DOI:
10.1039/D2AN00242F
(Paper)
Analyst, 2022,
147, 1859-1865
Ultrasensitive detection of tumor-derived small extracellular vesicles based on nonlinear hybridization chain reaction fluorescence signal amplification and immunomagnetic separation†
Received
9th February 2022
, Accepted 24th March 2022
First published on 30th March 2022
Abstract
Small extracellular vesicles (sEVs) have attracted wide attention as a promising tumor biomarker. However, sensitive and selective detection of sEVs is challenging due to the low levels of sEVs in the early stage of cancers. Herein, a novel fluorescent sensor was developed for the detection of sEVs with high sensitivity and selectivity based on nonlinear hybridization chain reaction (nHCR) signal amplification and immunomagnetic separation. Firstly, sEVs were captured and enriched by CD63 antibody conjugated magnetic beads via antibody–antigen reactions. Then, cholesterol-modified DNA probes were anchored spontaneously on lipid membranes of sEVs through efficient hydrophobic interactions between the cholesterol moiety and the phospholipid bilayer of sEVs. The simultaneous recognition of the transmembrane protein and the phospholipid bilayer structure of the sEVs could effectively eliminate interferences from free proteins. The sticky ends of the cholesterol-modified DNA probes acted as the initiator to trigger nHCR to form a hyperbranched network of DNA structure that could recruit more fluorescent signal molecules for signal amplification. Under the optimal conditions, the nHCR-based strategy showed high sensitivity for the detection of sEVs with a limit of detection of 80 particles per μL. In addition, the as-constructed method was successfully applied for the analysis of clinical samples. It provides a sensitive and selective platform for the isolation and detection of sEVs in the early diagnosis of cancers.
Introduction
Small extracellular vesicles (sEVs), a subgroup of extracellular phospholipid membranous vesicles with a diameter of 50–200 nm, are secreted by most cells.1,2 sEVs shed by cancer cells carry diverse biomolecules (e.g., proteins, bioactive lipids, nucleic acids, and metabolites), which play major roles in the regulation of the tumor microenvironment and pathogenesis of human malignancies, including cancer metastasis and tumor progression.3,4 It has been reported that tumor cells can produce a higher concentration of sEVs than normal cells, suggesting that cancer samples contain a high abundance of sEVs.5,6 Relatively high abundance and excellent stability of sEVs offer significant advantages in cancer diagnosis compared to other markers such as circulating tumor cells and cell-free DNA that are short-lived and rare in body fluids.7,8 Therefore, sEVs hold great promise as important noninvasive biomarkers for early cancer diagnostics.9
So far, various analytical methods have been developed to detect cancer cell-derived sEVs, including flow cytometry, western blot (WB), and enzyme-linked immunosorbent assay. However, the relatively low sensitivity of these methods severely hinders early cancer diagnosis. In addition, they are limited by complex instrument operation and high cost.10–12 To overcome these shortcomings, some DNA signal amplification technologies have been developed for quantitative detection of tumor sEVs, such as rolling circle amplification,13,14 DNA walkers,15,16 and terminal deoxynucleotidyl transferase-initiated DNA amplification.17,18 Although these strategies can enhance analytical performance to a certain extent, they are mostly enzyme-assisted signal amplification reactions, which might suffer from poor reproducibility. Compared with enzymatic amplification, the enzyme-free signal amplification has great advantages due to its stability, low cost, and simplicity.19–21 The development of an enzyme-free amplification platform with high sensitivity and selectivity is in ever-increasing demand for the detection of sEVs.
Recently, hybridization chain reaction (HCR) has attracted great attention for its enzyme-free, isothermal, entropy-driven spontaneous DNA assembly process. In a traditional linear HCR process, a DNA initiator triggers the cross-opening of two DNA hairpins to form strand-like DNA nanowires.22 In recent years, a variety of exponential expansion nHCR amplification strategies with high dimensions have been reported, including dendritic HCR, branched HCR, and clamped HCR.23–25 The formation of a super-chain structure through nHCR in the presence of an initiator results in an exponential increase in signal intensity and thereby improves the sensitivity of biosensing platforms.26 It is worth noting that fluorescence assays based on nHCR are becoming a popular detection technique due to simple operation and high sensitivity.23,27 Recently Liu and coworkers constructed an nHCR-based fluorescent DNA dendrimer–streptavidin (SA) complex for tumor cell sensing. The hyperbranched nanostructure recruited the multimolecule labeling of SYBR Green I, further improving the sensitivity and selectivity.28 In view of the advantages of nHCR, such as enzyme free, fast reaction kinetics, and exponential isothermal amplification, the development of nHCR-based fluorescence strategies is of great necessity for the construction of sEV biosensors.
Herein, we developed a novel biosensing platform for the sensitive and selective detection of sEVs based on immunomagnetic separation-assisted nHCR fluorescence signal amplification. As described in Scheme 1(A), the biotin-modified DNA hairpins (H1 and H2) are respectively connected to the four binding sites of SA to form four-armed protein scaffolds (tetrads-H1 and tetrads-H2) by utilizing the strong covalent interaction through SA and biotin. The hyperbranched DNA structures are activated in the presence of an initiator. FAM labeled on both DNA hairpins acts as the fluorescence signal output. As shown in Scheme 1(B), the method consists of three steps. Firstly, MCF-7 cell-derived sEVs were captured and enriched using CD63 antibody conjugated magnetic beads (MBs–CD63). Then, the MB-binding sEVs (MB–sEVs) were obtained by magnetic separation. Secondly, a cholesterol-modified DNA probe (CP) was inserted into the sEV membrane and it served as the nHCR trigger for signal transmission. The MBs–sEVs–CP complex was obtained through a second magnetic separation. Thirdly, tetrads-H1 and tetrads-H2 were mixed with the MBs–sEVs–CP complex. nHCR was triggered and hyperbranched DNA networks were formed. The generation of MBs–sEVs–hyperbranched DNA network (MBs–sEVs–HDN) enabled the signal amplification due to numerous repeat units marked by FAM based on alternating H1 and H2 linkages. Thus, the sensitive and selective detection of sEVs was realized by measuring the fluorescence signal of FAM labeled on the sEVs. The change in fluorescence intensity was proportional to the concentration of sEVs enabling the quantitative analysis of sEVs. In this work, the combination of immunomagnetic separation with nHCR signal amplification provides a versatile and general strategy for sEV detection. The simultaneous recognition of the bilayer membrane structure and transmembrane protein of sEVs can effectively eliminate the interference of free proteins and enhance the specificity of capturing sEVs. In addition, the hyperbranched structure generated by nHCR can recruit more signal molecules, effectively improving the detection sensitivity. Overall, the biosensing system exhibits high specificity and sensitivity for the detection of tumor cell-derived sEVs.
 |
| | Scheme 1 Schematic representation of the (A) DNA tetrahedral hairpins (tetrads-H1 and tetrads-H2) enabled nHCR and (B) ultrasensitive sEVs detection strategy based on nHCR and immunomagnetic separation. | |
Experimental section
Materials and reagent
Sodium chloride (NaCl) and 3,3′,5,5′-tetramethylbenzidine (TMB) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Streptavidin-modified magnetic beads (10 mg mL−1, 1 μm diameter) were purchased from Thermo Fisher Scientific (MA, USA). Tween-20, Fetal bovine serum (FBS), radio immunoprecipitation assay (RIPA) lysis buffer, penicillin–streptomycin (P/S), trypsin–EDTA solution, CD63 monoclonal antibody, CD9 monoclonal antibody, and HRP labeled secondary antibody were offered by the Beyotime Biotechnology Co. Ltd (Shanghai, China). MCF-7 cells were provided by Procell Life Science Co., Ltd (Wuhan, China). DMEM medium, streptavidin, and the DNA sequences given in Table S1† were purchased from Sangon Biotech Co., Ltd (Shanghai, China). The MagExoTM sEV isolation and purification kit was purchased from Mag-Gene Nano Tech Co., Ltd (Shanghai, China).
Instruments
Fluorescence spectra were measured on an F-7000 fluorescence spectrometer (Hitachi, Japan). Transmission electron microscopy (TEM) images used to observe the morphology of sEVs were obtained on a Hitachi-7700 instrument (Tokyo, Japan). Gel electrophoresis was carried out using an EPS 300 apparatus (Tanon, Shanghai) and gel imaging system (Tanon, China). UV-vis absorption spectra were obtained with a UV-2550 spectrophotometer (Shimadzu, Japan).
Cell culture and sEV extraction by ultracentrifugation
MCF-7 cells were cultured in DMEM medium (10% FBS, 1% penicillin–streptomycin) at 37 °C under a humidified atmosphere containing 5% CO2 and 95% air. sEVs are collected from the cell supernatant by the following steps. After reaching 70%–80% confluence, the MCF-7 cells were transferred to sEV-depleted FBS DMEM medium after being washed with PBS twice (37 °C) and the cell supernatant was harvested after 48 h of incubation. Then, it was further centrifuged at 4 °C (300g and 2000g for 10 min, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min) to remove cellular debris and larger vesicles. The supernatant was then filtered with a 0.22 μm pore size filter, and centrifuged twice at 100000g for 70 min to collect sEVs. After resuspension in PBS, it was placed in a −80 °C refrigerator until used.
000g for 30 min) to remove cellular debris and larger vesicles. The supernatant was then filtered with a 0.22 μm pore size filter, and centrifuged twice at 100000g for 70 min to collect sEVs. After resuspension in PBS, it was placed in a −80 °C refrigerator until used.
sEV extraction from whole blood samples
The whole blood samples of healthy and breast cancer patients were obtained from LongHua Hospital of Shanghai University of Traditional Chinese Medicine. All experiments were performed in accordance with the national guidelines (Ethical Guidelines for Biomedical Research on Human Participants, provided by China National Health and Family Planning commission) and approved by the ethics committee of LongHua Hospital of Shanghai University of Traditional Chinese Medicine (2021LCSY026). Informed consent was obtained from human participants of this study. The serum samples were centrifuged at 4 °C (2000g for 20 min, 10000g for 30 min) to remove the debris. The MagExoTM sEV isolation and purification kit was further employed for sEV extraction from the serum samples. Then, the final serum derived sEVs were stored at −80 °C for later use.
sEV characterization and quantification
The characterization of sEVs with TEM was based on previous literature.29 10 μL of sEVs was dispersed on the carbon grids for 10 min, stained with 2% uranyl acetate for 30 s, and then rinsed with water. After drying for 30 min, sEVs were observed on H-7700 TEM with the voltage of 100 kV. Nanoparticle tracking analysis (NTA) was used to detect the concentration and particle size of the purified MCF-7 sEVs. The WB experiment was performed to confirm the presence of the CD63 protein on the sEVs according to a previous protocol.30 sEVs and MCF-7 cells were lysed in RIPA buffer at high temperature and boiled for 10 min in order to denature the proteins. The proteins were separated by 12% SDS-PAGE, and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane. After sealing the PVDF for 1 h with 5% milk, the membranes were incubated overnight at 4 °C with a special antibody. Finally, the membrane was incubated with HRP labeled secondary antibody for 1.5 h and then imaged in a gel imaging system.
Preparation of CD63 antibody modified MBs (MBs–CD63)
The biotin-modified CD63 antibody was bound to streptavidin-modified magnetic beads (MBs) for specific capture of sEVs. First, 50 μL of MBs was washed three times with 500 μl bead washing buffer (containing 1 mM EDTA, 1 M NaCl, 10 mM Tris-HCl, and 0.02% Tween-20, pH = 7.4), and then re-suspended in PBS. Then, 20 μL of biotin-modified CD63 antibodies were added into the above solution with gentle shaking for 1 h. Finally, the obtained MBs–CD63 were washed three times with the bead washing buffer and resuspended in PBS for later use.
Preparation of tetrads-H1 and tetrads-H2
Tetrads-H1 and tetrads-H2 were synthesized as described previously.31 Briefly, biotin-modified hairpins DNA (H1 and H2) were dissolved in PBSN buffer (containing 8 mM Na2HPO4, 2.6 mM KCl, 2 mM NaH2PO4, and 500 mM NaCl, pH = 7.2), separately. The hairpin structures of H1 and H2 were formed by heating for 5 min at 95 °C and slowly annealing to 25 °C. H1 and H2 were separately mixed with SA at a molar ratio of 1:4 and then shaken lightly for 1 h at room temperature to form tetrads-H1 and tetrads-H2, respectively.
sEV detection
Different concentrations of sEVs were added to 10 μL of MBs–CD63 and incubated at room temperature for 1 h. After incubation, the mixture was washed three times by magnetic separation with PBST buffer (containing 0.01% Tween-20 in PBS buffer) and used as sEV-bound beads. Next, 20 μL of 1.0 μM CP was used to resuspend the sEV-bound beads, and cultured with gentle shaking at room temperature for 45 min. Then, the MBs–sEVs–CP complexes were washed three times with PBST buffer. After that, a solution of 2 μM tetrads-H1 and 2 μM tetrads-H2 was added to the above MBs–sEVs–CP complex solution and incubated for 30 min. Finally, the formation of the MBs–sEVs–HDN complex after magnetic separation was measured at an excitation wavelength of 488 nm.
Native PAGE analysis
Different DNA solutions were separately loaded into the holes of 8% nondenaturing polyacrylamide gel with 6× loading buffer running at 110 V for 1.5 h in 1× TAE buffer (containing 40 mM Tris, 20 mM acetic acid, and 1 mM EDTA, pH = 8.3). The gels were stained with Gel Red for about 20 min and photographed by a gel image system after being washed three times with distilled water.
Results and discussion
Characterization of sEVs
sEVs were isolated from the MCF-7 cells culture supernatant and characterized by TEM (Fig. 1A). The TEM image shows that the sEVs maintain a typical cup-shaped structure with the diameter ranging from 50 to 100 nm, which is consistent with the previous literature.32Fig. 1B shows the results of WB analysis of sEVs, indicating the enrichment of the protein markers CD9 and CD63. The concentration of the extracted sEVs is found to be approximately 3.5 × 107 particles per μL by NTA (Fig. 1C). In particular, the particle size of sEVs measured by NTA is concentrated at 120 nm, which is larger than that shown in the TEM image due to the effect of light scattering in the aqueous solution. These results confirm the successful extraction and isolation of sEVs.
 |
| | Fig. 1 (A) TEM image of the sEVs derived from MCF-7 cells (B) WB analysis of CD63 and CD9 proteins from MCF-7 cell lysates and the purified sEVs. (C) The concentration and size distribution of the extracted sEVs characterized by NTA. | |
Feasibility of the fluorescence signal amplification strategy
In our proposed signal amplification platform, the successful coupling of CD63 antibody to magnetic beads is the critical step to capture and isolate sEVs. We verified the conjugation via the chromogenic reaction of TMB. HRP labeled secondary antibody was added to bare MBs and MBs–CD63 solution, respectively. TMB and TMB stop solution were added sequentially after washing and magnetic separation. As shown in Fig. S1,† compared with the bare MBs solution, the MBs–CD63 solution turned yellow and had a significant absorbance at 450 nm, indicating that CD63 antibody had successfully bound with MBs due to the high affinity of streptavidin and biotin.
As one of the key factors of the signal amplification strategy, the insertion of CP into the lipid membrane of sEVs was also verified by fluorescence microscopy. We introduced FAM modified DNA sequence (CCP) which could hybridize with the sticky ends of CP. As shown in Fig. 2A, a bright green fluorescence around MBs–CD63 was observed with the addition of sEVs. In contrast, fluorescence signal could be hardly observed in the absence of sEVs (Fig. 2B). It indicated that CP probes were successfully inserted into the lipid membrane of sEVs. Furthermore, fluorescence spectroscopy was subsequently performed with or without sEVs using the nHCR amplification method (Fig. 2C). As expected, there was a significant increase in fluorescence intensity after the insertion of CP into sEVs to trigger the nHCR. As for the system without sEVs, only a quite weak fluorescence signal was obtained. The above results show that CP probes have been grafted on the surface of sEVs. In addition, we investigated the effect of free CD63 protein in solution for sEV detection based on the signal amplification strategy. The recombinant CD63 protein was added to the solution containing 2.0 × 105 particles per μL sEVs. the fluorescence spectra in Fig. S2† showed that the recombinant CD63 protein produced a negligible increase in fluorescence signal, indicating that the interference of free CD63 protein in complex samples could be effectively avoided.
 |
| | Fig. 2 Confocal fluorescence images of (A) capture sEVs with MBs–CD63 and (B) MBs–CD63 in the absence of sEVs. Scale bar = 25 μm (C) fluorescence spectra of the nHCR reaction system without and with sEVs (2.0 × 105 particles per μL). | |
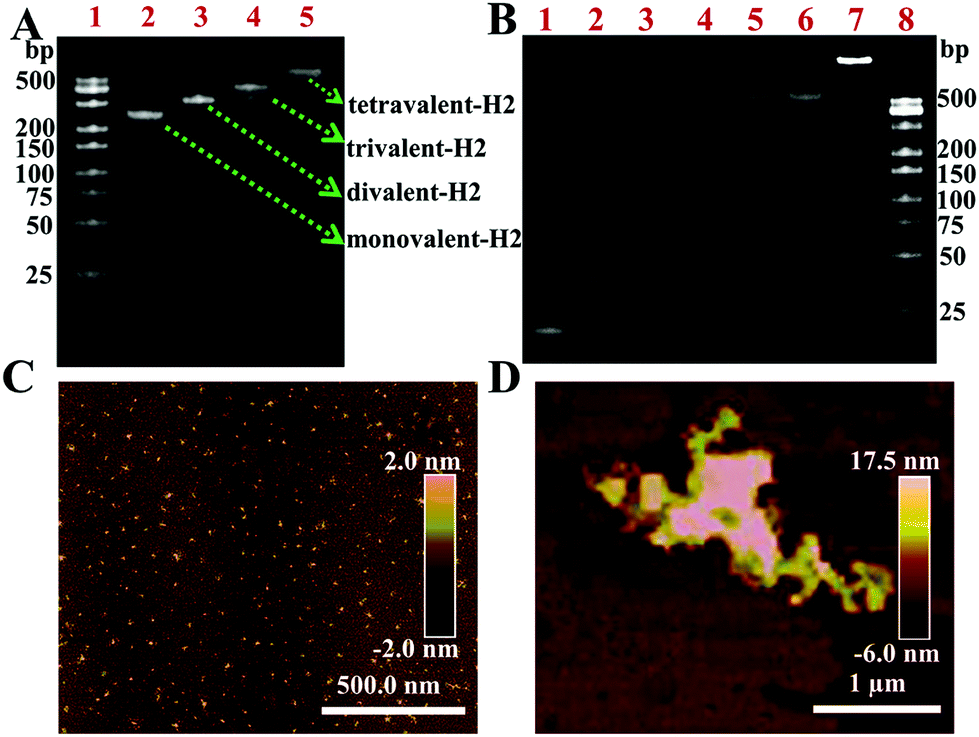
To examine the feasibility of the biosensing platform, the triggering of nHCR was further evaluated. Fig. 3A exhibited the distinct bands of formation of SA and H2 complexes by native PAGE. Four SA–H2 complexes with different molecular weights could be obtained under different concentration ratios. The lines 2–5 indicated the monovalent-H2, divalent-H2, trivalent-H2, and tetravalent-H2 (tetrads-H2) complexes, respectively. The results were consistent with the literature.31 Tetrads-H2 was used as a hairpin to trigger nHCR. Similarly, the tetrads-H1 was also formed as described above. The formation of a hyperbranched DNA structure by nHCR was also characterized by PAGE. Clearly, as shown in Fig. 3B, the gel electrophoresis images of lanes 1–5 were CP, H1, H2, tetrads-H1, and tetrads-H2, respectively. No new band appeared when tetrads-H1 was mixed with tetrads-H2 (lane 6), indicating that the mixture of tetrads-H1 and tetrads-H2 was very stable in the solution without CP. After incubating CP with the mixture of tetrads-H1 and tetrads-H2 at 37 °C for 30 min, a new bright band with a lower migration rate was produced, indicating that nHCR had been successfully triggered. Additionally, the signal amplification process was verified by fluorescence analysis. As shown in Fig. S3,† the nHCR amplification reaction produced a significant fluorescence signal in the presence of CP. However, only a very weak fluorescence signal was observed in the absence of CP. The results strongly confirmed that the signal amplification system through the nHCR was successfully triggered in the presence of CP. The atomic force microscope (AFM) image shows that the diameter of a mixture of DNA tetrads was about 5 nm in the absence of CP (Fig. 3C). But the nHCR product was in micron-sized clumps after adding CP (Fig. 3D). The formation of a large-sized structure also demonstrated the successful occurrence of nHCR.
 |
| | Fig. 3 (A) PAGE analysis of the formation of four types of SA–H2 complexes at various molar ratios (lane 1, DNA marker; lane 2–lane 5: SA:H2 = 1:1, 1:2, 1:3, and 1:4, respectively). (B) PAGE image for nHCR assay. Lane 1, CP; lane 2, H1; lane 3, H2; lane 4, tetrads-H1; lane 5, tetrads-H2; lane 6, tetrads-H1 + tetrads-H2; lane 7, CP + tetrads-H1 + tetrads-H2; lane 8, DNA marker; the AFM images of (C) tetrads-H1 + tetrads-H2 and (D) nHCR products. | |
Optimization of experimental conditions
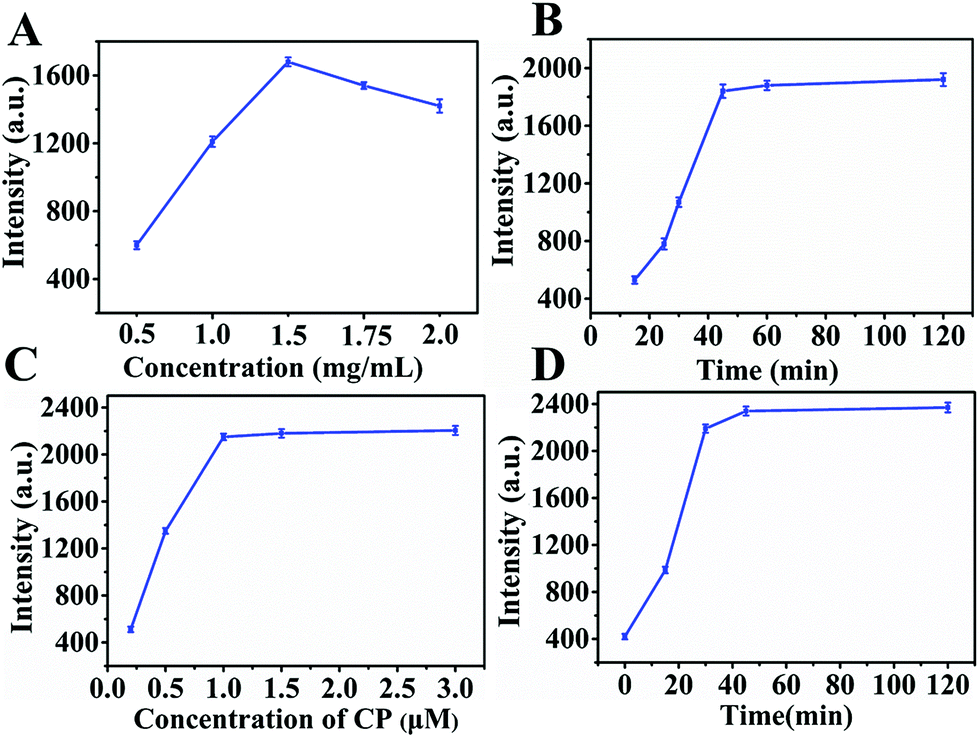
For sensitive detection of sEVs, we optimized the experimental conditions, including the concentration of MBs–CD63 and CP, incubation and triggering time. MBs–CD63 was used to capture sEVs, and its capture efficiency had a great impact on the nHCR signal amplification systems. Therefore, the concentration of MBs–CD63 was optimized first. The same amount of sEVs was added to different concentrations of MBs–CD63 and shaken gently at room temperature. The corresponding fluorescence intensity was measured, respectively. As shown in Fig. 4A, by increasing the concentration of MBs–CD63s from 0.5 mg mL−1 to 2.0 mg mL−1, the system could obtain a maximum signal at 1.5 mg mL−1. Thus, 1.5 mg mL−1 was selected as the optimal concentration of MBs–CD63. The incubation time and concentration of the CP probe were also key factors for signal amplification. As depicted in Fig. 4B, the fluorescence intensity increased rapidly within the initial 45 min and then remained stable, indicating that the optimal incubation time was 45 min. Fig. 4C exhibited the effect of the concentration of CP on fluorescence response. Clearly, the fluorescence intensity rose rapidly in the range of 0–1.0 μM, and then, it remained almost constant. Therefore, 1.0 μM was selected to be the appropriate CP concentration. In addition, the nHCR reaction time was a very important factor for the successful formation of a non-linear structure. Fig. 4D shows that the fluorescence intensity increased rapidly within 0–30 min, and then there was no significant change over time. Therefore, the triggering time for nHCR was set as 30 min.
 |
| | Fig. 4 Optimization of detection conditions. Effects of (A) concentration of MBs–CD63, (B) CP anchoring time, (C) CP concentration, and (D) nHCR reaction time on the fluorescence signal intensities. | |
Analytical performance of the nHCR signal amplification method
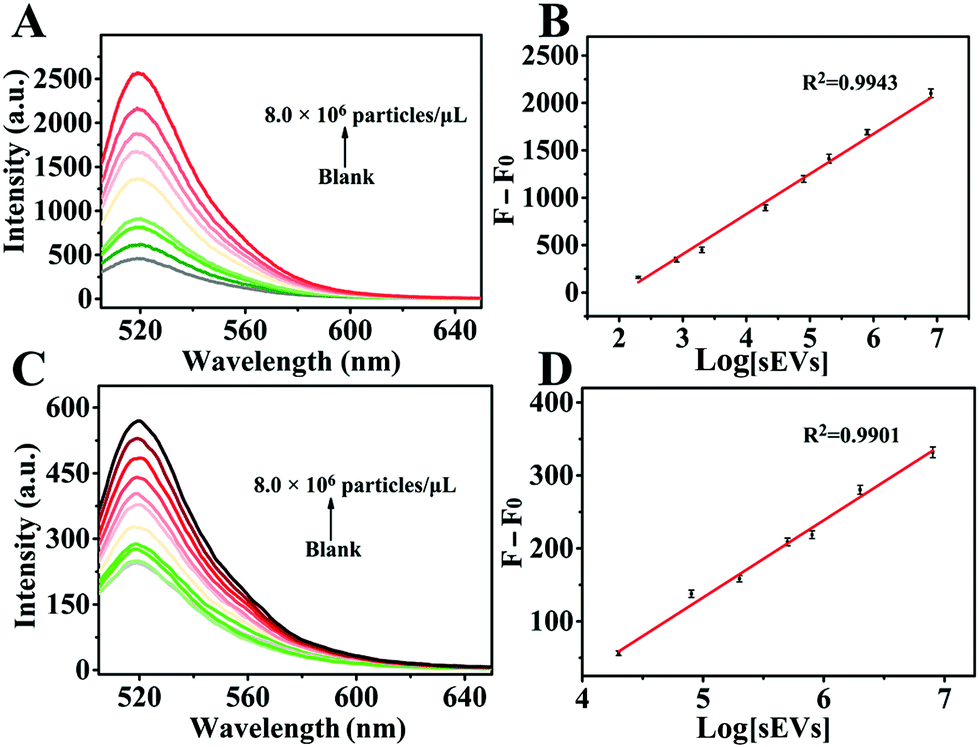
Under the optimal experimental conditions, we evaluated the linear range and sensitivity of the nHCR-based fluorescence signal amplification strategy for the detection of MCF-7-derived sEVs in PBS buffer. As shown in Fig. 5A, the fluorescence intensity gradually enhanced with the increase of the sEV concentration. As shown in Fig. 5B, a linear relationship between the increased fluorescence signal intensity and the logarithm of the sEV concentration ranging from 2.0 × 102 to 8.0 × 106 particles per μL is apparent. The linear correlation equation was ΔF = 423.03 × lg[sEVs] − 864.03 (R2 = 0.9943), where ΔF represents the increase of fluorescence signal after the addition of sEVs. The limit of detection (LOD) was calculated to be 80 particles per μL according to a 3σ/slope. However, as for the CCP method, a linear relationship was obtained within the concentration ranging from 2.0 × 104 to 8.0 × 106 particles per μL. The fitted linear equation was ΔF = 104.80 × lg[sEVs] − 388.20 (R2 = 0.9901) with an LOD of 4.0 × 103 particles per μL. The LOD of nHCR signal amplification was about 50 times lower than that of the CCP method. The sensitivity of our developed biosensing system is superior or comparable to that of most sEV biosensing platforms (Table S2†). This is mainly because the tetrads-H1 and tetrads-H2 stretch in different directions, and the amplification through nHCR can be carried out in different orientations. The superbranched nanostructure formed by nHCR can be loaded with a large number of signal molecules, generating “an sEV-numerous signal molecule” amplification effect.
 |
| | Fig. 5 Fluorescence spectra for the detection of different concentrations of sEVs originating from MCF-7 cells based on (A) nHCR signal amplification (concentration of sEVs: 0, 2.0 × 102, 8.0 × 102, 2.0 × 103, 2.0 × 104, 8.0 × 104, 2.0 × 105, 8.0 × 105, and 8.0 × 106 particles per μL) and (C) CCP method (concentration of sEVs: 0, 2.0 × 102, 8.0 × 102, 2.0 × 103, 2.0 × 104, 8.0 × 104, 2.0 × 105, 5.0 × 105, 8.0 × 105, 2.0 × 106, and 8.0 × 106 particles per μL). (B) and (D) are corresponding linear relationships of the increase of fluorescence intensity versus logarithm of sEV concentration. | |
Clinical feasibility
In order to verify the practical applicability of the proposed strategy, we studied the anti-interference ability of the method. Different concentrations of sEVs were spiked into 10% sEV-free serum (SS) and 10% ultracentrifuged serum (US), respectively and then detected through an nHCR based biosensing system. 10% US was centrifuged to remove EVs by 4 °C, 120000g for 24 h. As shown in Fig. 6A, the fluorescence signal intensity was similar in the PBS, 10% SS and 10% US samples, indicating that the complex biological matrix had little effect on the performance of the biosensing system. This was mainly due to the specificity of the simultaneous recognition of sEV transmembrane proteins and bilayer structures, as well as the excellent separation ability of MBs–CD63. In addition, we analyzed sEVs in fresh human serum samples, in which clinical serum samples were obtained from eight healthy people and eight breast cancer patients. As shown in Fig. 6B, there was a significant difference in fluorescence intensity between healthy individuals and cancer patients. The results were consistent with previous reports that cancer patients might secrete more sEVs.6,33 The real sample analysis showed that the combination of nHCR with magnetic separation is an excellent strategy for early cancer diagnosis.
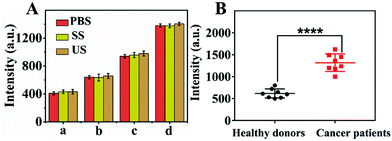 |
| | Fig. 6 (A) The fluorescence intensity of different concentrations of MCF-7 cell-derived sEVs spiked into PBS, 10% SS, and 10% diluted US by the proposed assay. (a) Blank, (b) 2.0 × 102 particles per μL, (c) 2.0 × 103 particles per μL, (d) 2.0 × 104 particles per μL. (B) The expression level of sEVs in the clinical samples obtained from healthy persons (n = 8) and breast cancer patients (n = 8) (****P < 0.0001). | |
Conclusions
In summary, an enzyme-free method was developed to detect sEVs. The method combines the advantages of immunomagnetic separation, hydrophobic interaction and nHCR fluorescence signal amplification. Based on the magnetic bead platform, the separation and enrichment of sEVs can be simplified. The specific CD63 antibody recognition and cholesterol anchor can efficiently eliminate the interference of free proteins and enhance the specificity of the sensing platform. Meanwhile, the nHCR signal amplification strategy takes full advantage of enzyme-free, isothermal properties and achieves the “an sEV-numerous signal molecule” signal amplification effect, which greatly improves the sensitivity of fluorescence detection. In addition, this method was further applied for the isolation and detection of sEVs from spiked biological samples, and can also distinguish the difference in sEV concentration between healthy individuals and cancer patients. Our method provides a robust and sensitive platform for early cancer diagnosis.
Author contributions
Qianqian Kong: conceptualization, methodology, formal analysis, software, investigation, validation, data curation, writing – original draft, writing – review and editing. Shasha Cheng: conceptualization, methodology, software, formal analysis, supervision. Xinyu Hu: methodology, validation, formal analysis, investigation. Jia You: software, investigation, supervision. Cuiling Zhang and Yuezhong Xian: conceptualization, formal analysis, funding acquisition, methodology, resources, writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (21974050, 11727810), the Natural Science Foundation of Shanghai (20ZR1418000), and the Fundamental Research Funds for the Central Universities. The authors gratefully thank Prof. Di Li of East China Normal University and LongHua Hospital of Shanghai University of Traditional Chinese Medicine for sharing the whole blood samples.
Notes and references
- Y. Yu, Q. Guo, W. Jiang, H. Zhang and C. Cai, Anal. Chem., 2021, 93, 11298–11304 CrossRef CAS PubMed.
- H. Yan, Y. Li, S. Cheng and Y. Zeng, Anal. Chem., 2021, 93, 4739–4774 CrossRef CAS PubMed.
- A. Thind and C. Wilson, J. Extracell. Vesicles, 2016, 5, 31292 CrossRef PubMed.
- M. Wu, G. Wang, W. Hu, Y. Yao and X.-F. Yu, Mol. Cancer, 2019, 18, 53 CrossRef PubMed.
- L. Zhang, H. Wang, G. Zhao, N. Li, X. Wang, Y. Li, Y. Jia and X. Qiao, Anal. Chem., 2021, 93, 6534–6543 CrossRef CAS PubMed.
- A. M. Friel, C. Corcoran, J. Crown and L. O'Driscoll, Breast Cancer Res. Treat., 2010, 123, 613–625 CrossRef CAS PubMed.
- T. L. Whiteside, Nat. Rev. Clin. Oncol., 2020, 17, 719–720 CrossRef PubMed.
- R. Xu, A. Rai, M. Chen, W. Suwakulsiri, D. W. Greening and R. J. Simpson, Nat. Rev. Clin. Oncol., 2018, 15, 617–638 CrossRef CAS PubMed.
- H. Shao, H. Im, C. M. Castro, X. Breakefield, R. Weissleder and H. Lee, Chem. Rev., 2018, 118, 1917–1950 CrossRef CAS PubMed.
- Y. Yoshioka, N. Kosaka, Y. Konishi, H. Ohta, H. Okamoto, H. Sonoda, R. Nonaka, H. Yamamoto, H. Ishii, M. Mori, K. Furuta, T. Nakajima, H. Hayashi, H. Sugisaki, H. Higashimoto, T. Kato, F. Takeshita and T. Ochiya, Nat. Commun., 2014, 5, 3591 CrossRef PubMed.
- D. Choi, L. Montermini, H. Jeong, S. Sharma, B. Meehan and J. Rak, ACS Nano, 2019, 13, 10499–10511 CrossRef CAS PubMed.
- K. Ueda, N. Ishikawa, A. Tatsuguchi, N. Saichi, R. Fujii and H. Nakagawa, Sci. Rep., 2014, 4, 6232 CrossRef CAS PubMed.
- R. R. Huang, L. He, Y. Y. Xia, H. P. Xu, C. Liu, H. Xie, S. Wang, L. J. Peng, Y. F. Liu, Y. Liu, N. Y. He and Z. Y. Li, Small, 2019, 15, 1900735 CrossRef PubMed.
- R. Huang, L. He, S. Li, H. Liu, L. Jin, Z. Chen, Y. Zhao, Z. Li, Y. Deng and N. He, Nanoscale, 2020, 12, 2445–2451 RSC.
- L. Zhao, R. Sun, P. He and X. Zhang, Anal. Chem., 2019, 91, 14773–14779 CrossRef CAS PubMed.
- Y. Yu, W. S. Zhang, Y. Guo, H. Peng, M. Zhu, D. Miao and G. Su, Biosens. Bioelectron., 2020, 167, 112482 CrossRef CAS PubMed.
- L. Wang, Y. Deng, J. Wei, Y. Huang, Z. Wang and G. Li, Biosens. Bioelectron., 2021, 191, 113465 CrossRef CAS PubMed.
- Y. Pan, L. Wang, Y. Deng, M. Wang, Y. Peng, J. Yang and G. Li, Chem. Commun., 2020, 56, 13768–13771 RSC.
- J. Wu, J. Lv, X. Zheng and Z.-S. Wu, Talanta, 2021, 234, 122637 CrossRef CAS PubMed.
- H. Wang, C. Li, X. Liu, X. Zhou and F. Wang, Chem. Sci., 2018, 9, 5842–5849 RSC.
- H. Ye, K. Yang, J. Tao, Y. Liu, Q. Zhang, S. Habibi, Z. Nie and X. Xia, ACS Nano, 2017, 11, 2052–2059 CrossRef CAS PubMed.
- S. Bi, S. Z. Yue and S. S. Zhang, Chem. Soc. Rev., 2017, 46, 4281–4298 RSC.
- F. Xuan and I. M. Hsing, J. Am. Chem. Soc., 2014, 136, 9810–9813 CrossRef CAS PubMed.
- Y. Tang, X.-L. Zhang, L.-J. Tang, R.-Q. Yu and J.-H. Jiang, Anal. Chem., 2017, 89, 3445–3451 CrossRef CAS PubMed.
- J. B. Wang, J. Chao, H. J. Liu, S. Su, L. H. Wang, W. Huang, I. Willner and C. H. Fan, Angew. Chem., Int. Ed., 2017, 56, 2171–2175 CrossRef CAS PubMed.
- Z. Zeng, R. Zhou, R. Sun, X. Zhang, Z. Cheng, C. Chen and Q. Zhu, Biosens. Bioelectron., 2021, 173, 112814 CrossRef CAS PubMed.
- Q. Xue, C. Liu, X. Li, L. Dai and H. Wang, Bioconjugate Chem., 2018, 29, 1399–1405 CrossRef CAS PubMed.
- Y. Zhao, S. Hu, H. Wang, K. Yu, Y. Guan, X. Liu, N. Li and F. Liu, Anal. Chem., 2017, 89, 6907–6914 CrossRef CAS PubMed.
- Q. Li, G. K. Tofaris and J. J. Davis, Anal. Chem., 2017, 89, 3184–3190 CrossRef CAS PubMed.
- Y. Guo, J. Tao, Y. Li, Y. Feng, H. Ju, Z. Wang and L. Ding, J. Am. Chem. Soc., 2020, 142, 7404–7412 CrossRef CAS PubMed.
- D. J. Huang, Z. M. Huang, H. Y. Xiao, Z. K. Wu, L. J. Tang and J. H. Jiang, Chem. Sci., 2018, 9, 4892–4897 RSC.
- Y. Zhang, D. Wang, S. Yue, Y. Lu, C. Yang, J. Fang and Z. Xu, ACS Sens., 2019, 4, 3210–3218 CrossRef CAS PubMed.
- C. Sotiriou and L. Pusztai, N. Engl. J. Med., 2009, 360, 790–800 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*