteiro, Cyclic Polymers: Methods and Strategies, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085–2097, DOI:10.1002/pola.25999.
- R. Liénard, J. De Winter and O. Coulembier, Cyclic Polymers: Advances in Their Synthesis, Properties, and Biomedical Applications, J. Polym. Sci., 2020, 58, 1481–1502, DOI:10.1002/pol.20200236.
- P. Ruggli, A Ring with Triple Bonds, Justus Liebigs Ann. Chem., 1912, 392, 92–100, DOI:10.1002/jlac.1912392010.
- B. A. Laurent and S. M. Grayson, Synthetic Approaches for the Preparation of Cyclic Polymers, Chem. Soc. Rev., 2009, 38, 2202–2213, 10.1039/b809916m.
- H. R. Kricheldorf, Cyclic Polymers: Synthetic Strategies and Physical Properties, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284, DOI:10.1002/pola.23755.
- H. A. Brown and R. M. Waymouth, Zwitterionic Ring-Opening Polymerization for the Synthesis of High Molecular Weight Cyclic Polymers, Acc. Chem. Res., 2013, 46, 2585–2596, DOI:10.1021/ar400072z.
- Y. Zhu and N. S. Hosmane, Advanced Developments in Cyclic Polymers: Synthesis, Applications, and Perspectives, ChemistryOpen, 2015, 4, 408–417, DOI:10.1002/open.201402172.
- Y. A. Chang and R. M. Waymouth, Recent Progress on the Synthesis of Cyclic Polymers via Ring-Expansion Strategies, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2892–2902, DOI:10.1002/pola.28635.
- F. M. Haque and S. M. Grayson, The Synthesis, Properties and Potential Applications of Cyclic Polymers, Nat. Chem., 2020, 12, 433–444, DOI:10.1038/s41557-020-0440-5.
- R. Elupula, J. Oh, F. M. Haque, T. Chang and S. M. Grayson, Determining the Origins of Impurities during Azide-Alkyne Click Cyclization of Polystyrene, Macromolecules, 2016, 49, 4369–4372, DOI:10.1021/acs.macromol.6b00968.
- T. Josse, J. De Winter, P. Dubois, O. Coulembier, P. Gerbaux and A. Memboeuf, A Tandem Mass Spectrometry-Based Method to Assess the Architectural Purity of Synthetic Polymers: A Case of a Cyclic Polylactide Obtained by Click Chemistry, Polym. Chem., 2015, 6, 64–69, 10.1039/c4py01087f.
- R. Liénard, Q. Duez, S. M. Grayson, P. Gerbaux, O. Coulembier and J. De Winter, Limitations of Ion Mobility Spectrometry–mass Spectrometry for the Relative Quantification of Architectural Isomeric Polymers: A Case Study, Rapid Commun. Mass Spectrom., 2020, 34, e8660, DOI:10.1002/rcm.8660.
- F. M. Haque, A. Alegria, S. M. Grayson and F. Barroso-Bujans, Detection, Quantification, and “Click-Scavenging” of Impurities in Cyclic Poly(Glycidyl Phenyl Ether) Obtained by Zwitterionic Ring-Expansion Polymerization with B(C6F5)3, Macromolecules, 2017, 50, 1870–1881, DOI:10.1021/acs.macromol.6b02755.
- F. M. Haque, C. M. Schexnayder, J. M. Matxain, F. Barroso-Bujans and S. M. Grayson, MALDI-ToF MS Study of Macrocyclic Polyethers Generated by Electrophilic Zwitterionic Ring Expansion Polymerization of Monosubstituted Epoxides with B(C6F5)3, Macromolecules, 2019, 52, 6369–6381, DOI:10.1021/acs.macromol.9b01050.
- C. Wesdemiotis, Multidimensional Mass Spectrometry of Synthetic Polymers and Advanced Materials, Angew. Chem., Int. Ed., 2017, 56, 1452–1464, DOI:10.1002/anie.201607003.
- C. Wesdemiotis, N. Solak, M. J. Polce, D. E. Dabney, K. Chaicharoen and B. C. Katzenmeyer, Fragmentation Pathways of Polymer Ions, Mass Spectrom. Rev., 2011, 30, 523–539, DOI:10.1002/mas.20282.
- T. Fouquet, J. Petersen, J. A. S. Bomfim, J. Bour, F. Ziarelli, D. Ruch and L. Charles, Electrospray Tandem Mass Spectrometry Combined with Authentic Compound Synthesis for Structural Characterization of an Octamethylcyclotetrasiloxane Plasma Polymer, Int. J. Mass Spectrom., 2012, 313, 58–67, DOI:10.1016/j.ijms.2012.01.007.
- A. M. Yol, D. E. Dabney, S.-F. Wang, B. A. Laurent, M. D. Foster, R. P. Quirk, S. M. Grayson and C. Wesdemiotis, Differentiation of Linear and Cyclic Polymer Architectures by MALDI Tandem Mass Spectrometry (MALDI-MS2), J. Am. Soc. Mass Spectrom., 2013, 24, 74–82, DOI:10.1007/s13361-012-0497-5.
- S. Crotty, S. Gerişlioǧlu, K. J. Endres, C. Wesdemiotis and U. S. Schubert, Polymer Architectures via Mass Spectrometry and Hyphenated Techniques, Anal. Chim. Acta, 2016, 932, 1–21, DOI:10.1016/j.aca.2016.05.024.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an00208f |
| This journal is © The Royal Society of Chemistry 2022 |