
Dendritic lipopeptide liposomes decorated with dual-targeted proteins†
Sensen
Zhou
a,
Cheng
Li
a,
Yang
Yuan
a,
Lei
Jiang
 *ab,
Weizhi
Chen
*a and
Xiqun
Jiang
*a
*ab,
Weizhi
Chen
*a and
Xiqun
Jiang
*a
aDepartment of Polymer Science & Engineering, College of Chemistry & Chemical Engineering, and Jiangsu Key Laboratory for Nanotechnology, Nanjing University, Nanjing, 210093, P.R. China. E-mail: jiangx@nju.edu.cn; chenwz@nju.edu.cn; jianglei0453@163.com
bState Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China
First published on 21st October 2022
Abstract
Due to their homing effects, cell and cell membrane-derived nanocarriers have been widely used to enhance drug target delivery. Inspired by the protein-anchored cell membrane architecture, we here report a tumor-targeted liposome, dtDLP, which was constructed through the electrostatic interaction between dendritic lipopeptide liposomes and a dual-targeted recombinant protein, achieving superior tumor homing, cellular endocytotic and penetration abilities. The dual-targeted recombinant protein consists of an anti-epidermal growth factor receptor single domain antibody and a peptide ligand for the integrin αvβ3. dtDLPs substantially reduced macrophage phagocytosis and increased drug internalization in both 4T1 cells and HeLa cells by providing more endocytic pathways. In addition, the dtDLPs showed great penetration ability in both multicellular spheroids and tumor tissues. Due to the improved cancer cellular uptake and tumor penetration, the dtDLPs exhibited a superior anticancer effect in both HeLa and 4T1 tumor-bearing mice. This work will be helpful for the design of cell-specific liposomes with admirable tumor targeting, endocytotic and penetration abilities.
Introduction
Most chemical drugs used for cancer therapy are non-targeted and tend to distribute evenly throughout the body and usually clean quickly. Thus, a large dosage or multiple administration is needed, leading to high side effects and poor bioavailability. To improve the therapeutic index of drugs, various drug formulations including polymer or nanocarrier-based drug delivery systems have been developed for delivering drugs, especially anticancer drugs to disease sites.1–4 Among these drug delivery systems, cell membrane mimicking liposomes have attracted much more attention.5–9 Traditionally, phospholipid-based liposomes have been decorated with polyethylene glycol (PEG) to offer the characteristics of long circulation and enhanced accumulation in disease sites.10–12 In fact, PEG-decorated liposomes have been used to encapsulate doxorubicin (DOX) for the clinical treatment of cancers and is trademarked as Doxil.13 However, in most cases, PEG-decorated liposomes do not provide enough accumulation and sufficient cellular uptake in disease sites due to the lack of active targeting function and poor interactions with target cells.14–17 Thus, the development of liposomes with active targeting function and enhanced cellular uptake for overcoming traditional liposomal shortcomings is highly desirable. So far, many efforts have been made to endow liposomes with active targeting function by covalently conjugating ligands such as antibodies, peptides and aptamers at the end of the PEG chain.17–20 Alternatively, active targeting liposomes could also be prepared by the co-assembly of single targeting lipids in solution.21,22 More recently, cancer cell membranes have been used to decorate liposomes to enhance tumor-targeting function since these cancer cell membranes carry some receptors expressed by cancer cells. However, the receptors and membrane proteins on cancer cell membranes are very complex. It is hard to identify which proteins are desired and which proteins are harmful.Inspired by the membrane protein architecture, we here report dual-targeted protein decorated liposomes to enhance liposome targeting, penetration and cellular uptake. Different membrane proteins anchor in the cell membrane in a transmembrane fashion, and we decorated liposomes using dual-targeted proteins through electrostatic interactions. To achieve enough electrostatic interactions between the dual-targeted proteins and liposomes, the liposomes reported consist of positively charged dendritic lipopeptides which provide plentiful positive charges at the surface of liposomes. The dual-targeted protein we used here was a recombinant protein containing a single domain antibody against the epidermal growth factor receptor (EGFR) and an integrin αvβ3-bound peptide, C-RGD-KGPDC (Fig. 1a). The EGFR is overexpressed in various cancers and several drugs targeting the EGFR have been approved.23,24 Integrins play vital roles in connecting cells with the extracellular matrix and are upregulated in many cancers.25,26 Using a recombinant protein that could bind the EGFR and integrin simultaneously would provide more opportunities to target cancer cells as compared to single targeting.27,28 In addition, this recombinant protein bears negative charges. Thus, the positively charged liposomes can be decorated by the negatively charged recombinant protein through electrostatic interactions. Such liposomes not only provide an opportunity to overcome tumoral heterogeneity by targeting multiple receptors but also enhance tumor penetration ability.
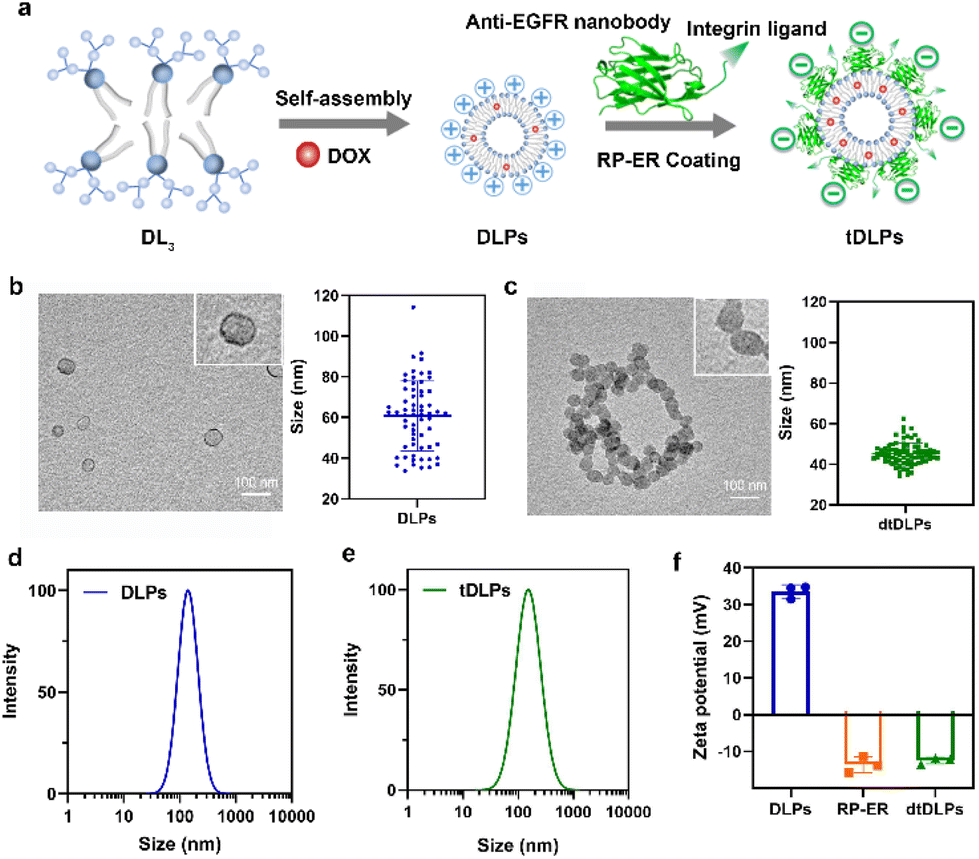 | ||
| Fig. 1 (a) Schematic diagram of dendritic lipopeptide liposomes self-assembled from third generation dendritic phosphoilipid (DL3) and the process of recombinant protein RP-ER coating on the surface of DLPs and the resulting dtDLPs. TEM images of DLPs (b) and dtDLPs (c) as well as the sizes measured by TEM, n = 63 for (b), 79 for (c); the hydrodynamic sizes of DLPs (d) and dtDLPs (e) measured by DLS (in 0.01 M PBS); (f) zeta potentials of DLPs and dtDLPs, n = 3. Data are presented as mean ± s.d. | ||
Materials and methods
Materials
1-Hydroxybenzotriazole hydrate (HOBT) and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from GL Biochem. Ltd (Shanghai, China). N,N-Diisopropylethylamine (DIPEA) and trifluoroacetic acid (TFA) were purchased from Alfa Aesar. The third-generation dendritic poly(L-lysine) molecule G3 (Lys) was from our lab.29 Doxorubicin hydrochloride (DOX·HCl) was purchased from Hisun Pharmaceutical (Zhejiang, China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) was purchased from CordenPharma (Switzerland). All other solvents were purchased from Sinopharm Chemical Reagent Co. Ltd, China. DMEM medium, RPMI-1640 medium, streptomycin and penicillin were purchased from Hyclone (USA). Fetal bovine serum (FBS) was purchased from Gibco (USA). Human cervical cancer cells HeLa, mouse breast cancer cells 4T1 and murine macrophage RAW 264.7 cells were obtained from the Chinese Academy of Science Cell Bank for Type Culture Collection (Shanghai, China). All media were supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were grown in a humidified incubator (Heal Force) with 5% CO2 at 37 °C.Synthesis of third-generation dendritic phospholipids (DL3)
DSPE (0.75 g, 1.00 mmol), G3 (Lys) (2.57 g, 1.5 mmol), HOBT (0.26 g, 2 mmol) and HBTU (0.57 g, 2 mmol) were dissolved in anhydrous trichloromethane (25 mL) and anhydrous DMF (3 mL) under a nitrogen atmosphere. DIPEA (2.00 mL, 14.00 mmol) was added to the above mixed solution under stirring at 0 °C. The solution was stirred under nitrogen for another 48 h at room temperature. The mixture was washed with saturated NaHCO3, NaHSO4, and NaCl solution several times. The mixture was dried with MgSO4 for 2 h. After the removal of solvents, the mixture was purified by silica gel column chromatography (DCM/MeOH, 12/1, v/v) to obtain BOC-DL (yield: 79%). BOC-DL was dried in a vacuum and dissolved in anhydrous dichloromethane (DCM)/TFA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mL) for 4 h to remove tert-butyl groups. The mixture was concentrated, and the product was treated with anhydrous diethyl ether to obtain DL3 (yield: 91%).
:1, 10 mL) for 4 h to remove tert-butyl groups. The mixture was concentrated, and the product was treated with anhydrous diethyl ether to obtain DL3 (yield: 91%).
Expression of recombinant protein
The recombinant protein RP-E and RP-ER gene with His tag were synthesized by Generay Biotech Co., Ltd (Shanghai, China). Then the pET-25b vector containing RP-E or RP-ER gene was transformed into E. coli strain BL21(DE3) cells by heat shock, and grown for 12 h at 37 °C in agar plates with LB media supplemented with 100 μg mL−1 of ampicillin. A single colony was picked out and transferred into a shake flask culture with TB media, followed by incubation at 37 °C with 200 rpm for 24 h. After harvesting by centrifugation at 6000 rpm, 4 °C for 10 min, the E. coli cells were resuspended into binding buffer (500 mM NaCl, 20 mM PB, 20 mM imidazole, pH = 7.4) followed by ultrasonication on ice. Then the supernatant was collected by centrifugation at 11000 rpm, 4 °C for 30 min. The crude protein was purified through HisTrap column using an AKTA purification system. Briefly, the protein in the buffer flowed through the HisTrap column at 3 mL min−1. After washing with binding buffer for 3 column volumes, the resulting proteins were gradient eluted with washing buffer (500 mM NaCl, 20 mM PB, 800 mM imidazole, pH = 7.4). The imidazole salts were removed by dialysis and the pure proteins were then concentrated by ultrafiltration and preserved at 4 °C for further use.
Preparation of DLPs and targeted DLPs
DLPs based on DL3 were prepared via thin film hydration.29 Briefly, DL3 (or with hydrophobic DOX) was dissolved in methanol and formed dried lipid films after removal of residual organic solvent. After then, 10 mM PBS (pH 7.4) or ultrapure water was added and sonicated in a sonication bath. The unencapsulated hydrophobic DOX was removed by centrifugation at 3000 rpm for 15 min. The drug-loading capacity was calculated by the following formula:| DOX loading capacity = WDOX/WDLPs × 100%, |
A certain amount of DLPs was dropped into the recombinant protein solution (320 μg mL−1) in 10 mM PBS with continuous stirring. After stirring for 1 h, the unbound proteins were removed by ultrafiltration (MWCO 100 kDa). The resulted stDLPs with RP-E coating and dtDLPs with RP-ER coating were stored at 4 °C for further use.
Characterization of DLPs and dtDLPs
The molecular weights of dendritic phospholipids and recombinant protein were analyzed by matrix assisted laser desorption ionization time of flight mass spectroscopy (MALDI-TOF MS, Bruker Autoflex III) and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), respectively. Dynamic light scattering (DLS) and zeta potentials of DLPs and dtDLPs were performed on a Brookhaven Zetasizer Nano ZS at 25 °C. Each measurement was performed in triplicate and an average value was reported. UV-Vis absorbance spectra were obtained with a UV3100 (Shimadzu, Japan). The morphology and structure of DLPs and dtDLPs were observed with a transmission electron microscope (TEM, JEM-100S, JEOL, Japan).In vitro drug release
1 mL of DLPs and dtDLPs in a dialysis bag (MWCO 3500 Da) were suspended into 5 mL of release medium (10 mM PBS, pH 7.4 with 0.5% tween-20), with agitation at about 200 rpm at 37 °C. To evaluate the drug release behaviors under the storage conditions, 1 mL of DLPs and tDLPs in a dialysis bag (MWCO 3500 Da) was suspended into 5 mL of release medium (10 mM PBS, pH 7.4), with agitation about 200 rpm at 4 °C. Periodically, the medium outside of the dialysis bag was collected and another 5 mL of medium was added. The concentration of DOX in the release medium was measured using a fluorescence spectrometer (RF-5301PC, Shimadzu, Japan). A standard curve of free DOX was initially obtained (excitation at 480 nm and emission at 590 nm).In vitro cytotoxicity
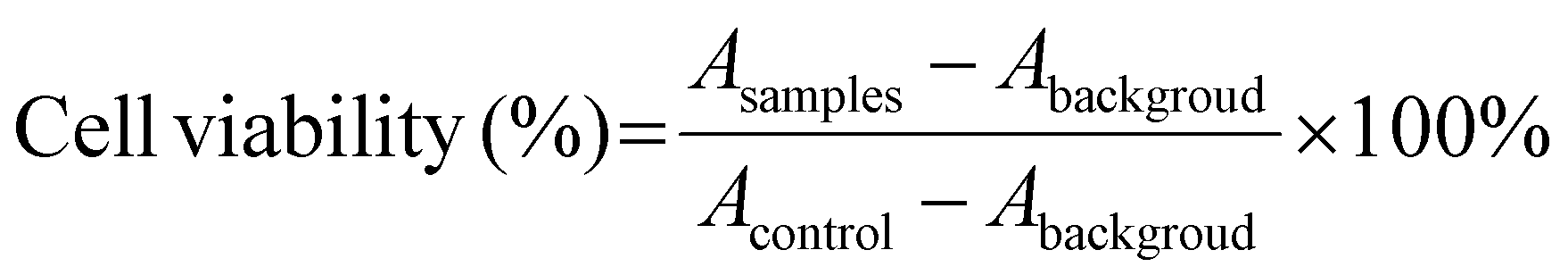
The cell cytotoxicity of DLPs, stDLPs and dtDLPs as well as free DOX was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays. HeLa, 4T1 or RAW 264.7 cells at a density of 5 × 103 were seeded into a 96-well plate and subsequently incubated at 37 °C overnight. The medium was then replaced with 200 μL of fresh medium including free DOX, DLPs, stDLPs and dtDLPs at 0.5, 1, 2, 4, 8 and 16 μg mL−1 (DOX concentration eq.), respectively. Wells with only medium were considered as background and wells with only cells served as the control. After incubation for 24 h, all the medium was withdrawn and 180 μL of fresh medium with 20 μL of MTT solution (5 mg mL−1) was added in each well, and incubated for another 4 h. The absorbance of each well at 570 nm was measured by a microplate reader (Molecular Devices CMax Plus). Cell viability was calculated by the following equation:where A is the absorbance.
To evaluate the cytocompatibility of the materials, LPs and dtLPs were incubated with macrophages RAW 264.7 at different gradient concentrations (10, 50, 100, 200, 500 and 1000 μg mL−1). The cellular viability was evaluated using the MTT assay as described above.
WB analysis
Protein lysates from cancer cells were prepared using RIPA buffer, separated by gel electrophoresis and transferred to polyvinylidene difluoride membranes. After being incubated with the block solution (5% skim milk powder in TBST) for 1 h at room temperature, the membranes were incubated with primary antibodies at 4 °C overnight, followed by incubation with the HRP-conjugated goat anti-rabbit secondary antibody (Proteintech, SA00001-2, USA) at room temperature for 1 h. Proteins on the membranes were imaged using ab electro-chemi-luminescence (ECL) instrument (Tanon-5200, Tanon Science & Technology Co., Ltd Shanghai, China). Primary antibodies used were anti-EGFR (Cell Signaling Technology, 4267, USA), anti-integrin αvβ3 (Novus biologicals, NBP2-67557, USA) and anti-GAPDH (Proteintech, 10494-1-AP, USA).Cellular uptake
HeLa cells were cultured in DMEM medium. 4T1 cells and RAW 264.7 cells were cultured in RPMI-1640 medium. For evaluating the cellular uptake of DLPs, stDLPs and dtDLPs, 1 × 105 cells were seeded into a 6-well plate with cover-glasses inside. After the cells were adhered onto the cover-glasses, 100 μL of DLPs, stDLPs and tDLPs with equal DOX concentration was added and incubated for 4 h at 37 °C, followed by washing with PBS to remove any DLPs, stDLPs and tDLPs. Subsequently, 4% paraformaldehyde was used to fix the cells for 10 min at room temperature. After washing with PBS, cells onto the coverslips were then stained with DAPI. Confocal laser scanning microscopy (CLSM) was then performed to observe the fluorescence in cells in the cover-glasses.For quantitative analysis of the fluorescence intensity by flow cytometry, about 1 × 104 cells were seeded into a 24-well plate and incubated for one night. A predetermined amount of DLPs, stDLPs and tDLPs with equal DOX concentration was added. The wells with only cells were used as the control. After incubation for 4 h in the cell incubator, the cells were rinsed with PBS several times. Then the cells were trypsinized. After removing trypsin by centrifugation, the cells were collected and analyzed using a flow cytometer (Accuri C6, BD Biosciences, USA). To further evaluate the endocytosis pathways of DLPs and tDLPs in HeLa and 4T1 cells, endocytosis inhibitors were added 1 hour before the cells were treated with DLPs and tDLPs for 2 h at 37 °C. Chlorpromazine (CPZ) (10 μM), methyl-β-cyclodextrin (MβCD) (300 μM), and 5-(N-ethyl-N-isopropyl) amiloride (EIPA) (100 μM) were used as inhibitors of clathrin-mediated endocytosis, caveolae-mediated endocytosis, and micropinocytosis.
Penetration in multicellular spheroids (MCs)
The preparation of HeLa and 4T1 MCs was similar to that of SY5Y MCs.30 Briefly, a certain amount of poly(2-hydroxyethyl methacrylate) (poly-HEMA) solution was put into a culture flask. After the solvent was volatilized, a thin film was formed at the bottom of the culture flask. Then the poly-HEMA coated flask was sterilized by ultraviolet light for at least 1 h. 1 × 106 cells in 5 mL of medium were placed in the poly-HEMA coated flask, followed by incubation at 37 °C with 5% carbon dioxide in a humidified atmosphere. Every other day, the culture medium was replaced.After a week, HeLa MCs with 500 μm and 4T1 MCs with about 150 μm were collected and divided equally into several centrifugation tubes. A certain amount of DLPs and dtDLPs with an equal concentration of DOX was added and allowed to incubate in the cell incubator. After 24 h of incubation, MCs were collected, washed, and finally observed by CLSM. Migration index (MI), retention index (RI) and distribution index (DI) were defined to quantify the retention and migration capacity of the DLPs and dtDLPs, from the previous report with some modification.27 Since we used MCs as a system to simulate the tumors, MCs were scanned by CLSM at the maximum focal plane and each section was segmented into 50 μm thick ring elements located at radial locations of Ri from the surface of the tumor. MI is a measurement of the intratumoral penetration depth of the therapeutic agents toward the center of the MC or a simulated tumor. It is defined as:
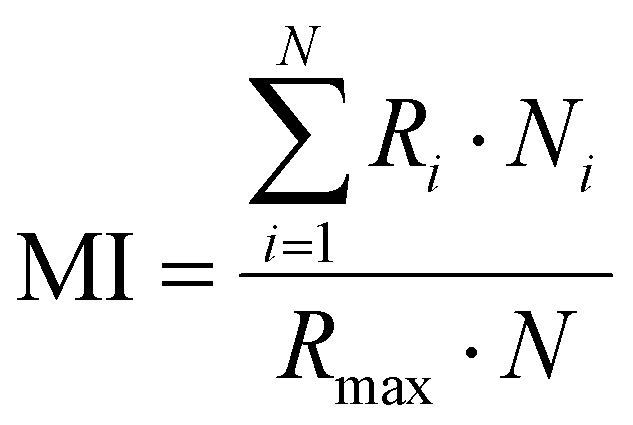
where S is the area of the entire MC slice. The value of RI describes the tumor retention of the drug without consideration of its spatial distribution. DI represents a composite normalized index of migration and retention and is defined as:
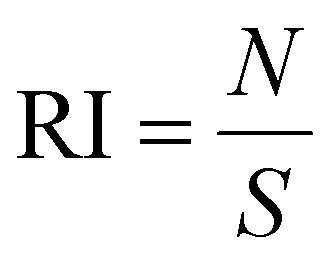
| DI = MI × RI |
Thus, the DI provides a measurement of the intratumoral distribution efficacy.
Near-infrared fluorescence (NIRF) imaging and biodistribution
To establish a subcutaneous 4T1 tumor model, 5 × 106 cells in 100 μL of saline were subcutaneously inoculated into Balb/c mice at the right hind leg. The HeLa tumor model was established in the same way, 1 × 107 cells in 100 μL of saline were subcutaneously inoculated into Balb/c nude mice at the right hind leg. After about a week, the tumors were established and 200 μL of free NIR-797 and NIR-797 labeled DLPs and tDLPs (NIR-797 concentration eq.) were intravenously injected into the mice. At every predetermined time, the mice were visualized with the MaestroTM in vivo imaging system (CRi, USA), with exposure to the anesthetic and initial depilation. At 72 h post-injection for 4T1 tumors and 120 h post-injection for HeLa tumors, the organs were taken out and visualized with the MaestroTM in vivo imaging system (CRi, USA). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Nanjing University and approved by the Medical Ethics Committee of Nanjing University.Penetration in tumor tissue
To evaluate the penetration abilities of DLPs and dtDLPs in tumor tissue, 100 μg of free DOX, DLPs and dtDLPs (with equal DOX concentration) was intravenously injected into 4T1 and HeLa tumor-bearing mice. At 24 h post-injection, mice were sacrificed and tumors were collected. The tumors were fixed in 4% paraformaldehyde for 4 h, immersed in 25% sucrose aqueous solution at 4 °C overnight and frozen in the optimal cutting temperature (O.C.T.) embedding medium. About 9 μm sections were cut for immunofluorescence analysis. Next, the sections were rehydrated in 0.1% (v/v in PBS) Triton X-100 for 10 min, then exposed to 3% BSA at room temperature for 1 h, and subsequently incubated with the primary antibody (1:400, rat anti-mouse CD31, BD Pharmingen, San Jose, California) for 1 h at room temperature. After being washed three times with 0.05% (v/v, PBS) tween 20 for 5 minutes, the sections were stained with an Alexa-488 conjugated secondary antibody (1:1000, donkey anti-rat, Molecular Probes, Eugene, OR) at room temperature shielded from light for 0.5 h. Finally, after the sections were washed, DAPI was used to label the nucleus. These sections were then covered with Fluoromount™ aqueous mounting medium (Invitrogen, Carlsbad, USA) and observed by CLSM.
In vivo antitumor potency
Mice bearing HeLa tumors and 4T1 tumors were used. To establish a subcutaneous tumor model, 1 × 107 HeLa cells or 5 × 106 4T1 cells in 100 μL of saline were subcutaneously inoculated into Balb/c nude or Balb/c mice at the right hind leg. When the tumors grew to a predetermined size, the tumor-bearing mice were randomly divided into four groups. Free DOX (5 mg kg−1), DLPs and dtDLPs (5 mg kg−1 eq.) were injected intravenously, respectively. And PBS was used as the control. This day was designated as Day 1. All the agents were injected once. The body weights of mice were weighed every two days. And a vernier caliper was used to record the diameters of tumors every second day and the tumor volume (V) was calculated using the following equation:where “a” and “b” corresponded to the maximal diameter of the tumors and the vertical diameter to “a”, respectively.
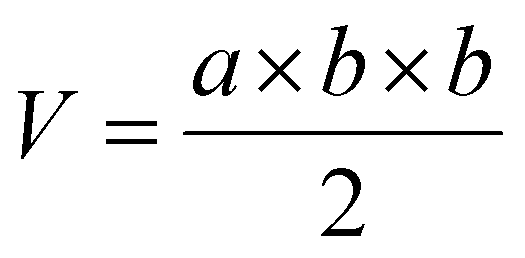
The tumor growth inhibition (TGI) was calculated using the following equation:
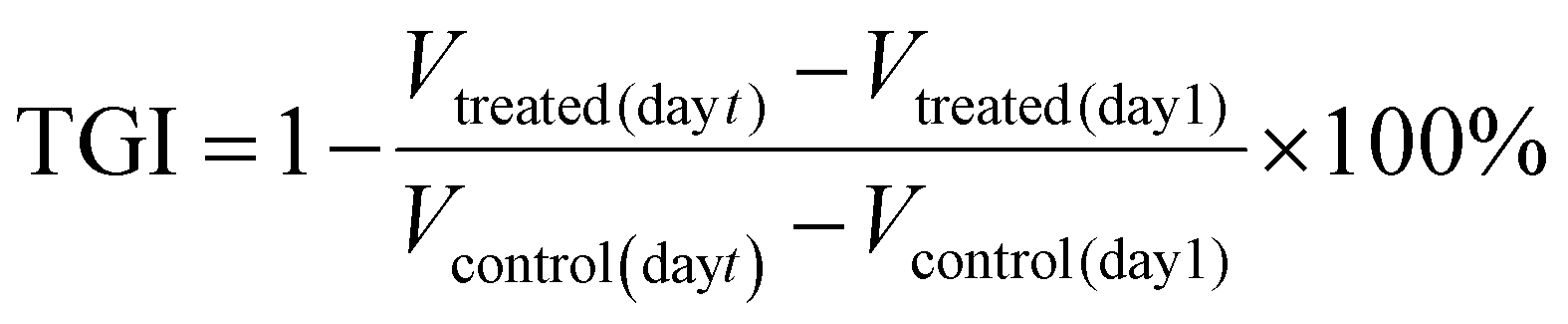
Body weights of the animals were recorded for 15 days. At the end of the experiment, the mice were euthanized and the tumors were taken out, weighed and imaged. To further evaluate the tissue damage, the heart, liver, spleen, lungs, kidneys and tumors were taken out for H&E staining.
Statistical analysis
Results are expressed as means ± s.d. The sample sizes are shown in the figure legends. Significant differences in the mean values were evaluated by the multiple t-test. The differences were considered significant for P values less than 0.05.Results and discussion
Synthesis of dendritic phospholipids and expression of recombinant protein
The lysine-based third-generation dendritic phospholipids (DL3) were synthesized, as shown in Fig. S1.† In brief, the carboxyl groups of the third-generation dendritic poly(L-lysine) molecule reacted with the amino groups of DSPE to form DL3. The molecular structure of DL3 was confirmed by the 1H NMR spectrum (Fig. S2†). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) measurement revealed that the molecular weight of DL3 was 1645.4 Da (Fig. S3†), which was consistent with the theoretical value, 1645.4 Da.The gene sequence of the recombinant protein containing a single domain antibody against the epidermal growth factor receptor (EGFR) and an integrin αvβ3-bound peptide was examined by DNA sequencing and no mutation and frameshift were found. Then, the dual-targeting recombinant protein was expressed from the pET-25b plasmid in E. coli strain BL21(DE3) cells and purified through a HisTrap column. The sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and MALDI-TOF MS characterization (Fig. S4†) confirmed that the dual-targeting recombinant protein with a molecular weight of about 18 kDa was successfully expressed and purified with a desirable purity. The resulting dual-targeting recombinant protein was named RP-ER.
Preparation of dendritic lipopeptide liposomes decorated with a dual-targeted protein
Due to its amphiphilic property, the third-generation dendritic lipopeptide could self-assemble into liposomes in an aqueous solution. As shown in Fig. 1a, a model antitumor agent DOX, which was premixed with the lipopeptide, was loaded into the self-assembled liposomes, resulting in drug-loaded liposomes (DLPs). The dual-targeted recombinant protein RP-ER was then decorated on the surface of DLPs through electrostatic interaction and the dual-targeting liposomes (dtDLPs) were obtained. To confirm whether DLPs and dtDLPs were encapsulated with DOX, the UV-Vis absorption spectra of free DOX, DLPs and dtDLPs were measured. DLPs and dtDLPs shared the same absorption spectra with free DOX (Fig. S5a†), suggesting that DOX had been successfully loaded into liposomes. The drug loading content of DLPs and dtDLPs was determined to be about 12% by UV-Vis absorption spectra.The vesicle structure of DLPs and dtDLPs was confirmed by transmission electron microscopy (TEM), as shown in Fig. 1b and c. The mean sizes of DLPs and dtDLPs measured by TEM were about 60 nm and 45 nm, respectively. Compared to DLPs, the size of dtDLPs decreased and size distribution was more uniform, which means that RP-ER decoration makes the structure of dtDLPs much denser than that of DLPs. In contrast, the hydrodynamic sizes of DLPs and dtDLPs were 142 nm and 151 nm, respectively, measured by dynamic light scattering (DLS) (Fig. 1d and e), indicating that RP-ER coating increases the hydrodynamic size of dtDLPs. The changes of the sizes of DLPs and dtDLPs measured by DLS and TEM were different mainly because DLS measured the hydrodynamic sizes while TEM detected the morphologies in the dry state. Both DLPs and dtDLPs showed great stability (Fig. S5b†). For DLPs, the cationic lysine exposed at the surface of liposomes with high density provided a high surface potential and the zeta potential was +33.6 mV (Fig. 1f). After RP-ER decoration on the surface of DLPs through electrostatic interaction, the zeta potential of the dtDLPs switched from a positive value to negative value (−12.4 mV).
In vitro drug release and cytotoxicity of dtDLPs
The drug release behaviors of DLPs and dtDLPs were evaluated in 10 mM PBS with 0.5% tween-20 at 37 °C with 200 rpm. The release profiles of DOX from DLPs and dtDLPs had a similar trend (Fig. S6a†), indicating that the protein decoration does not affect the release behavior of DOX from liposomes. As time elapsed, more DOX was released. Both DLPs and dtDLPs revealed a burst release behavior at the initial period which may be due to the addition of 0.5% tween-20 disrupting the liposome structures, leading to drug release. However, less than 5% DOX was released from both DLPs and dtDLPs after incubated for two weeks at 4 °C (Fig. S6b†), suggesting the stability of DLPs and dtDLPs.The cytocompatibilities of blank liposomes LPs and dtLPs were first analyzed in the mouse macrophage cells RAW 264.7 (Fig. S7a†). Even at the highest concentration (500 μg mL−1), LPs and dtLPs showed no cytotoxicity, suggesting their great cytocompatibility. Different from HeLa cells and 4T1 cells, DLPs showed a higher cytotoxicity than dtDLPs in RAW 264.7 cells (Fig. S7b†). The IC50 values for DLPs and dtDLPs in RAW 264.7 cells were 0.31 and 0.46 μg mL−1, respectively. To better understand the targeting ability of dtDLPs, we further prepared a single-targeting liposome with RP-E coating and named them stDLPs. RP-E was a single domain antibody against the EGFR. RP-E was expressed in the same way as RP-ER and characterized by SDS-PAGE and MALDI-TOF MS with a molecular weight about 17 kDa (Fig. S8†). The cytotoxicity of DLPs, stDLPs and dtDLPs in human cervical carcinoma HeLa cell and mouse breast cancer 4T1 cells were examined using the MTT assay. As shown in Fig. 2a and b, similar to free DOX, DLPs, stDLPs and dtDLPs revealed a dose-dependent cytotoxicity. At the same concentration, all the pharmacological activities of DLPs, stDLPs and dtDLPs were lower than that of free DOX which is mainly because DOX loaded in the liposomes needs to be released to exert its therapeutic effect. Although dtDLPs showed a superior cytotoxicity than DLPs and stDLPs in both HeLa cells and 4T1 cells, the enhancement level was different. The IC50 values for free DOX, DLPs, stDLPs and dtDLPs were 0.32, 1.11, 0.69 and 0.41 μg mL−1, respectively, in HeLa cells and 1.09, 4.27, 4.18 and 2.34 μg mL−1, respectively, in 4T1 cells (Fig. 2c). Dual-targeted dtDLPs showed the lowest IC50 values in both HeLa cells and 4T1 cells. And single-targeted stDLPs exhibited a lower IC50 value than non-targeted DLPs in HeLa cells but not in 4T1 cells.
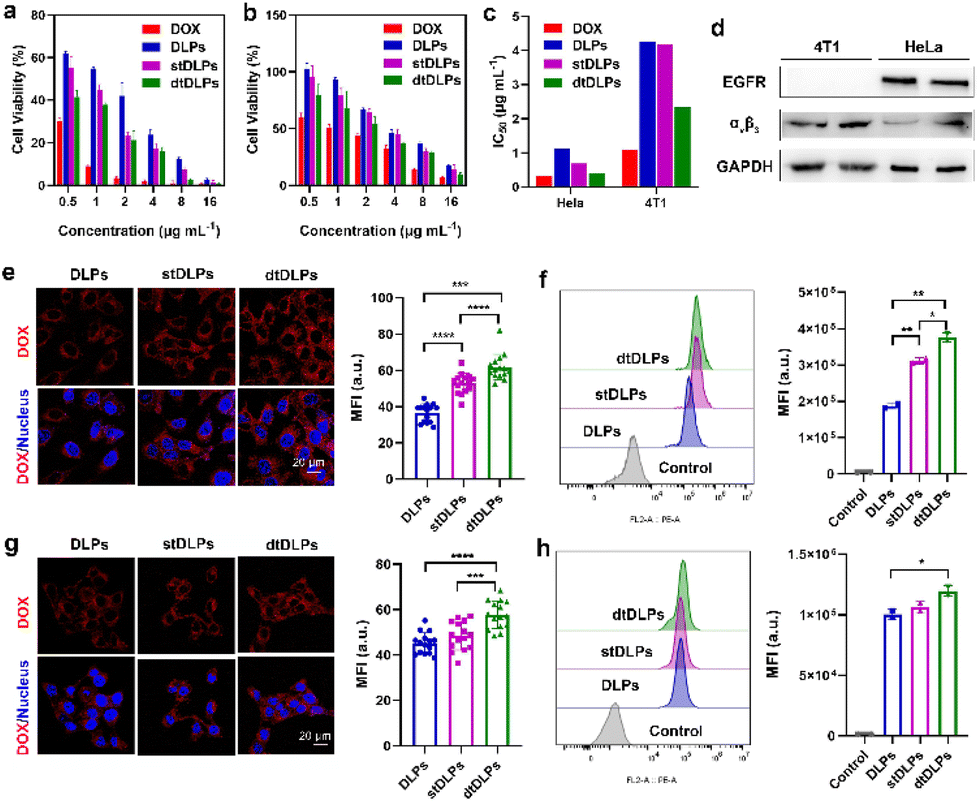 | ||
| Fig. 2 In vitro cytotoxicity of DLPs, stDLPs and dtDLPs in comparison with free DOX against HeLa cells (a) and 4T1 cells (b). (c) IC50 values of free DOX, DLPs, stDLPs and dtDLPs in both HeLa and 4T1 cells. (d) The expression levels of the EGFR and integrin αvβ3 in HeLa and 4T1 cells were evaluated by WB. The cellular internalization abilities of DLPs, stDLPs and dtDLPs in HeLa cells were evaluated by CLSM (e) and flow cytometry (f). The cellular internalization abilities of DLPs, stDLPs and tDLPs in 4T1 cells were evaluated by CLSM (g) and flow cytometry (h). Data are presented as mean ± s.d., n = 3 for (a) and (b), 15 for (e) and (g), 2 for (f) and (h). Statistical significances were calculated using the multiple t-test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. | ||
Cellular endocytosis and macrophage uptake of dtDLPs
To observe cellular endocytosis of dtDLP, we first evaluated the expression levels of the EGFR and integrin αvβ3 in these two cancer cells through western blotting (WB) analysis. As shown in Fig. 2d, HeLa cells expressed both the EGFR and integrin αvβ3, and 4T1 cells only expressed integrin αvβ3.The cellular internalization of DLPs and dtDLPs in HeLa and 4T1 cells was investigated by confocal laser scanning microscopy (CLSM) and flow cytometry. From the CLSM images (Fig. 2e), we could see that most of DOX fluorescence was confined in the cytoplasm of HeLa cells for all DLPs, stDLPs and dtDLPs, suggesting that much of DOX resides in the liposomes. Comparing to DLPs, the mean fluorescence intensity (MFI) of the cells incubated with dtDLPs was the strongest, indicating that dtDLPs have the best cellular internalization ability. The fluorescence intensity of cells incubated with the three types of liposomes was further quantified by flow cytometry. The MFI of HeLa cells treated with dtDLPs and stDLPs was 2.0-fold and 1.6-fold of the cells treated with DLPs respectively (Fig. 2f). The improvement of cellular uptake of dtDLPs in 4T1 cells was similar to that of HeLa cells but in a lower degree since 4T1 cells only express integrin αvβ3. The MFI of 4T1 cells incubated with dtDLPs and stDLPs was 1.2-fold and 1.1-fold of that for DLPs respectively (Fig. 2g and h). The difference in cellular internalization ability between DLPs, stDLPs and dtDLPs mainly derived from RP-ER coated on the surface of dtDLPs, which could bind the EGFR and integrin αvβ3 in HeLa cells and 4T1 cells.
The endocytosis pathways for DLPs and dtDLPs to enter HeLa cells and 4T1 cells were further studied by flow cytometry. Chlorpromazine (CPZ), methyl-β-cyclodextrin (MβCD), 5-(N-ethyl-N-isopropyl) amiloride (EIPA), which are used as inhibitors of clathrin-mediated endocytosis, caveolae-mediated endocytosis, and micropinocytosis, were incubated with HeLa cells and 4T1, respectively, 1 h before the incubation of DLPs or dtDLPs. DLPs could enter HeLa cells through both clathrin-mediated endocytosis and micropinocytosis (Fig. 3a). On the other hand, there were three endocytosis pathways for dtDLPs, including clathrin-mediated endocytosis, caveolae-mediated endocytosis, and micropinocytosis (Fig. 3b). Similarly, there were more endocytosis pathways for dtDLPs to enter 4T1 cells compared to those in DLPs. DLPs entered 4T1 cells through only micropinocytosis (Fig. 3c). However, dtDLPs could enter 4T1 cells through both clathrin-mediated endocytosis and micropinocytosis (Fig. 3d). DLPs had more endocytosis pathways to enter HeLa cells than 4T1 cells, suggesting it was easier for DLPs to enter into HeLa cells than 4T1 cells. In addition, dtDLPs could enter both HeLa cells and 4T1 cells through clathrin-mediated endocytosis, consistent with the receptor-mediated endocytosis from the coated dual-targeted RP-ER. These results may be could explain that, compared to DLPs, dtDLPs showed a higher degree of enhancement of the cellular uptake and cytotoxicity in HeLa cells than in 4T1 cells.
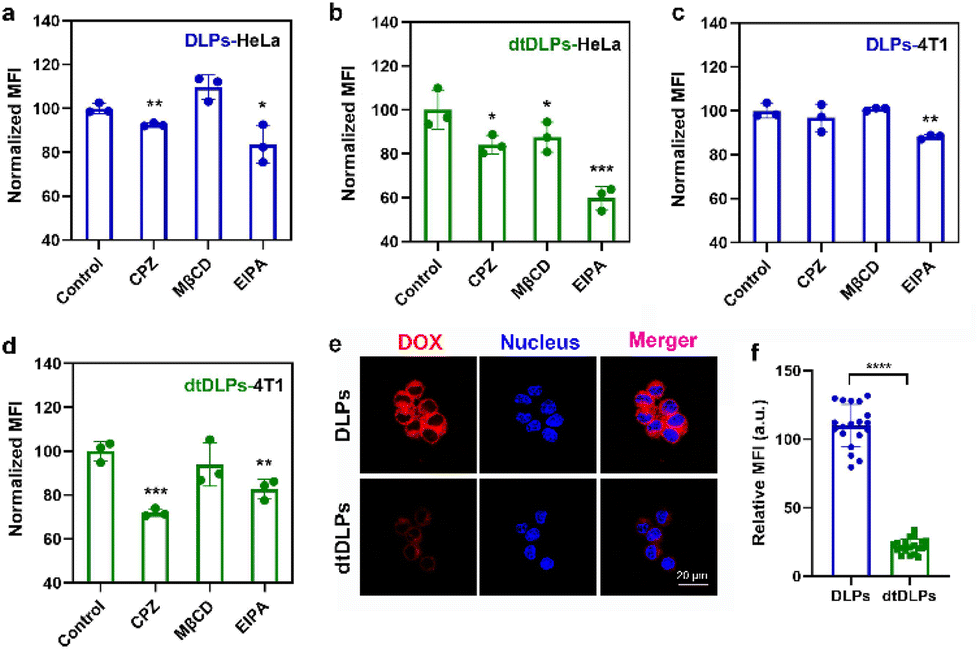 | ||
| Fig. 3 The endocytosis pathways of DLPs (a and c) and dtDLPs (b and d) in HeLa cells (a and b) and 4T1 (c and d) cells were evaluated by flow cytometry. Chlorpromazine (CPZ) (10 μM), methyl-β-cyclodextrin (MβCD) (300 μM), and 5-(N-ethyl-N-isopropyl) amiloride (EIPA) (100 μM) were used as inhibitors of clathrin-mediated endocytosis, caveolae-mediated endocytosis, and micropinocytosis. (e) CLSM images of RAW 264.7 cell uptake of DLPs and dtDLPs. (f) The relative MFI of RAW 264.7 cells incubated with DLPs and dtDLPs measured by the CLSM software. Data are presented as mean ± s.d., n = 3 for (a–d), 19–20 for (f). Statistical significances were calculated using the multiple t-test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. | ||
The cellular uptake of DLPs and dtDLPs in RAW 264.7 cells was also evaluated. Since cationic liposomes were easily taken up by macrophages, recombinant protein coating reversed the positive potential of DLPs to negative potential, leading to a reduced macrophage uptake (Fig. 3e and f). It was noted that the cells treated with DLPs showed a significantly stronger fluorescence than those treated with dtDLPs (Fig. 3e). The MFI of RAW 264.7 cells incubated with dtDLPs was reduced 5 times compared to that of DLPs (Fig. 3f), indicating that RP-ER decoration decreases the interaction between macrophages and the liposomes.
Drug penetration in multicellular spheroids (MCs)
Tumor penetration of liposomes is one of the major obstacles to achieving desirable antitumor efficiency due to the large size and poor diffusion in tumors.31–33 Three-dimensional multicellular spheroids (MCs) are considered the most ideal model to mimic the compact tumor microenvironment in vitro. Here, we incubated DLPs and dtDLPs with HeLa and 4T1 MCs to evaluate their penetration abilities (Fig. 4). To further evaluate the penetration behaviors of DLPs and dtDLPs, the HeLa and 4T1 MCs were scanned every 30 and 10 μm, respectively, from top to bottom. For HeLa MCs treated with DLPs (Fig. 4a), most of the DOX fluorescence was around at the periphery. However, some DOX fluorescence appeared in the center of the MCs treated with dtDLPs. To further calculate the penetration distance, the fluorescence intensity distribution from the edge to the center of the HeLa MCs along the equatorial line was analyzed and the MFI threshold for penetration distance measurement was set as 50 a.u. DLPs could penetrate a distance of 180 μm (Fig. 4b). In contrast, dtDLPs were found to penetrate over a distance of 300 μm (Fig. 4c) much farther than that for DLPs, suggesting that dtDLPs have a better penetration ability than DLPs. To better quantitatively analyse the penetration ability of DLPs and dtDLPs, we used retention index (RI), migration index (MI) and distribution index (DI) to describe the spheroid retention, spatial distribution and the whole distribution within MCs.27 dtDLPs had a higher RI, MI and DI than DLPs in HeLa MCs (Fig. S9†), indicating that dtDLPs have superior retention, migration and distribution performances over DLPs.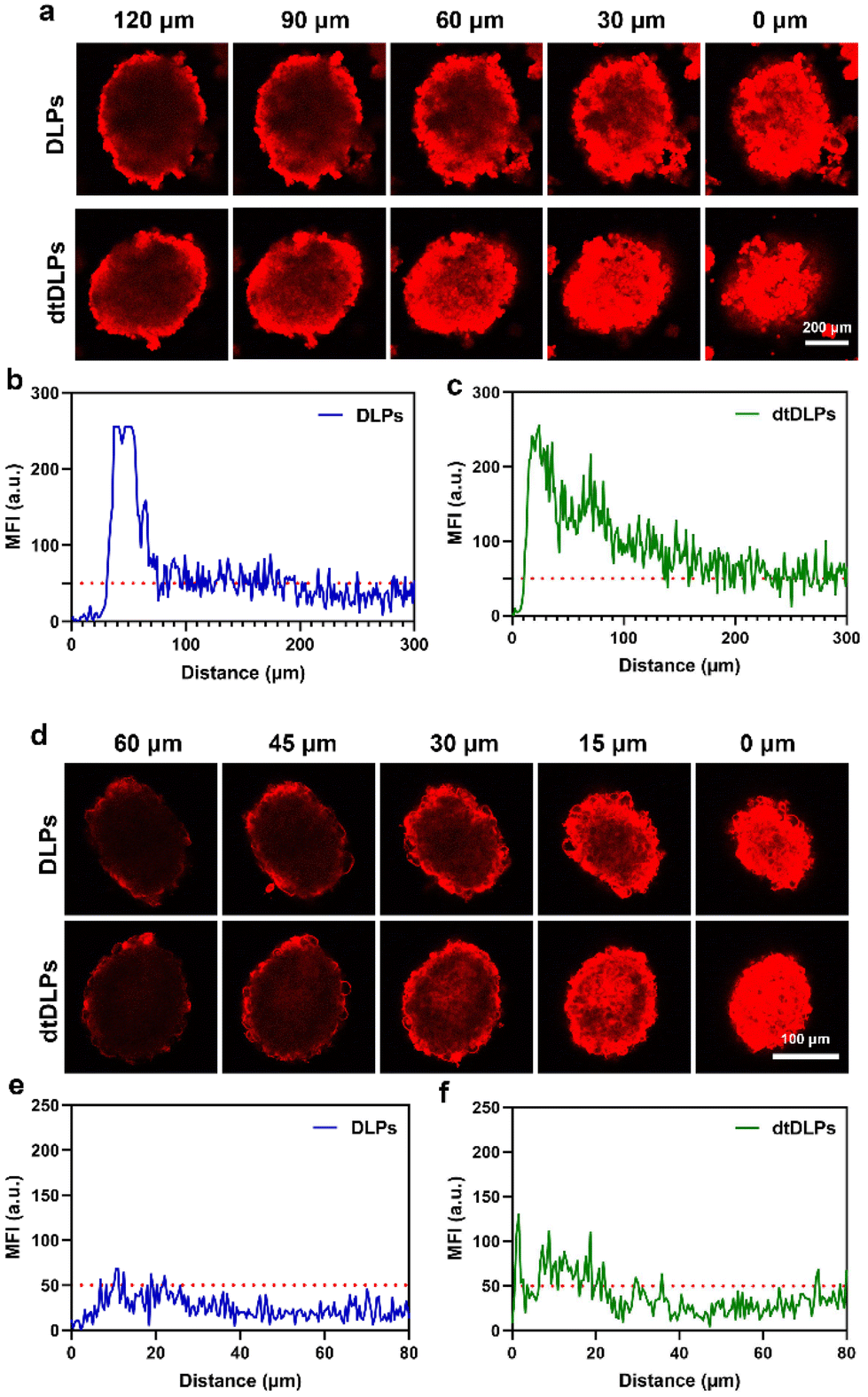 | ||
| Fig. 4 (a) CLSM images of HeLa MCs incubated with DLPs, dtDLPs for 24 h at 37 °C and the representative Z-stack images. The MFI distribution along the equatorial line of HeLa MCs for DLPs (b) and dtDLPs (c). (d) CLSM images of 4T1 MCs incubated with DLPs, dtDLPs for 24 h at 37 °C and the representative Z-stack images. The MFI distribution along the equatorial line of 4T1 MCs for DLPs (e) and dtDLPs (f). | ||
Compared to HeLa MCs, 4T1 MCs were hard to be penetrated by DLPs although their diameter was much smaller than that of HeLa MCs (Fig. 4d). The penetration distances for DLPs (Fig. 4e) and dtDLPs (Fig. 4f) in 4T1 MCs were only 15 and 20 μm, respectively, which were much lower than the values in HeLa MCs. In addition, we found that the MI of dtDLPs was equivalent to that of DLPs and the improvement of DI for dtDLPs was mainly derived from the enhanced RI (Fig. S10†). The different penetration behaviors in HeLa and 4T1 MCs reflect the difference of the receptor expression on the surface of HeLa cells and 4T1 cells as well as the different endocytosis pathways for DLPs and dtDLPs in these two cancer cells.
Tumor accumulation and biodistribution
In order to evaluate the biodistribution of DLPs and dtDLPs in vivo, DLPs and dtDLPs were labeled with NIR-797. Then DLP-NIR-797 and dtDLP-NIR-797 were intravenously injected into mice with 4T1 or HeLa tumors, and near infrared fluorescence (NIRF) imaging of the mice was performed at predetermined times. We could see that the fluorescence signals were very weak for the mice treated with free NIR-797 for all times, which was mainly due to the short blood circulation time of free NIR-797 (Fig. 5a). For DLP-NIR-797, the fluorescence signal was stronger than that of free NIR-797. However, the signal in the tumor decreased with time and the major signal was from the liver. This is mainly due to the positive zeta potential of DLPs, leading to a rapid uptake by the reticuloendothelial system (RES) in the liver. Mice treated with dtDLP-NIR-797 displayed the strongest fluorescence signal among the three samples in tumors, suggesting that recombinant protein decoration can effectively make liposomes target tumors and improve the tumor drug accumulation. The MFIs in tumors and the liver at different times indicate that dtDLP-NIR-797 showed the strongest MFI in both the tumor and liver. Different from the monotonic reduction in MFI for free NIR-797 and DLPs-NIR-797, the MFI of tumors for dtDLP-NIR-797 had a maximum at 8 h post-injection and then gradually decreased (Fig. 5b). Moreover, the MFIs in the liver for dtDLP-NIR-797 also increased from 4 h to 8 h and then gradually reduced (Fig. 5c). For HeLa tumors, mice treated with dtDLP-NIR-797 displayed the stronger fluorescence signal in tumors than that treated with DLP-NIR-797 (Fig. S12a†). The MFI in tumors for dtDLP-NIR-797 had a maximum from 48 h to 96 h post-injection and then decreased (Fig. S12b†). Moreover, the MFIs in the liver for dtDLP-NIR-797 also increased at 96 h and then decreased (Fig. S12c†).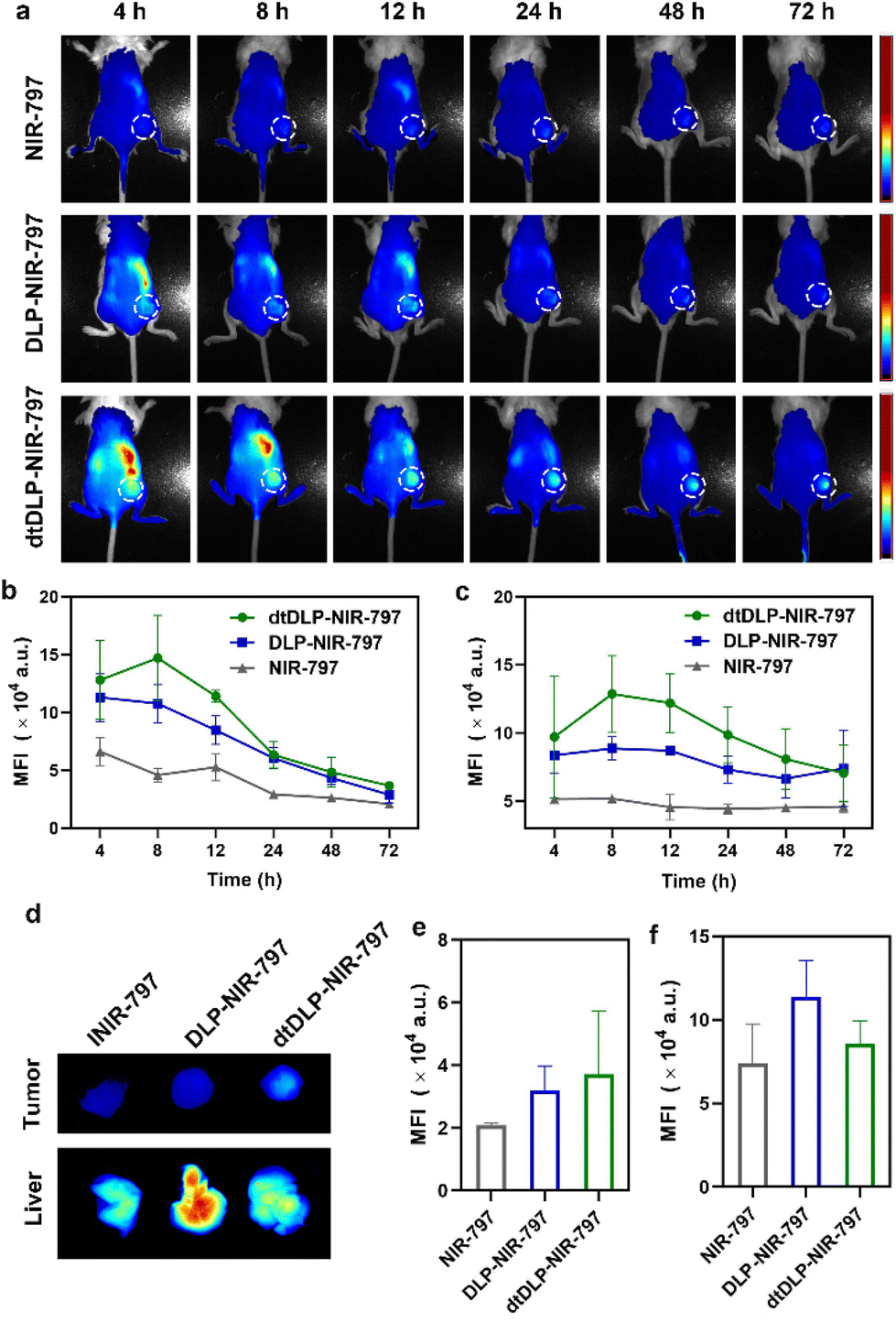 | ||
| Fig. 5 (a) The in vivo NIRF images of 4T1 tumor-bearing mice following intravenous injection of free NIR-797, DLP-NIR-797 and dtDLP-NIR-797 at different time points. The tumors were surrounded with dotted lines. The MFIs in tumors (b) and livers (c) for different agents at different times were measured from the NIRF images. (d) At the end of the experiment, livers and tumors were taken out for NIRF imaging. The MFIs for livers (e) and tumors (f) collected from different groups were measured. Data are presented as mean ± s.d., n = 2 for NIR-797 group, 3 for DLPs and dtDLPs group. | ||
Ex vivo NIRF imaging at 72 h post-injection for 4T1 tumors and 120 h post-injection for HeLa tumors further confirmed that dtDLPs had greater accumulation in tumors compared to DLPs (Fig. 5d–f and Fig. S11 and S12d–f†). From the NIRF images of the organs from the mice treated with different agents, we could see that free NIR-797 was mainly accumulated in the liver and tumors (Fig. S11a and S11b†), while DLP-NIR-797 and dtDLP-NIR-797 could accumulate in tumors, liver, spleen, lungs, kidneys and intestine (Fig. S11c, S11d and S13†), which was mainly due to the extended circulation time of liposomes.
Drug penetration in tumor tissue
To evaluate the penetration of dtDLPs in tumor, dtDLPs were intravenously injected into 4T1 and HeLa tumor-bearing mice and the mice injected with free DOX and DLPs were used as controls. At 24 h post-injection, the tumors were resected and the blood vessels in tumors were stained with Alexa-488 through immunofluorescence. It was noted that at 24 h post-injection, the DOX fluorescence signal in tumors was hardly detected for free DOX and DLPs (Fig. S14 and S15†). However, the DOX fluorescence signal in tumor was still quite strong for dtDLPs, suggesting that dtDLPs improved the DOX accumulation in tumors. On the other hand, the DOX fluorescence signal from dtDLPs was not colocalized with the vessels, implying that dtDLPs could penetrate far from blood vessels and deliver drugs into deep tumors.Antitumor efficiency in both HeLa and 4T1 tumors
The antitumor efficiency of DLPs and dtDLPs in vivo was finally examined in both HeLa and 4T1 tumor-bearing mice. When the tumor size reached a predetermined value, the mice were treated with free DOX, DLPs and dtDLPs, respectively, at 5 mg kg−1 dose (DOX concentration eq.), and we defined this day as Day 1. Meanwhile, the mice treated with PBS are designated as the control. Fig. 6a exhibits the HeLa tumor growth curves of the mice that received the free DOX, DLP and dtDLP treatments. The tumors of mice that received PBS grew rapidly while those treated with free DOX grew slowly. In contrast to DLPs and free DOX which had limited antitumor effects, dtDLPs showed the best antitumor activity. The mice that received dtDLP treatment displayed the slowest tumor growth rate, indicating that dtDLPs had a prominent impact on inhibiting tumor growth. At Day 15, the average tumor volumes for control, free DOX, DLPs and dtDLPs groups were 364, 212, 250 and 103 mm3, respectively. The difference between free DOX and dtDLP groups was highly significant (P < 0.01). And for free DOX, DLPs and dtDLPs, the tumor growth inhibition (TGI) at Day 15 were 45%, 34% and 78%, respectively. Body weight changes of the mice receiving various treatments are shown in Fig. 6b. The mice treated with free DOX presented a decreasing body weight in the first week because of the side effect of free DOX.34 On the other hand, DLP and dtDLP groups showed a steady weight increase, suggesting that liposomal formulation reduces the side effects of free DOX. At the end of the anticancer experiment, the tumors in all groups were taken out and weighed. The average weights of tumors for control, free DOX, DLP and dtDLP groups were 186, 100, 132 and 84 mg, respectively (Fig. 6c).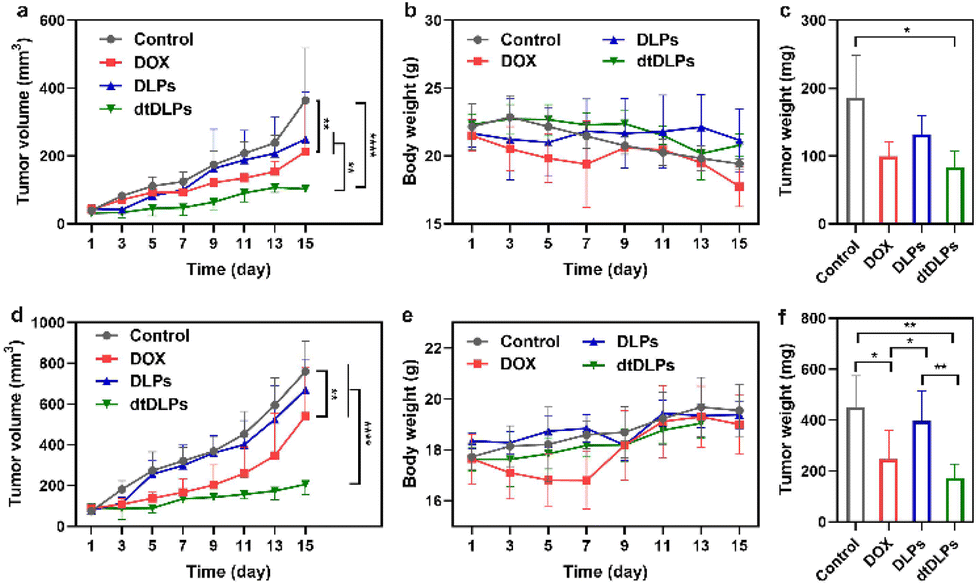 | ||
| Fig. 6 The anti-cancer effect of dtDLPs in both HeLa tumor (a–c) and 4T1 tumor (d–f) were evaluated. Tumor volume curves of HeLa (a) and 4T1 (d) tumor-bearing mice receiving different treatments. Evolution of body weight change of mice bearing HeLa tumor (b) or 4T1 tumor (e) receiving different treatments during the experiments. The weights of HeLa (c) and 4T1 (f) tumors collected from the treated mice at the end of the experiment. Data are presented as mean ± s.d., n = 4–6. Statistical significances were calculated using the multiple t-test, *P < 0.05, **P < 0.01, ****P < 0.0001. | ||
Similar anticancer effect of dtDLPs was found in 4T1 tumors. dtDLPs showed the most effective anticancer activity with a negligible side effect (Fig. 6d and e). At Day 15, the tumor volumes for control, free DOX, DLP and dtDLP groups were 760, 541, 672 and 206 mm3, respectively. And for free DOX, DLPs and dtDLPs, the TGI was 34%, 15% and 83%, respectively. The average weights of tumors from control, free DOX, DLP and dtDLP groups were 452, 247, 397 and 173 mg, respectively (Fig. 6f). For further analyzing the biosafety and antitumor effect of dtDLPs, the major organs and tumors were stained by hematoxylin–eosin (H&E). No significant tissue damage was found for all the groups (Fig. S16†). Meanwhile, the tumors in the dtDLP treated group exhibited the largest areas of necrosis (Fig. S17 and S18†). All the results indicate that dtDLPs have the greatest antitumor effect, while positively charged DLPs have a moderate antitumor effect. Thus, dual-targeting recombinant protein decoration is a desirable strategy to design biomimetic liposomes to improve drug delivery efficacy through improving tumor targeting, endocytosis and penetration.
Conclusions
Due to their cell membrane-like structure, targeted protein-anchored liposomes showed great potential to improve drug delivery to tumors through active targeting.35,36 Different from traditional chemical modification, electrostatic adsorption was applied in this work to develop an EGFR and integrin αvβ3 dual-targeted recombinant protein decorated dtDLPs. The dual-targeted recombinant protein was prepared through genetic engineering, which provides a precise control of the two targeting moieties.27 Due to the intratumor heterogeneity, dual-targeted biomimetic liposomes increase the possibility to efficiently deliver drugs to tumors. Through targeting two receptors on the cancer cells simultaneously, dtDLPs provide more endocytic pathways, leading to enhanced cellular endocytosis in both HeLa cells and 4T1 cells, despite the significantly different receptor expression levels in these two cells. Meanwhile, dtDLPs improved tumor penetration. Thanks to these features, dual-targeted dtDLPs have a superior antitumor effect in both HeLa and 4T1 tumors. Our work provides a design strategy to fabricate bispecific biomimetic liposomes with great tumor accumulation and penetration abilities.Author contributions
X. Jiang, S. Zhou, W. Chen, and L. Jiang conceived the idea and designed the experiments. S. Zhou, W. Chen, L. Jiang, C. Li and Y. Yuan conducted the experiments. The manuscript was written through contributions of all authors. All the authors have given their approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFA0701301), Natural Science Foundation of China (No. 92163214, 51803089, 51690153 and 21720102005), the Natural Science Foundation of Jiangsu Province (No. BK20202002) and the Fundamental Research Funds for the Central Universities (0205-14380264).References
- A. M. Vargason, A. C. Anselmo and S. Mitragotri, Nat. Biomed. Eng., 2021, 5, 951–967 CrossRef PubMed.
- S. A. A. Kooijmans, O. G. de Jong and R. M. Schiffelers, Adv. Drug Delivery Rev., 2021, 173, 252–278 CrossRef CAS PubMed.
- J. Majumder, O. Taratula and T. Minko, Adv. Drug Delivery Rev., 2019, 144, 57–77 CrossRef CAS PubMed.
- L. Wang, X. You, C. Dai, Y. Fang and J. Wu, Biomater. Sci., 2022, 10, 2263–2274 RSC.
- S. Tan, T. Wu, D. Zhang and Z. Zhang, Theranostics, 2015, 5, 863–881 CrossRef CAS PubMed.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2018, 30, 1706759 CrossRef PubMed.
- D. E. Large, R. G. Abdelmessih, E. A. Fink and D. T. Auguste, Adv. Drug Delivery Rev., 2021, 176, 113851 CrossRef CAS PubMed.
- P. E. Saw, X. Xu, M. Zhang, S. Cao, O. C. Farokhzad and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 6249–6252 CrossRef CAS PubMed.
- H. Zhang, X. You, X. Wang, L. Cui, Z. Wang, F. Xu, M. Li, Z. Yang, J. Liu, P. Huang, Y. Kang, J. Wu and X. Xia, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2005191118 CrossRef CAS PubMed.
- Y. Sadzuka, K. Kishi, S. Hirota and T. Sonobe, J. Liposome Res., 2003, 13, 157–172 CrossRef CAS PubMed.
- R. Eliasen, T. L. Andresen and J. B. Larsen, Adv. Mater. Interfaces, 2019, 6, 1801807 CrossRef.
- N. Dos Santos, C. Allen, A.-M. Doppen, M. Anantha, K. A. K. Cox, R. C. Gallagher, G. Karlsson, K. Edwards, G. Kenner, L. Samuels, M. S. Webb and M. B. Bally, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1367–1377 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- J. Tang, L. Zhang, H. Fu, Q. Kuang, H. Gao, Z. Zhang and Q. He, Acta Pharm. Sin. B, 2014, 4, 67–73 CrossRef PubMed.
- H. Hatakeyama, H. Akita and H. Harashima, Biol. Pharm. Bull., 2013, 36, 892–899 CrossRef CAS PubMed.
- J. M. Tuscano, S. M. Martin, Y. Ma, W. Zamboni and R. T. O'Donnell, Clin. Cancer Res., 2010, 16, 2760–2768 CrossRef CAS PubMed.
- L. Belfiore, D. N. Saunders, M. Ranson, K. J. Thurecht, G. Storm and K. L. Vine, J. Controlled Release, 2018, 277, 1–13 CrossRef CAS PubMed.
- L. Zhu, P. Kate and V. P. Torchilin, ACS Nano, 2012, 6, 3491–3498 CrossRef CAS PubMed.
- G. T. Noble, J. F. Stefanick, J. D. Ashley, T. Kiziltepe and B. Bilgicer, Trends Biotechnol., 2014, 32, 32–45 CrossRef CAS PubMed.
- S. E. Baek, K. H. Lee, Y. S. Park, D.-K. Oh, S. Oh, K.-S. Kim and D.-E. Kim, J. Controlled Release, 2014, 196, 234–242 CrossRef CAS PubMed.
- L. Mei, L. Fu, K. Shi, Q. Zhang, Y. Liu, J. Tang, H. Gao, Z. Zhang and Q. He, Int. J. Pharm., 2014, 468, 26–38 CrossRef CAS PubMed.
- P. M. Peiris, F. He, G. Covarrubias, S. Raghunathan, O. Turan, M. Lorkowski, B. Gnanasambandam, C. Wu, W. P. Schiemann and E. Karathanasis, Nanoscale, 2018, 10, 6861–6871 RSC.
- A. De Luca, A. Carotenuto, A. Rachiglio, M. Gallo, M. R. Maiello, D. Aldinucci, A. Pinto and N. Normanno, J. Cell. Physiol., 2008, 214, 559–567 CrossRef CAS PubMed.
- M. A. S. Abourehab, A. M. Alqahtani, B. G. M. Youssif and A. M. Gouda, Molecules, 2021, 26, 6677 CrossRef CAS PubMed.
- J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9–22 CrossRef CAS PubMed.
- J. Xiong, L. Yan, C. Zou, K. Wang, M. Chen, B. Xu, Z. Zhou and D. Zhang, J. Hematol. Oncol., 2021, 14, 177 CrossRef CAS PubMed.
- W. Chen, Y. Yuan, C. Li, H. Mao, B. Liu and X. Jiang, Adv. Mater., 2022, 34, 2109376 CrossRef CAS PubMed.
- H. Sha, Z. Zou, K. Xin, X. Bian, X. Cai, W. Lu, J. Chen, G. Chen, L. Huang, A. M. Blair, P. Cao and B. Liu, J. Controlled Release, 2015, 200, 188–200 CrossRef CAS PubMed.
- L. Jiang, W. Chen, S. Zhou, C. Li, X. Zhang, W. Wu and X. Jiang, Biomater. Sci., 2018, 6, 774–778 RSC.
- W. Chen, S. Ji, X. Qian, Y. Zhang, C. Li, W. Wu, F. Wang and X. Jiang, Polym. Chem., 2017, 8, 2105–2114 RSC.
- M. W. Dewhirst and T. W. Secomb, Nat. Rev. Cancer, 2017, 17, 738 CrossRef CAS PubMed.
- Y.-J. Li, J.-Y. Wu, X.-B. Hu, T. Ding, T. Tang and D.-X. Xiang, Adv. Healthcare Mater., 2021, 10, 2100794 CrossRef CAS PubMed.
- G. Wang, B. Wu, Q. Li, S. Chen, X. Jin, Y. Liu, Z. Zhou, Y. Shen and P. Huang, Small, 2020, 16, 2004172 CrossRef CAS PubMed.
- A. B. E. Attia, C. Yang, J. P. K. Tan, S. Gao, D. F. Williams, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2013, 34, 3132–3140 CrossRef PubMed.
- N. Lozano, Z. S. Al-Ahmady, N. S. Beziere, V. Ntziachristos and K. Kostarelos, Int. J. Pharm., 2015, 482, 2–10 CrossRef CAS PubMed.
- M. Ying, Q. Shen, Y. Liu, Z. Yan, X. Wei, C. Zhan, J. Gao, C. Xie, B. Yao and W. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 13232–13241 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00952h |
| This journal is © The Royal Society of Chemistry 2022 |