
Aqueous synthesis and self-assembly of bioactive and thermo-responsive HA-b-ELP bioconjugates†
Manon
Levêque
,
Ye
Xiao
,
Laura
Durand
,
Louise
Massé
,
Elisabeth
Garanger
 * and
Sébastien
Lecommandoux
*
* and
Sébastien
Lecommandoux
*
Université de Bordeaux, CNRS, Bordeaux INP, LCPO, UMR 5629, Pessac F-33600, France. E-mail: garanger@enscbp.fr; lecommandoux@enscbp.fr
First published on 13th September 2022
Abstract
The design of synthetic (bio)macromolecules that combine biocompatibility, self-assembly and bioactivity properties at the molecular level is an intense field of research for biomedical applications such as (nano)medicine. In this contribution, we have designed and synthesized a library of bioactive and thermo-responsive bioconjugates from elastin-like polypeptides (ELPs) and hyaluronic acid (HA) in order to access bioactive self-assembled nanoparticles. These were prepared by a simple synthetic and purification strategy, compatible with the requirements for biological applications and industrial scale-up. A series of 9 HA-b-ELP bioconjugates with different compositions and block lengths was synthesized under aqueous conditions by strain-promoted azide–alkyne cycloaddition (SPAAC), avoiding the use of catalysts, co-reactants and organic solvents, and isolated by a simple centrifugation step. An extensive physico-chemical study was then performed on the whole library of bioconjugates in an attempt to establish structure–property relationships. In particular, the determination of the critical conditions for thermally driven self-assembly was carried out upon temperature (CMT) and concentration (CMC) gradients, leading to a phase diagram for each of these bioconjugates. These parameters and the size of nanoparticles were found to depend on the chemical composition of the bioconjugates, namely on the respective size of individual blocks. Understanding the mechanism underlying this dependency is a real asset for designing more effective experiments: with key criteria defined (e.g. concentration, temperature, salinity, and biological target), the composition of the best candidates can be rationalized. In particular, four of the bioconjugates (HA4.6k-ELPn80 or n100 and HA24k-ELPn80 or n100) were found to self-assemble into well-defined spherical core–shell nanoparticles, with a negative surface charge due to the HA block exposed at the surface, a hydrodynamic diameter between 40 and 200 nm under physiological conditions and a good stability over time at 37 °C. We therefore propose here a versatile and simple design of smart, controllable, and bioactive nanoparticles that present different behaviors depending on the diblocks’ composition.
Introduction
For many years, significant efforts have been dedicated to the design of synthetic (bio)macromolecules that combine biocompatibility, self-assembly and bioactivity properties at the molecular level for specific applications, in particular for the field of (nano)medicine. Among these, elastin-like polypeptides (ELPs) have arisen as promising biocompatible and stimuli-responsive protein-like polymer scaffolds.1,2 Inspired from a natural structural protein (i.e., elastin), ELPs are artificial recombinant polypeptides,3 behaving simultaneously as intrinsically disordered proteins (IDPs) and random-coil polymers in θ-solvent in their soluble form in aqueous media.4,5 Most common ELPs are based on consecutive repeats of “Val-Pro-Gly-Xaa-Gly” pentapeptides, Xaa being a guest residue that can be any natural or non-natural residue except for proline.6 Efficiently produced in Escherichia coli (E. coli) bacteria and purified by a chromatography-free technique termed Inverse Transition Cycling (ITC),7,8 recombinantly produced ELPs are polypeptides characterized by an exquisite control in the primary sequence (including the monomer sequence and chain length) unachievable with current polymerization techniques. Perfect batch-to-batch reproducibility from bacterial clones and exact molar masses allow the establishment of precise structure–property relationships. A major characteristic of ELPs is their lower critical solubility temperature (LCST) transition behavior in aqueous media.9,10 At a specific concentration, an ELP of a defined sequence and chain length is soluble below a cloud point temperature (Tcp) and insoluble above it. This cloud point temperature depends on several macromolecular and environmental parameters, such as the primary sequence of the ELP (guest residue composition), overall hydrophilicity/-phobicity of the polypeptide chain,10–13 the presence and concentration of salts,14,15 ionic strength, and ELP molar concentration following a logarithmic law.12 Importantly, the LCST is the lowest Tcp among all concentrations considered and is defined as a critical point (Cc; Tcp,c) characteristic of a family of ELPs with a defined monomer sequence but of any chain length.11 At any concentration, the aggregation of an ELP above the Tcp is perfectly reversible, with a re-solubilization into free chains upon cooling the aqueous solution below the Tcp. This property represents a real advantage in the design of “smart” polymer materials, and in particular for the formulation of self-assembled nanoparticles. In multiblock copolymer systems, the presence of an ELP block can induce a thermo-dependent amphiphilicity, leading to self-assembly into different (nano)objects.16 The resulting self-assemblies strongly depend on the nature of the other block(s). For instance, in the case of a hybrid diblock copolymer, where the ELP is combined with a hydrophilic polymer segment (e.g., polysaccharide,17 PEG,18 and PLA19), the resulting bioconjugate is soluble below the Tcp of the ELP and self-assembled into micelle-like nanoparticles above it (the Tcp is therefore termed critical micellar temperature, CMT). In contrast, the coupling of an ELP to more hydrophobic species (e.g., drugs,20,21 lipids,22–24 and polymers25) leads to an assembled system at low temperature that macroscopically aggregates upon heating of the solution and dehydration-induced aggregation of the ELP. In the case of an ELP diblock copolymer, in which the Tcp of the two ELPs are significantly different, three physical states can be observed with increasing temperature: free chains in cold aqueous medium (i.e., below the Tcp of the most hydrophilic block), micelles above the CMT and below the Tcp of the most hydrophobic block, and macroscopic aggregates above the latter (termed bulk Tcp).5,26–29 Finally, a copolymer composed of an ELP block and a UCST-polymer block (e.g., resilin-like polypeptide) will present an inversion of micelle behavior: at low temperature, the UCST-block is hydrophobic while the ELP is hydrophilic, therefore assembling into micelles with a UCST-polymer core and an ELP corona.30 The situation is reversed at higher temperature, with a hydrophobic ELP core and a hydrophilic UCST-polymer corona. In between, the system can be either all soluble or all aggregated depending on the relative values of the cloud point and clearing point temperatures (Tcp and Tcl) of the two blocks. This behavior is of significant interest as it enables the selective exposure of different ligands by coupling them to one or the other block. ELPs are therefore particularly relevant precision polymers for biomedical applications, in particular for nanomedicine: they are bioinspired, biocompatible, and stimuli-responsive, and their integration in multiblock polymer systems allows a precise control of their self-assembly properties.1,31–34 However, because they lack intrinsic bioactivity, several strategies have been explored for their use as effective drug-delivery nanocarriers.1,34 In particular, targeting moieties have been added either by genetic engineering or by chemical post-modifications of recombinant ELPs.35–44In the present work, we have particularly explored the possibility of conjugating ELPs to hyaluronan (HA) as a CD44 targeting ligand.45,46 CD44 is a biological receptor that is overexpressed at the surface of tumor cells in many cancers.47–52 Notably, it is considered as a reliable marker for cancerous stem cells (CSCs) that are suspected to be responsible for relapses after treatment.53–58 Its main ligand is HA,59 a polysaccharide naturally present in the organism, mostly in the skin and cartilage, hence its high biocompatibility and biodegradability.45 Its chemical and physicochemical properties make it an excellent candidate for the design of drug delivery systems,60,61 among other biological applications. In particular, its high hydrophilicity provides it with great hydration capacities that have made it an inescapable ingredient in the cosmetic field.62,63 It is thus already approved by the FDA for cosmetic and pharmaceutical use and produced at industrial scale.64 Interestingly, ELPs and HA have already been combined either in mixed formulations or as covalent conjugates, but mainly for tissue engineering and cell culture applications. Their association for the design of smart nanocarriers is to our knowledge still unexplored.
In a previous study, we have described the synthesis of a small library of oligo- or polysaccharide-b-ELP bioconjugates, including a HA-b-ELP bioconjugate based on a 7 kDa HA and 17 kDa ELP.17 These bioconjugates were synthesized by copper-catalyzed azide–alkyne cycloaddition (CuAAC) in organic solvents. Once isolated, these were shown to spontaneously self-assemble in water into nanometer-sized particles upon heating and to reversibly disassemble upon cooling back the solution. In particular, the HA-b-ELP bioconjugate presented a CMT ranging between 37 °C at 800 μM and 58 °C at 30 μM, and formed sub-micron sized particles (ca. 200–300 nm) above the CMT, values that were poorly relevant to our target application (i.e., nanomedicine). In this contribution, we therefore aimed at providing a straightforward synthetic design to afford well-defined HA-b-ELP bioconjugates and explore their self-assembly. The synthetic strategy was first developed to be carried out in water and without the use of catalysts and co-reactants. ELPs with various chain lengths were explored to lower the CMT in a physiologically relevant temperature domain and to minimize its dependence on the molar concentration of the bioconjugates. Different sizes of HA were also considered to study the effect of the size of the hydrophilic block on the nanoparticles’ properties. A total of 9 HA-b-ELP bioconjugates were therefore synthesized and characterized and their self-assembly in aqueous media was extensively explored. In particular, the determination of CMCs at different temperatures and CMTs at different concentrations allowed the establishment of phase diagrams for all bioconjugates, allowing the prediction of their soluble or self-assembled state in subsequent biological studies.
Results and discussion
The diblock HA-b-ELP bioconjugate previously reported by our group comprised a 7 kDa HA segment covalently linked to a 40 repeat unit-ELP (ELP[M1V3-40]) of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 035 g mol−1.17 For the present work, the primary sequence of the ELP was conserved, i.e., [(VPGVG)(VPGMG)(VPGVG)2]m, noted as ELP[M1V3-n] with n being the total number of (VPGXG) pentapeptide repeats (n = 4 × m). However, this HA7k-b-ELP[M1V3-40] displays a high CMT and a strong dependency of the CMT on molar concentration, which is typical of short ELPs, and can compromise the stability of self-assembled nanoparticles over a large concentration range. We have thus decided to increase the length (and therefore molar mass) of the ELPs used for this study, and explored three different ELP[M1V3-n] with n = 60, 80, and 100 (shortened as ELPn60, ELPn80 and ELPn100 in this manuscript). These ELPs were produced recombinantly in E. coli bacteria following previously reported procedures.65 Their detailed characterization is provided in Fig. S1–S5.† Three sizes of HA were also selected, namely 4.6, 24 and 42 kDa. The synthesis of the 9 HA-b-ELP bioconjugates (thereafter denoted as HAxk-ELPnZ, with x corresponding to the molar mass of the HA segment and Z corresponding to the number of pentapeptide repeats) was achieved in water by strain promoted azide–alkyne cycloaddition (SPAAC) in order to avoid the use of a copper catalyst, co-reactants and organic solvents as compared to our previous synthetic strategy.17 To this end, the different HA segments were modified at the reducing chain end to introduce an azido group, while the three ELPs were modified at the N-terminal chain end to provide them with a dibenzocyclooctyne (DBCO) moiety, the strain induced by the cyclic structure making the alkyne highly reactive and enabling a catalyst-free cycloaddition reaction66–68 (Fig. 1).
035 g mol−1.17 For the present work, the primary sequence of the ELP was conserved, i.e., [(VPGVG)(VPGMG)(VPGVG)2]m, noted as ELP[M1V3-n] with n being the total number of (VPGXG) pentapeptide repeats (n = 4 × m). However, this HA7k-b-ELP[M1V3-40] displays a high CMT and a strong dependency of the CMT on molar concentration, which is typical of short ELPs, and can compromise the stability of self-assembled nanoparticles over a large concentration range. We have thus decided to increase the length (and therefore molar mass) of the ELPs used for this study, and explored three different ELP[M1V3-n] with n = 60, 80, and 100 (shortened as ELPn60, ELPn80 and ELPn100 in this manuscript). These ELPs were produced recombinantly in E. coli bacteria following previously reported procedures.65 Their detailed characterization is provided in Fig. S1–S5.† Three sizes of HA were also selected, namely 4.6, 24 and 42 kDa. The synthesis of the 9 HA-b-ELP bioconjugates (thereafter denoted as HAxk-ELPnZ, with x corresponding to the molar mass of the HA segment and Z corresponding to the number of pentapeptide repeats) was achieved in water by strain promoted azide–alkyne cycloaddition (SPAAC) in order to avoid the use of a copper catalyst, co-reactants and organic solvents as compared to our previous synthetic strategy.17 To this end, the different HA segments were modified at the reducing chain end to introduce an azido group, while the three ELPs were modified at the N-terminal chain end to provide them with a dibenzocyclooctyne (DBCO) moiety, the strain induced by the cyclic structure making the alkyne highly reactive and enabling a catalyst-free cycloaddition reaction66–68 (Fig. 1).
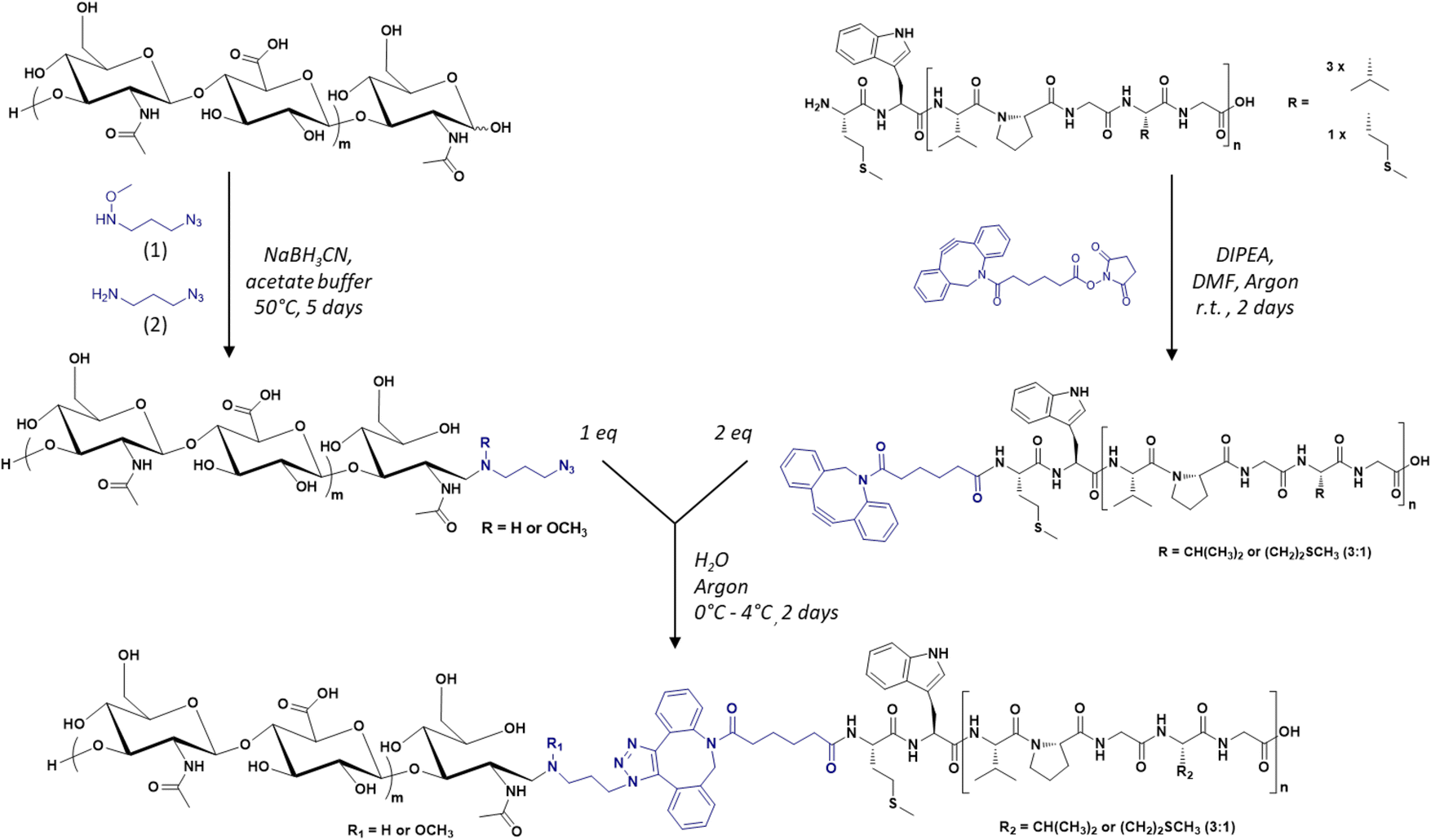 | ||
| Fig. 1 General synthetic scheme of the library of HA-b-ELP bioconjugates by SPAAC, using azido-functionalized HA segments (left) and DBCO-functionalized ELPs (right). | ||
In a first step, the anomeric position of the three HA segments was functionalized with an azido group through reductive amination in the presence of NaBH3CN at 50 °C (Fig. 1). N-(3-Azidopropyl)-O-methylhydroxylamine (1), a home-made reagent favoring the closed form of the last sugar unit,17,69 was used for HA 4.6 kDa, while the commercially available azidopropanamine (2) was used for HA 24 and 42 kDa. The ELP block was functionalized at the N-terminal chain end by amidation reaction using a commercially available heterobifunctional cross-linking agent containing a DBCO moiety and a N-hydroxysuccinimide-activated ester group. The choice of DBCO as a constrained alkyne was motivated by its stability, reactivity, and commercial availability. The three HA-N3 (i.e., 4.6, 24 and 42 kDa) and the three DBCO-ELP (n60, n80 and n100) were obtained in good yields and fully characterized by 1H NMR, MALDI and SEC (Fig. S6 and S7†). For the 9 HA-b-ELP bioconjugates, the consecutive SPAAC reactions were carried out in pure water, using a 2-fold excess of DBCO-ELP. In order to avoid ELP oxidation (at methionine residues) and aggregation, the reactions were performed at 4 °C under an inert atmosphere. After 2 days, HA-b-ELP bioconjugates were isolated and excess DBCO-ELP removed by a simple centrifugation at 40 °C, the diblock HA-b-ELP remaining in the supernatant while the aggregated excess ELP was eliminated in the pellet. The success of the reactions and purifications was confirmed by gel electrophoresis (SDS-PAGE) as illustrated in Fig. 2A for the HA4.6k-ELPn80 bioconjugate.
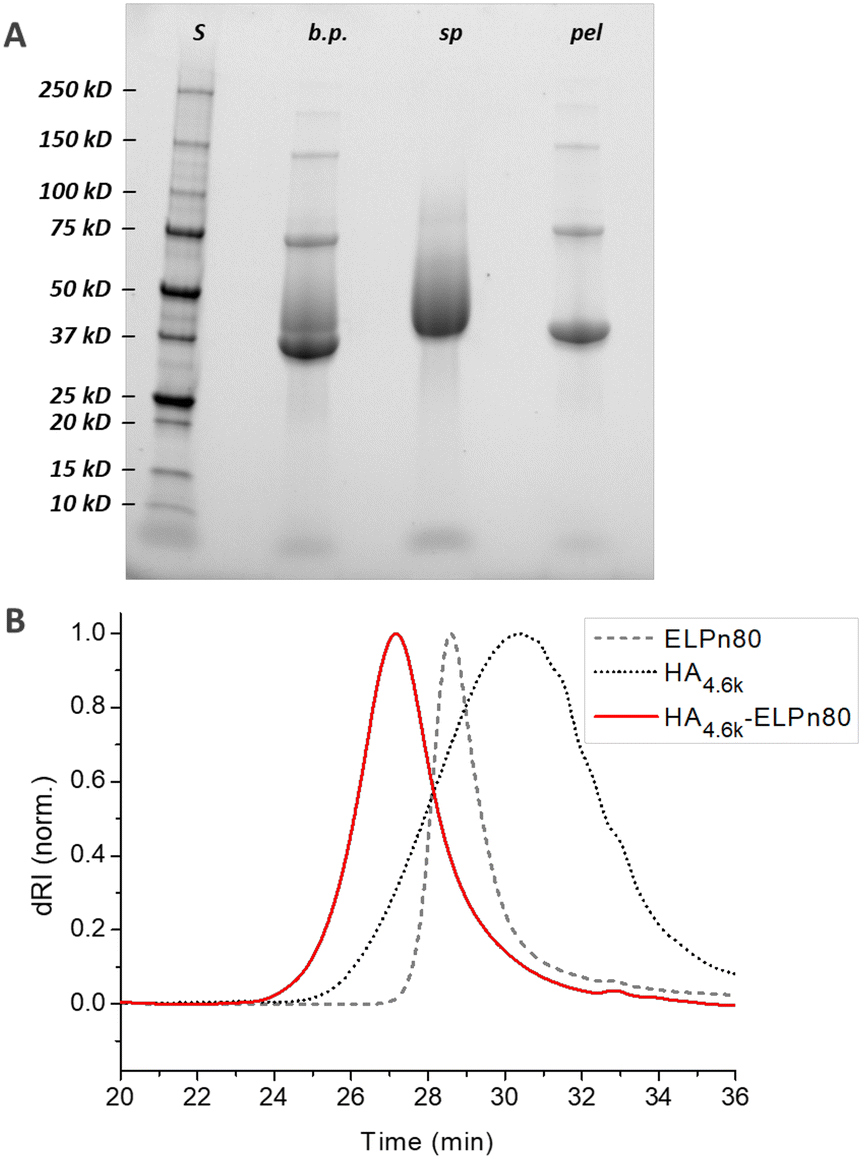 | ||
| Fig. 2 Characterization of the HA4.6k-ELPn80 bioconjugate by SDS-PAGE (A) and SEC (B). A: S: protein size markers; b.p.: reaction medium before purification; sp: supernatant; pel: pellet. B: samples in phosphate buffer. | ||
Before purification, the line on the protein gel (Fig. 2A, line b.p.) shows both neat black bands, characteristic of well-defined proteins, and a smear corresponding to polydisperse macromolecules. Indeed, while ELPn80 has a narrow dispersity due to its recombinant production (leading to neat bands on the gel), HA4.6k obtained by the degradation of longer HA chains presents a high dispersity (as noticed in the size exclusion chromatogram, Fig. 2B). The combination of the two blocks therefore leads to a polydisperse HA4.6k-ELPn80 bioconjugate appearing as a smear. After purification, this smear is observed only in the supernatant (Fig. 2A, line sp), whereas the precise bands corresponding to the excess ELPn80 used in the reaction (and to its non-covalent multimers) were recovered in the pellet (Fig. 2A, line pel). This confirmed the efficient separation of the HA4.6k-ELPn80 bioconjugate from DBCO-ELPn80 used in excess. Bioconjugates were also analysed by size exclusion chromatography (SEC). The chromatogram in Fig. 2B shows the elution times of the two individual blocks ELPn80 and HA4.6k in gray, confirming their difference in molar mass and molecular dispersity. Shown in red, the curve of the final product, HA4.6k-ELPn80, appears shifted towards shorter elution times, in agreement with the increased hydrodynamic diameter of the bioconjugate and greater molar mass. Characterization by SDS-PAGE and SEC for the entire library of bioconjugates is provided in Fig. S8 and S9,† respectively. Altogether these analyses confirmed the efficiency of both the SPAAC reaction and the simple purification method for the HA4.6k-ELPn80 bioconjugate. All bioconjugates, combining the three HA of different molar masses (4.6, 24, and 42 kDa) and the three ELPs of different molar masses (25.4, 33.7, and 42 kDa), were synthesized, purified and characterized following a similar procedure. A summary of the molecular characteristics and composition of the 9 HA-b-ELP bioconjugates of the library is provided in Fig. 3.
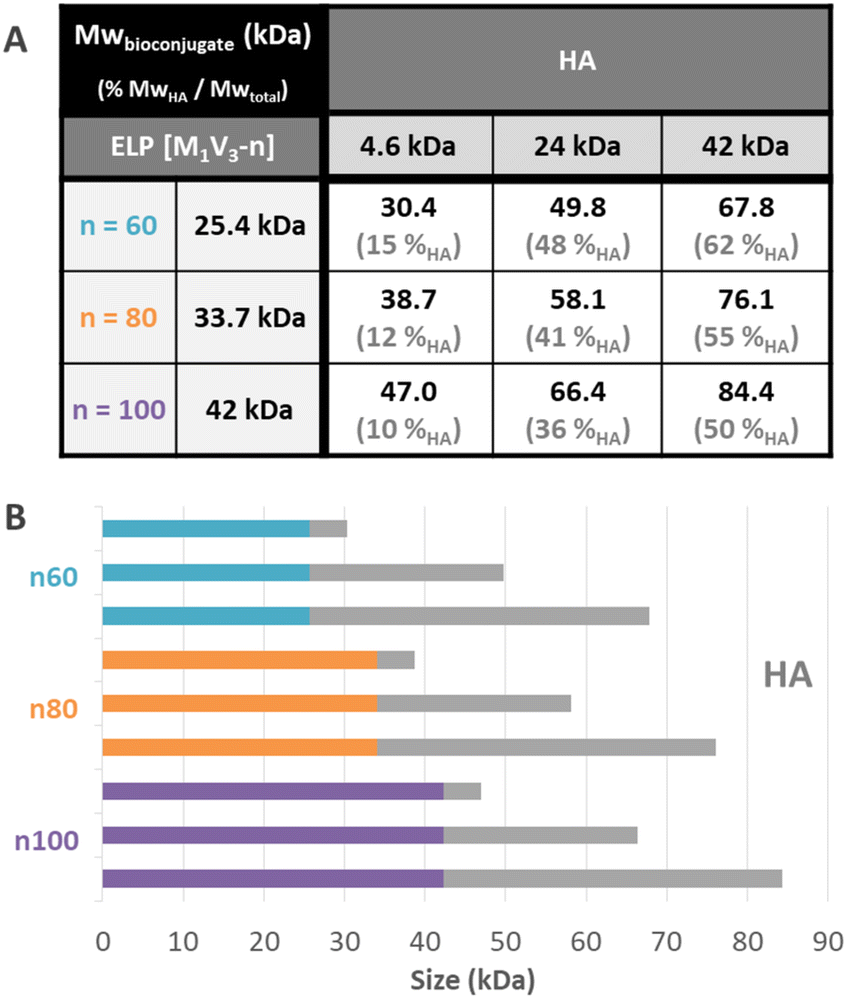 | ||
| Fig. 3 Description of the library of HA-b-ELP bioconjugates. (A) Table of molar masses and mass fractions of HA in the bioconjugates. The additional 0.4 kDa in the bioconjugate as compared to the addition of the molar mass of individual blocks corresponds to the molar mass of the cross-linker. (B) Schematic representation of the composition of the different bioconjugates. | ||
Most of the synthesized bioconjugates present a hydrophilic block (HA) shorter than the thermosensitive one (ELP). Two of them have rather equivalent composition (namely HA24k-ELPn60 and HA42k-ELPn100), and two others have a larger HA segment as compared to the ELP (namely HA42k-ELPn60 and HA42k-ELPn80). This library of HA-b-ELP bioconjugates presenting different molar masses and compositions allowed us to perform an extensive physicochemical characterization to understand their thermal behavior under aqueous conditions. We anticipated the difference in hydrophilicity to have a strong impact on the thermo-responsive behavior of the bioconjugates, influencing their self-assembly properties. From a biological perspective, the variation of HA molar masses implies a difference in the strength of its interaction with the CD44 receptor,70 which has a significant impact in the targeted nanomedicine applications.
The thermo-responsive properties of all bioconjugates were then studied by dynamic light scattering (DLS). A typical example for the bioconjugate HA4.6k-ELPn80 at 80 μM (concentration used for subsequent biological studies to be reported elsewhere) in PBS is given in Fig. 4. As illustrated in Fig. 4A, the scattered intensity, also referred to as the derived count rate (DCR), is very low below 24 °C, which is characteristic of small diffusive objects. The z-average varies around 60 nm, but the high polydispersity index (PdI) indicates the presence of few poorly defined aggregates. At this temperature, the system is therefore mainly composed of free chains in solution with the presence of some aggregates. Between 24 °C and 30 °C, a slight increase in the DCR can be observed, coupled with a small decrease of the PdI. But the major change is the evolution of the z-average, which increases sharply to reach 180 nm and decreases also abruptly to reach 40 nm. This phase is the hallmark of a coacervation process, with large objects highly hydrated, hence poorly scattering. Finally, from 30 °C, a 3rd phase is observed. The z-average stabilizes around 40 nm, with a very low PdI (<0.1), indicating the presence of only one population of objects. The DCR increases and is followed by a stabilization around 110000 kcps, which relates to the densification of nanoparticles’ core by dehydration of the ELP, in agreement with previously reported examples on the self-assembly of ELP diblock copolymers.5 At 30 °C and beyond, the solution therefore contains monodisperse nanoparticles with a hydrodynamic diameter (DH) of 40 nm. The reversibility of the self-assembly process is illustrated in Fig. 4B, with an inversed behaviour upon cooling. A schematic representation of the observed self-assembly behavior is illustrated in Fig. 4C. The previous conclusions are also supported by the representation of the size distribution (expressed in intensity) and correlograms obtained for this solution at 5 °C and 50 °C (Fig. S10†): while several populations and small objects are observed at low temperature, there is only one population of 40 nm-sized nanoparticles upon heating above the CMT. The temperature characteristic of the self-assembly (CMT) is here defined as the temperature at which only nanoparticles are present in solution. It is thus determined as the first temperature value at which both the z-average and the PdI stabilize. As shown in Fig. 4A, the CMT is therefore estimated at 30 °C (±1) for the HA4.6k-ELPn80 bioconjugate. This CMT was determined at different concentrations and is plotted in Fig. 4D. Interestingly, a similar trend was observed for HA-b-ELP bioconjugates to that for pure ELPs and in concordance with our previous observations on HA7k-ELPn40:17 the CMT increases with dilution, following a logarithmic trend previously described by Chilkoti's group.12 A similar self-assembly behaviour was observed for all of the 9 HA-b-ELP bioconjugates (Fig. S11†). For all bioconjugates, the evolution of CMT with molar concentration also followed a similar logarithmic law (Fig. S12†).
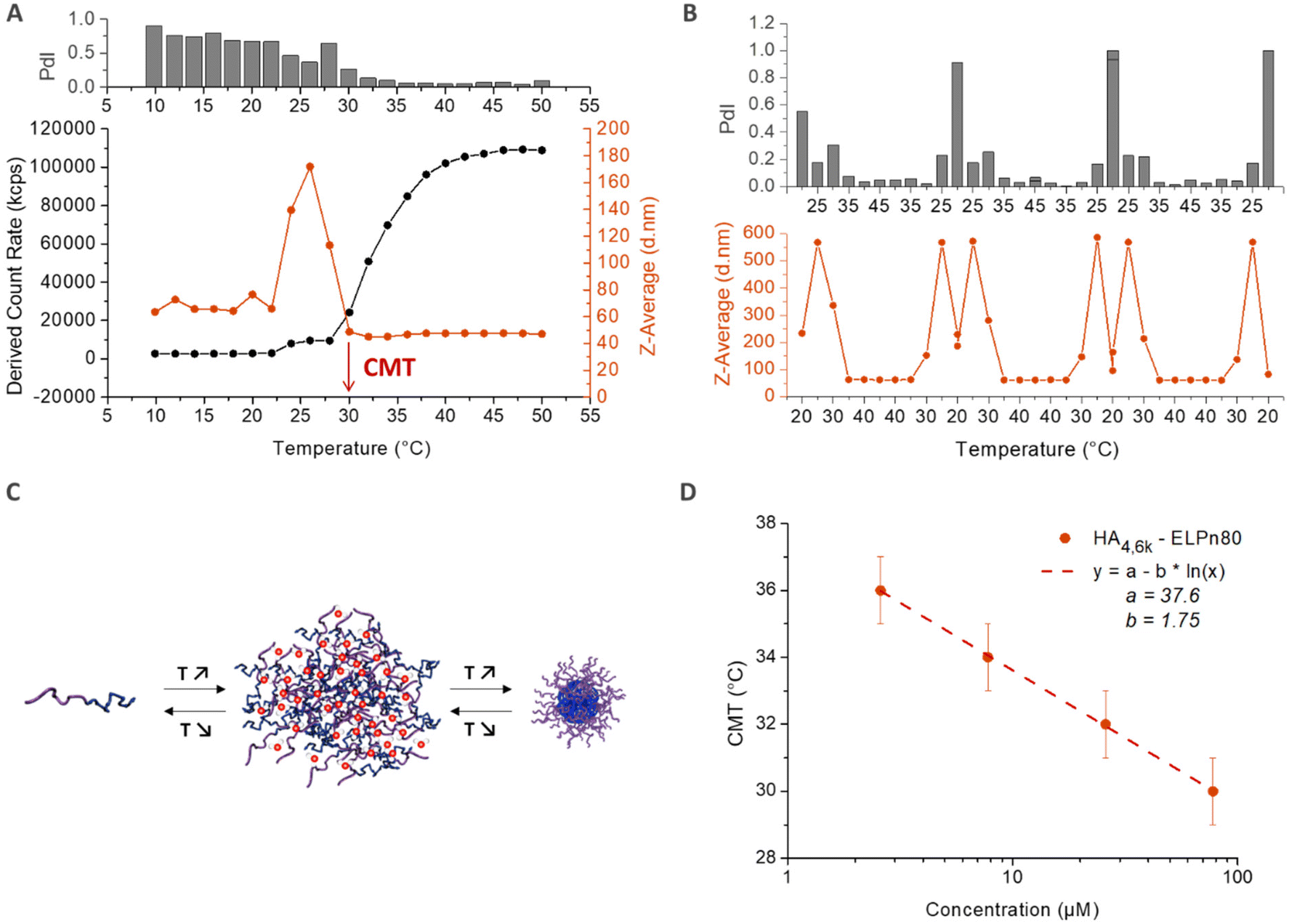 | ||
| Fig. 4 Characterization of the self-assembly behavior of the HA4.6k-ELPn80 bioconjugate in PBS. (A) Determination of the critical micellar temperature (CMT) by DLS analysis of a solution at 78 μM in PBS upon a heating ramp (10 °C–50 °C). (B) Monitoring of the reversibility of the self-assembly during 3 cycles of temperature ramps (20 °C–45 °C) for a solution at 80 μM in PBS. (C) Representation of the reversible bioconjugates’ self-assembly with temperature. (D) Evolution of the CMT with concentration. Fit: CMT = a − b × ln(C). | ||
The CMT was also determined by fluorescence spectroscopy using Nile Red. Indeed, the fluorescence properties of Nile Red change as a function of the polarity of the medium.71 In aqueous medium, Nile Red forms poorly soluble aggregates via Π–Π interactions that induce the quenching of the fluorescence.72,73 In more hydrophobic medium, Nile Red interacts mainly with the solvent, dissolving the aggregates and recovering its fluorescence.74,75 Nile Red is therefore often used to characterize the conditions of the appearance of hydrophobic microdomains in solution, characteristic of micellization behavior.72,75,76 Here, the self-assembly in nanoparticles above the CMT induces the formation of such hydrophobic microdomains in their core, leading to an increase of the fluorescence intensity of Nile Red. The simultaneous determination of the critical micellar temperature (CMT) and critical micellar concentration (CMC) is detailed in Fig. 5 for the HA4.6k-ELPn80 bioconjugate. Fluorescence spectra were acquired every 2 °C upon a heating ramp from 4 °C to 50 °C for solutions at 8 different concentrations. (Fig. 5A illustrates the fluorescence spectra obtained from 4 °C to 50 °C for a 100 μM concentration.) The intensity of fluorescence at 637 nm was then plotted as a function of temperature (Fig. 5B) or concentration (Fig. 5C). The CMT and CMC were respectively defined as the inflexion point of the curves or as the intersection of two logarithmic fits. This method was applied at each temperature and each concentration allowing access to complete phase diagrams.
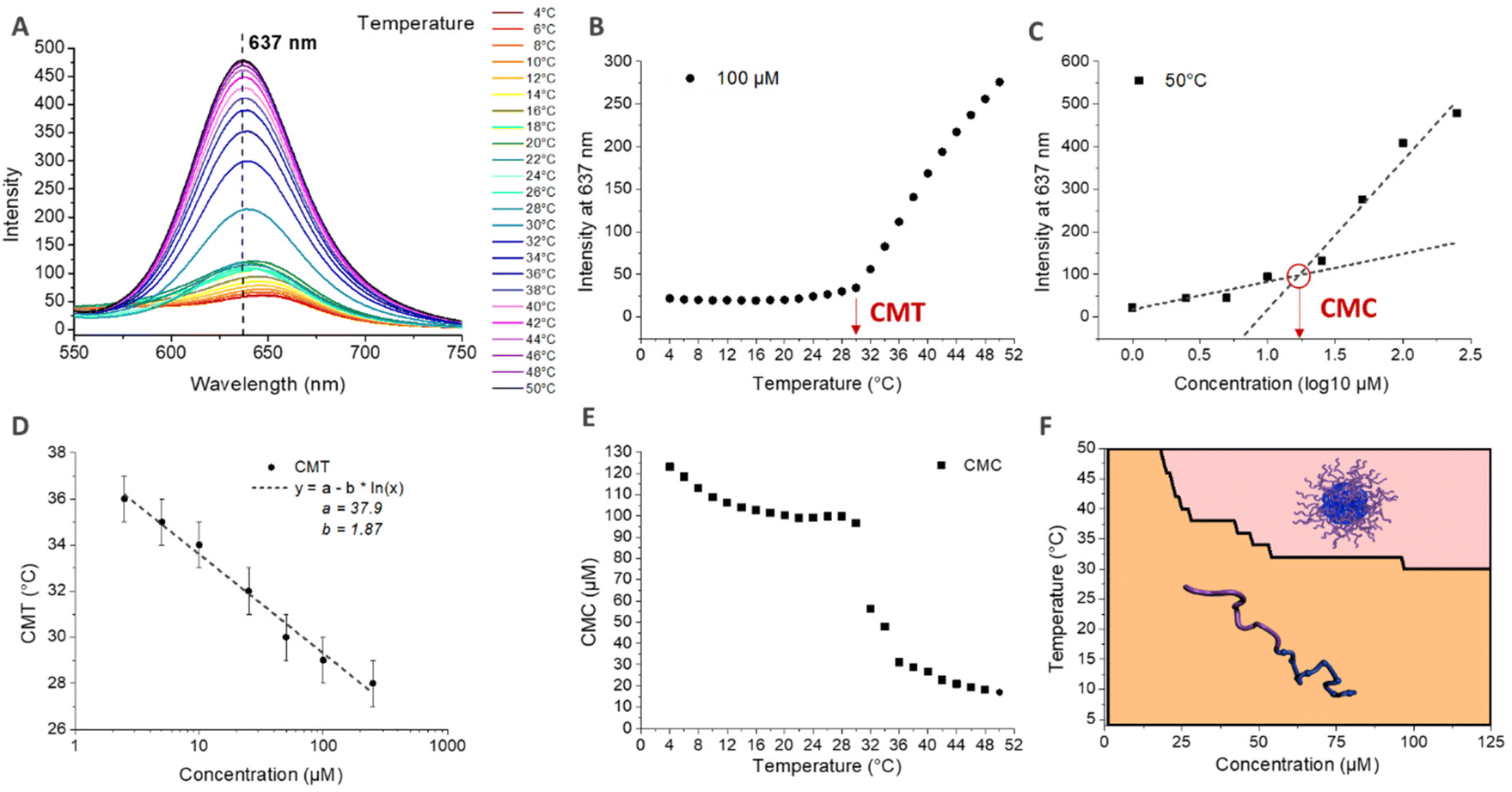 | ||
| Fig. 5 Determination of the critical micellar temperature (CMT) and concentration (CMC) by fluorescence spectroscopy (polarity probe: Nile Red) for HA4,6k-ELPn80 in PBS. (A) Stacking of the fluorescence emission spectra during a heating ramp (4 °C–50 °C) for a solution at 100 μM. λref = 637 nm → wavelength at the maximal fluorescence intensity. (B) Evolution of the fluorescence intensity at λref with the temperature at 100 μM. CMT = temperature at the inflexion point. (C) Evolution of the fluorescence intensity at λref with the concentration at 50 °C. Fit with two logarithmic models. CMC = concentration at the intersection. (D) Evolution of the CMT with concentration. Fit: CMT = a − b × ln(C). (E) Evolution of the CMC with temperature. (F) Representation of the corresponding phase diagram (coacervate phase non-represented). | ||
In Fig. 5D, the CMT is plotted as a function of concentration. If the values here are similar to those determined by DLS, those for the other bioconjugates are somewhat lower. These observations are consistent, considering that for DLS, the CMT corresponds to the temperature at which there are only nanoparticles in solution (end of the transition), while by fluorescence, the CMT is the temperature at which the first hydrophobic clusters are detected (beginning of the transition). Nevertheless, similar logarithmic dependence of CMT on concentration is observed (Fig. 4D and Fig. S13†). Fig. 5E shows the evolution of the CMC with temperature. At low temperature, the CMC is high (above 100 μM) and slowly decreases with increasing temperature. At about 28 °C, an abrupt decrease of the CMC is observed, up to about 36 °C, consistent with the range of the corresponding CMT for this system. Above 36 °C, the CMC continues its slow decrease, with values in the range 10–20 μM. The combination of these data allowed the elaboration of a phase diagram as a function of both molar concentration and temperature, circumscribing the domains of HA4.6k-ELPn80 present as free chains or as self-assembled nanoparticles (Fig. 5F). All of the 9 HA-b-ELP bioconjugates were studied and analysed in a similar manner (Fig. S13–S15†) and characteristic values of interest (CMT, CMC, and DH) are summarized in Table 1.
| CMT at 80 μM (°C) | CMC at 37 °C (μM) | D H at 50 °C (nm) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Bioconjugate | HA 4.6k | HA 24k | HA 42k | HA 4.6k | HA 24k | HA 42k | HA 4.6k | HA 24k | HA 42k |
| CMT: N = 3 except for * N = 4. CMC: N = 1. DH: N = 4–8. | |||||||||
| ELPn60 | 36 (±1.6)* | 39.7(±2.1) | 42.3 (±1.7) | 23 | 32 | 46 | 77 (±25) | 117 (±30) | 239 (±38) |
| ELPn80 | 31 (±0.7)* | 34 (±0.8) | 35.7 (±0.5) | 35 | 24 | 32 | 48 (±3) | 98 (±9) | 182 (±39) |
| ELPnl00 | 30.3 (±1.2) | 31.3 (±2.5) | 32.7 (±0.9) | 18 | 25 | 28 | 60 (±3) | 108 (±9) | 194 (±26) |
Two tendencies were observed concerning the CMTs. The CMTs increased with the length of the HA segment and decreased with the length of the ELP segment. This result is fully consistent with the contribution of these two blocks to the general hydrophilicity of the system. A longer HA increases the hydrophilicity, leading to a higher CMT, while a longer ELP increases the hydrophobicity, leading to a lower CMT. The variation of CMT with HA length and its dependency with the concentration seem of smaller amplitude for the bioconjugates with larger ELP blocks. At 80 μM, all the CMTs are between 30 °C and 42 °C, a perfectly suitable range of temperatures for forthcoming in vitro and in vivo biological studies where mild hypo- and hyperthermia shall be investigated. However, the influence of the bioconjugate's composition has to be considered: depending on the relative length of the two blocks, the system under physiological conditions can indeed be present either as coacervates (CMT > 37 °C) or nanoparticles (CMT < 37 °C). The hydrodynamic diameter (DH) of nanoparticles was measured by DLS (z-average) at 50 °C (high enough above the CMT of all bioconjugates) (Table 1). Hydrodynamic diameters evolved accordingly to the blocks' lengths: the size of nanoparticles decreased with decreasing ELP and HA block lengths (and molar masses). These results are in agreement with our expectation: a similar trend with ELP had been observed in a previous study,5 while the deployment on the surface of a longer hydrophilic HA segment logically increased nanoparticles’ size. Noteworthily, the values for HA-ELPn60 bioconjugates are artificially increased by the presence of aggregates in the samples. For all HA4.6k-ELP and HA24k-ELP bioconjugates, the size of nanoparticles was far below 200 nm and thus fully compatible with systemic administrations and accumulation in tumors through the enhanced permeation and retention effect.
Concerning the CMC, the same tendency was observed with the increase of the HA block leading to a higher CMC. However, there is no obvious tendency with the evolution of ELP block size. The values are all between 18 and 46 μM. An injection at such concentration could be contemplated in vivo in a murine model, but would be precluded in humans, justifying further improvement of the system.
A deeper characterization of HA4.6k-ELPn80 self-assembled nanoparticles was achieved using complementary techniques (Fig. 6).
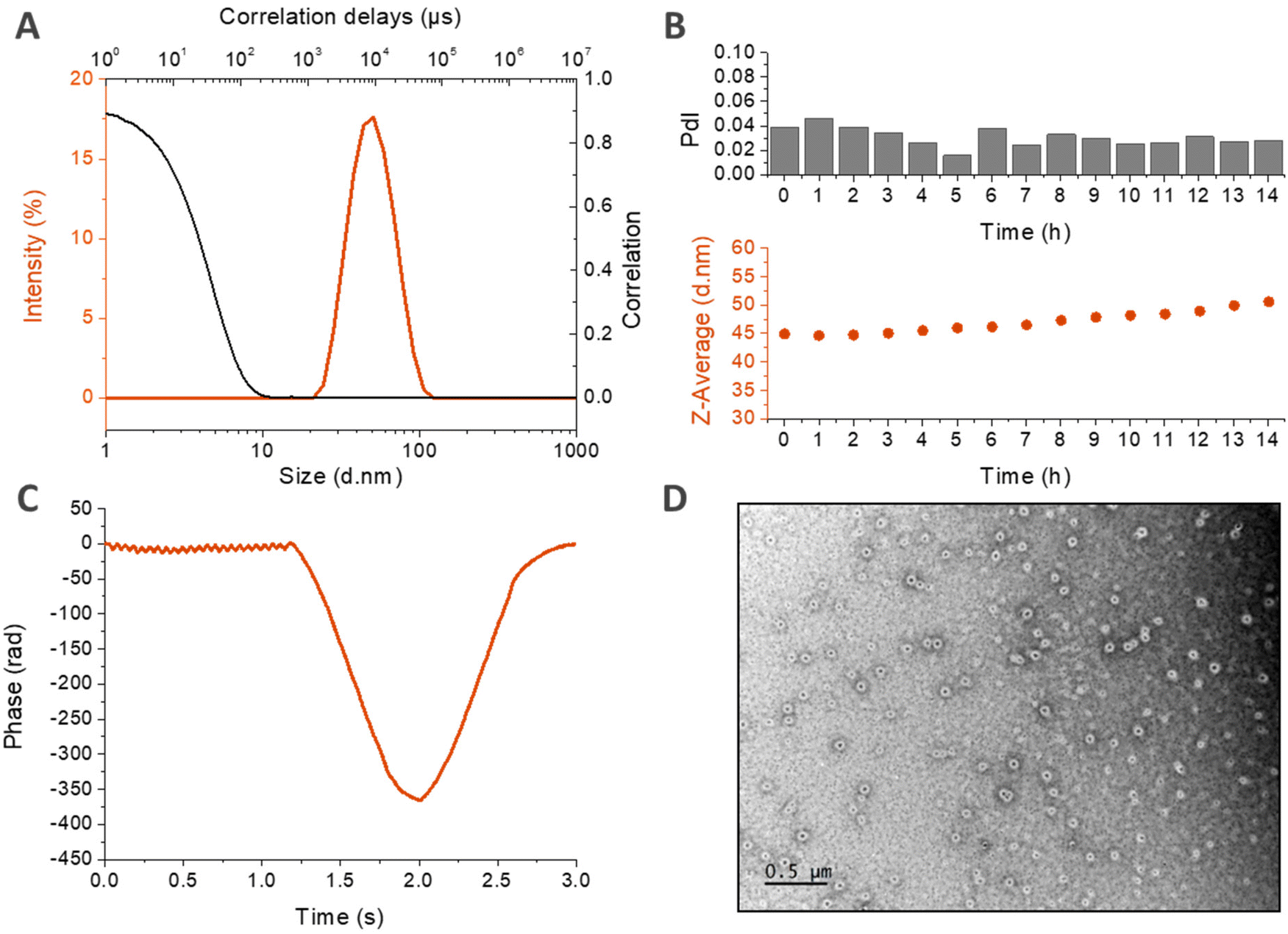 | ||
| Fig. 6 Physicochemical characterization of the self-assembled nanoparticles based on HA4,6k-ELPn80 bioconjugates. (A) Size distribution (in intensity) and correlogram from DLS analysis at 50 °C (78 μM in PBS). (B) Stability over time at 37 °C: evolution of the Z-Average and the PdI measured by DLS with time (78 μM in PBS). (C) Zeta potential measurements (78 μM in HEPES buffer, 60 °C). (D) TEM micrograph (2.6 μM in pure water, drop deposition at 80 °C, and samarium acetate staining). | ||
DLS measurements made in PBS at 80 μM are displayed in Fig. 6A and B. In panel A, the size distribution in intensity at 50 °C confirms the presence of a unique population at high temperature, with a narrow size dispersity centered around 40 nm. This population seems to be stable over time at 37 °C, as shown in Fig. 6B. If there is a slight increase of the z-average with time, the PdI retains its low value, which demonstrates the maintenance of a low dispersity and the absence of destabilization. The structure of the nanoparticles was then investigated by zeta potential measurements in HEPES buffer at 60 °C (Fig. 6C). The negative mean value suggests that the negatively charged HA is present at the surface of the nanoparticle, confirming a core–shell structure hypothesized earlier, with the hydrophobic ELP buried in the core and the hydrophilic HA deployed on the surface. Finally, the morphology of the nanoparticles was assessed by transmission electron microscopy (TEM). This time, the solution was prepared at a lower concentration (2.6 μM) in pure water in order to avoid the formation of salt crystals. Fig. 6D reveals spherical objects with a regular shape and homogeneous size, around 100 nm. This value is higher than the DH measured by DLS (Fig. 6B) due to the difference in the salinity of the solutions. In the absence of salts, the nanoparticles are more hydrated, which increases their size. This order of magnitude is however consistent with the observations previously made for HA7k-ELPn40 nanoparticles in pure water (200–300 nm at 52 °C).17
Conclusion
We have herein described an optimized synthetic strategy to access a library of HA-b-ELP bioconjugates presenting a bioactive, CD44-targeting HA segment, and a thermo-responsive biocompatible polypeptide block. The bioconjugation reaction was performed by SPAAC in water, and is a catalyst-, co-reactant- and organic solvent-free synthetic process being desirable for subsequent biological/biomedical applications. The efficient isolation of the bioconjugates by a simple centrifugation above the CMT in water adds to the straightforwardness of the process advantageous for a possible development at the industrial scale. A thorough physico-chemical study of the nine HA-b-ELP bioconjugates was then performed in an attempt to investigate the structure–property relationships. In particular, the determination of the critical conditions for thermally driven self-assembly has been carried out with temperature (CMT) and concentration (CMC) gradients, leading to a phase diagram for each of these bioconjugates. It has been observed that if the CMT values were fully compatible with in vivo applications, the CMCs were relatively high, indicating a compromised stability of the nanoparticles upon dilution, and therefore a specific issue to address for subsequent biological studies. Some general rules on physico-chemical characteristics (CMT, CMC, and DH) versus macromolecular parameters could be established and are found to be in agreement with previous studies: (i) an increase in the HA length, causing an increase of the general hydrophilicity of the bioconjugate, leads to an increase in the CMT, CMC and DH values; (ii) the increase of the ELP block length reduces the CMT as well as the dependence of the CMT over molar concentration, and increases the size of nanoparticles in agreement to our expectations.5 Four of the bioconjugates (HA4.6k-ELPn80 or n100 and HA24k-ELPn80 or n100) were found to self-assemble into well-defined spherical core–shell nanoparticles, with a negative surface charge due to the HA block constituting the shell, a DH between 40 and 200 under in physiological conditions and a good stability over time at 37 °C.Author contributions
ML was involved in investigations, project administration, resources and writing of the first manuscript draft. YX was involved in resources (HA4.6k-N3). LD (SPAAC experiments) and LM (Nile Red experiments) were involved in investigations. EG and SL were involved in conceptualization, funding acquisition, project administration, and writing of the final manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. Bertrand Garbay, Ms. Clara Toulouse, Ms. Chloé Ilos, and Ms. Pauline Coutand for their technical assistance in the recombinant production of the ELPs. The French Ministry for Higher Education and Research is gratefully acknowledged for the doctoral grant awarded to ML. China Scholarship Council and Université de Bordeaux (UB-CSC 2015) are acknowledged for the financial support to YX. This work was also supported by the French National Research Agency (ANR-15-CE07-0002). Continuous support from Univ. Bordeaux, CNRS and Bordeaux-INP is greatly acknowledged.References
- S. R. MacEwan and A. Chilkoti, Applications of Elastin-like Polypeptides in Drug Delivery, J. Controlled Release, 2014, 190, 314–330, DOI:10.1016/j.jconrel.2014.06.028
.
- I. C. Jenkins, J. J. Milligan and A. Chilkoti, Genetically Encoded Elastin–Like Polypeptides for Drug Delivery, Adv. Healthcare Mater., 2021, 2100209, DOI:10.1002/adhm.202100209
- C. Guda, X. Zhang, D. T. McPherson, J. Xu, J. H. Cherry, D. W. Urry and H. Daniell, Hyper Expression of an Environmentally Friendly Synthetic Polymer Gene, Biotechnol. Lett., 1995, 17(7), 745–750, DOI:10.1007/BF00130362
- S. Roberts, M. Dzuricky and A. Chilkoti, Elastin-like Polypeptides as Models of Intrinsically Disordered Proteins, FEBS Lett., 2015, 589(19PartA), 2477–2486, DOI:10.1016/j.febslet.2015.08.029
- E. Garanger, S. R. MacEwan, O. Sandre, A. Brûlet, L. Bataille, A. Chilkoti and S. Lecommandoux, Structural Evolution of a Stimulus-Responsive Diblock Polypeptide Micelle by Temperature Tunable Compaction of Its Core, Macromolecules, 2015, 48(18), 6617–6627, DOI:10.1021/acs.macromol.5b01371
- J. C. Rodríguez-Cabello, L. Martín, M. Alonso, F. J. Arias and A. M. Testera, “Recombinamers” as Advanced Materials for the Post-Oil Age, Polymer, 2009, 50(22), 5159–5169, DOI:10.1016/j.polymer.2009.08.032
- D. T. McPherson, J. Xu and D. W. Urry, Product Purification by Reversible Phase Transition FollowingEscherichia ColiExpression of Genes Encoding up to 251 Repeats of the Elastomeric Pentapeptide GVGVP, Protein Expression Purif., 1996, 7(1), 51–57, DOI:10.1006/prep.1996.0008
-
D. E. Meyer and A. Chilkoti, Protein Purification by Inverse Transition Cycling, In Protein-Protein Interaction: A Molecular Cloning Manual, 2002, ch. 18, pp. 329–343 Search PubMed
- D. W. Urry, M. M. Long, B. A. Cox, T. Ohnishi, L. W. Mitchell and M. Jacobs, The Synthetic Polypentapeptide of Elastin Coacervates and Forms Filamentous Aggregates, Biochim. Biophys. Acta, Protein Struct., 1974, 371(2), 597–602, DOI:10.1016/0005-2795(74)90057-9
- D. W. Urry, C. H. Luan, T. M. Parker, D. C. Gowda, K. U. Prasad, M. C. Reid and A. Safavy, Temperature of Polypeptide Inverse Temperature Transition Depends on Mean Residue Hydrophobicity, J. Am. Chem. Soc., 1991, 113(11), 4346–4348, DOI:10.1021/ja00011a057
- J. A. MacKay, D. J. Callahan, K. N. FitzGerald and A. Chilkoti, Quantitative Model of the Phase Behavior of Recombinant PH-Responsive Elastin-Like Polypeptides, Biomacromolecules, 2010, 11(11), 2873–2879, DOI:10.1021/bm100571j
- D. E. Meyer and A. Chilkoti, Quantification of the Effects of Chain Length and Concentration on the Thermal Behavior of Elastin-like Polypeptides, Biomacromolecules, 2004, 5(3), 846–851, DOI:10.1021/bm034215n
- J. R. McDaniel, D. C. Radford and A. Chilkoti, A Unified Model for De Novo Design of Elastin-like Polypeptides with Tunable Inverse Transition Temperatures, Biomacromolecules, 2013, 14(8), 2866–2872, DOI:10.1021/bm4007166
- J. Reguera, D. W. Urry, T. M. Parker, D. T. McPherson and J. C. Rodríguez-Cabello, Effect of NaCl on the Exothermic and Endothermic Components of the Inverse Temperature Transition of a Model Elastin-like Polymer, Biomacromolecules, 2007, 8(2), 354–358, DOI:10.1021/bm060936l
- Y. Cho, Y. Zhang, T. Christensen, L. B. Sagle, A. Chilkoti and P. S. Cremer, Effects of Hofmeister Anions on the Phase Transition Temperature of Elastin-like Polypeptides, J. Phys. Chem. B, 2008, 112(44), 13765–13771, DOI:10.1021/jp8062977
- S. Saha, S. Banskota, S. Roberts, N. Kirmani and A. Chilkoti, Engineering the Architecture of Elastin-Like Polypeptides: From Unimers to Hierarchical Self-Assembly, Adv. Ther., 2020, 3(3), 1900164, DOI:10.1002/adtp.201900164
- Y. Xiao, Z. S. Chinoy, G. Pecastaings, K. Bathany, E. Garanger and S. Lecommandoux, Design of Polysaccharide- b -Elastin-Like Polypeptide Bioconjugates and Their Thermoresponsive Self-Assembly, Biomacromolecules, 2020, 21(1), 114–115, DOI:10.1021/acs.biomac.9b01058
- M. B. van Eldijk, F. C. M. Smits, N. Vermue, M. F. Debets, S. Schoffelen and J. C. M. van Hest, Synthesis and Self-Assembly of Well-Defined Elastin-Like Polypeptide–Poly(Ethylene Glycol) Conjugates, Biomacromolecules, 2014, 15(7), 2751–2759, DOI:10.1021/bm5006195
- T.-H. H. Chen, Y. Bae, D. Y. Furgeson and G. S. Kwon, Biodegradable Hybrid Recombinant Block Copolymers for Non-Viral Gene Transfection, Int. J. Pharm., 2012, 427(1), 105–112, DOI:10.1016/j.ijpharm.2011.09.035
- D. Y. Furgeson, M. R. Dreher and A. Chilkoti, Structural Optimization of a “Smart” Doxorubicin–Polypeptide Conjugate for Thermally Targeted Delivery to Solid Tumors, J. Controlled Release, 2006, 110(2), 362–369, DOI:10.1016/j.jconrel.2005.10.006
- J. Bhattacharyya, J. J. Bellucci, I. Weitzhandler, J. R. McDaniel, I. Spasojevic, X. Li, C.-C. Lin, J.-T. A. Chi and A. Chilkoti, A Paclitaxel-Loaded Recombinant Polypeptide Nanoparticle Outperforms Abraxane in Multiple Murine Cancer Models, Nat. Commun., 2015, 6(1), 7939, DOI:10.1038/ncomms8939
- K. M. Luginbuhl, D. Mozhdehi, M. Dzuricky, P. Yousefpour, F. C. Huang, N. R. Mayne, K. L. Buehne and A. Chilkoti, Recombinant Synthesis of Hybrid Lipid–Peptide Polymer Fusions That Self-Assemble and Encapsulate Hydrophobic Drugs, Angew. Chem., Int. Ed., 2017, 56(45), 13979–13984, DOI:10.1002/anie.201704625
- D. Mozhdehi, K. M. Luginbuhl, J. R. Simon, M. Dzuricky, R. Berger, H. S. Varol, F. C. Huang, K. L. Buehne, N. R. Mayne, I. Weitzhandler, M. Bonn, S. H. Parekh and A. Chilkoti, Genetically Encoded Lipid–Polypeptide Hybrid Biomaterials That Exhibit Temperature-Triggered Hierarchical Self-Assembly, Nat. Chem., 2018, 10(5), 496–505, DOI:10.1038/s41557-018-0005-z
- D. Mozhdehi, K. M. Luginbuhl, M. Dzuricky, S. A. Costa, S. Xiong, F. C. Huang, M. M. Lewis, S. R. Zelenetz, C. D. Colby and A. Chilkoti, Genetically Encoded Cholesterol-Modified Polypeptides, J. Am. Chem. Soc., 2019, 141(2), 945–951, DOI:10.1021/jacs.8b10687
- G. Le Fer, D. Portes, G. Goudounet, J.-M. Guigner, E. Garanger and S. Lecommandoux, Design and Self-Assembly of PBLG- b -ELP Hybrid Diblock Copolymers Based on Synthetic and Elastin-like Polypeptides, Org. Biomol. Chem., 2017, 15(47), 10095–10104, 10.1039/C7OB01945A
- T. a. T. Lee, A. Cooper, R. P. Apkarian and V. P. Conticello, Thermo-Reversible Self-Assembly of Nanoparticles Derived from Elastin-Mimetic Polypeptides, Adv. Mater., 2000, 12(15), 1105–1110, DOI:10.1002/1521-4095(200008)12:15<1105::AID-ADMA1105>3.0.CO;2-1
- W. Hassouneh, E. B. Zhulina, A. Chilkoti and M. Rubinstein, Elastin-like Polypeptide Diblock Copolymers Self-Assemble into Weak Micelles, Macromolecules, 2015, 48(12), 4183–4195, DOI:10.1021/acs.macromol.5b00431
- S. M. Janib, M. F. Pastuszka, S. Aluri, Z. Folchman-Wagner, P. Y. Hsueh, P. Shi, Y. A. Lin, H. Cui and J. A. MacKay, A Quantitative Recipe for Engineering Protein Polymer Nanoparticles, Polym. Chem., 2014, 5(5), 1614–1625, 10.1039/C3PY00537B
- K. Widder, S. R. MacEwan, E. Garanger, V. Núñez, S. Lecommandoux, A. Chilkoti and D. Hinderberger, Characterisation of Hydration and Nanophase Separation during the Temperature Response in Hydrophobic/Hydrophilic Elastin-like Polypeptide (ELP) Diblock Copolymers, Soft Matter, 2017, 13(9), 1816–1822, 10.1039/C6SM02427K
- I. Weitzhandler, M. Dzuricky, I. Hoffmann, F. Garcia Quiroz, M. Gradzielski and A. Chilkoti, Micellar Self-Assembly of Recombinant Resilin-/Elastin-Like Block Copolypeptides, Biomacromolecules, 2017, 18(8), 2419–2426, DOI:10.1021/acs.biomac.7b00589
- F. C. M. Smits, B. C. Buddingh, M. B. van Eldijk and J. C. M. van Hest, Elastin-Like Polypeptide Based Nanoparticles: Design Rationale Toward Nanomedicine: Elastin-Like Polypeptide Based Nanoparticles: Design Rationale Toward Nanomedicine, Macromol. Biosci., 2015, 15(1), 36–51, DOI:10.1002/mabi.201400419
- S. R. MacEwan and A. Chilkoti, Elastin-like Polypeptides: Biomedical Applications of Tunable Biopolymers, Pept. Sci., 2010, 94(1), 60–77, DOI:10.1002/bip.21327
- A. K. Varanko, J. C. Su and A. Chilkoti, Elastin-Like Polypeptides for Biomedical Applications, Annu. Rev. Biomed. Eng., 2020, 22(1), 343–369, DOI:10.1146/annurev-bioeng-092419-061127
- E. Georgilis, M. Abdelghani, J. Pille, E. Aydinlioglu, J. C. M. van Hest, S. Lecommandoux and E. Garanger, Nanoparticles Based on Natural, Engineered or Synthetic Proteins and Polypeptides for Drug Delivery Applications, Int. J. Pharm., 2020, 586, 119537, DOI:10.1016/j.ijpharm.2020.119537
- P. Shi, S. Aluri, Y.-A. Lin, M. Shah, M. Edman, J. Dhandhukia, H. Cui and J. A. MacKay, Elastin-Based Protein Polymer Nanoparticles Carrying Drug at Both Corona and Core Suppress Tumor Growth in Vivo, J. Controlled Release, 2013, 171(3), 330–338, DOI:10.1016/j.jconrel.2013.05.013
- S. A. Costa, D. Mozhdehi, M. J. Dzuricky, F. J. Isaacs, E. M. Brustad and A. Chilkoti, Active Targeting of Cancer Cells by Nanobody Decorated Polypeptide Micelle with Bio-Orthogonally Conjugated Drug, Nano Lett., 2019, 19(1), 247–254, DOI:10.1021/acs.nanolett.8b03837
- G. L. Bidwell and D. Raucher, Application of Thermally Responsive Polypeptides Directed against C-Myc Transcriptional Function for Cancer Therapy, Mol. Cancer Ther., 2005, 4(7), 1076–1085, DOI:10.1158/1535-7163.MCT-04-0253
- V. Sarangthem, Y. Kim, T. D. Singh, B.-Y. Seo, S.-H. Cheon, Y.-J. Lee, B.-H. Lee and R.-W. Park, Multivalent Targeting Based Delivery of Therapeutic Peptide Using AP1-ELP Carrier for Effective Cancer Therapy, Theranostics, 2016, 6(12), 2235–2249, DOI:10.7150/thno.16425
- W. Zhang, S. Garg, P. Eldi, F. H. Zhou, I. R. D. Johnson, D. A. Brooks, F. Lam, G. Rychkov, J. Hayball and H. Albrecht, Targeting Prostate Cancer Cells with Genetically Engineered Polypeptide-Based Micelles Displaying Gastrin-Releasing Peptide, Int. J. Pharm., 2016, 513(1), 270–279, DOI:10.1016/j.ijpharm.2016.09.039
- M. Sun, J. Guo, H. Hao, T. Tong, K. Wang and W. Gao, Tumour-Homing Chimeric Polypeptide-Conjugated Polypyrrole Nanoparticles for Imaging-Guided Synergistic Photothermal and Chemical Therapy of Cancer, Theranostics, 2018, 8(10), 2634–2645, DOI:10.7150/thno.24705
- S. R. Aluri, P. Shi, J. A. Gustafson, W. Wang, Y.-A. Lin, H. Cui, S. Liu, P. S. Conti, Z. Li, P. Hu, A. L. Epstein and J. A. MacKay, A Hybrid Protein–Polymer Nanoworm Potentiates Apoptosis Better than a Monoclonal Antibody, ACS Nano, 2014, 8(3), 2064–2076, DOI:10.1021/nn403973g
- S. Fluegel, J. Buehler, K. Fischer, J. R. McDaniel, A. Chilkoti and M. Schmidt, Self-Assembly of Monodisperse Oligonucleotide-Elastin Block Copolymers into Stars and Compound Micelles, Chem. – Eur. J., 2011, 17(20), 5503–5506, DOI:10.1002/chem.201100436
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel, M. Schmidt and A. Chilkoti, Temperature Triggered Self-Assembly of Polypeptides into Multivalent Spherical Micelles, J. Am. Chem. Soc., 2008, 130(2), 687–694, DOI:10.1021/ja0764862
- J. Pille, S. A. M. van Lith, J. C. M. van Hest and W. P. J. Leenders, Self-Assembling VHH-Elastin-Like Peptides for Photodynamic Nanomedicine, Biomacromolecules, 2017, 18(4), 1302–1310, DOI:10.1021/acs.biomac.7b00064
- J. Necas, L. Bartosikova, P. Brauner and J. Kolar, Hyaluronic Acid (Hyaluronan): A Review, Vet. Med., 2008, 53(No. 8), 397–411, DOI:10.17221/1930-VETMED
- G. Mattheolabakis, L. Milane, A. Singh and M. M. Amiji, Hyaluronic Acid Targeting of CD44 for Cancer Therapy: From Receptor Biology to Nanomedicine, J. Drug Targeting, 2015, 23(7–8), 605–618, DOI:10.3109/1061186X.2015.1052072
- A. Y. Liu, Expression of CD44 in Prostate Cancer Cells, Cancer Lett., 1994, 76(1), 63–69, DOI:10.1016/0304-3835(94)90135-X
- M. B. Penno, J. T. August, S. B. Baylin, M. Mabry, R. I. Linnoila, V. S. Lee, D. Croteau, X. L. Yang and C. Rosada, Expression of CD44 in Human Lung Tumors, Cancer Res., 1994, 54(5), 1381–1387 CAS
- R. Kopp, M. Fichter, G. Schalhorn, J. Danescu and S. Classen, Frequent Expression of the High Molecular, 673-Bp CD44v3,v8-10 Variant in Colorectal Adenomas and Carcinomas, Int. J. Mol. Med., 2009, 24(5), 677–683, DOI:10.3892/ijmm_00000279
- E. Olsson, G. Honeth, P.-O. Bendahl, L. H. Saal, S. Gruvberger-Saal, M. Ringnér, J. Vallon-Christersson, G. Jönsson, K. Holm, K. Lövgren, M. Fernö, D. Grabau, Å. Borg and C. Hegardt, CD44 Isoforms Are Heterogeneously Expressed in Breast Cancer and Correlate with Tumor Subtypes and Cancer Stem Cell Markers, BMC Cancer, 2011, 11(1), 418, DOI:10.1186/1471-2407-11-418
- Y. Chen, Z. Fu, S. Xu, Y. Xu and P. Xu, The Prognostic Value of CD44 Expression in Gastric Cancer: A Meta-Analysis, Biomed. Pharmacother., 2014, 68(6), 693–697, DOI:10.1016/j.biopha.2014.08.001
- X.-P. Li, X.-W. Zhang, L.-Z. Zheng and W.-J. Guo, Expression of CD44 in Pancreatic Cancer and Its Significance, Int. J. Clin. Exp. Pathol., 2015, 8(6), 6724–6731 Search PubMed
- M. Al-Hajj, M. S. Wicha, A. Benito-Hernandez, S. J. Morrison and M. F. Clarke, Prospective Identification of Tumorigenic Breast Cancer Cells, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(7), 3983–3988, DOI:10.1073/pnas.0530291100
- M. Al-Hajj, M. W. Becker, M. Wicha, I. Weissman and M. F. Clarke, Therapeutic Implications of Cancer Stem Cells, Curr. Opin. Genet. Dev., 2004, 14(1), 43–47, DOI:10.1016/j.gde.2003.11.007
- M. O. Idowu, M. Kmieciak, C. Dumur, R. S. Burton, M. M. Grimes, C. N. Powers and M. H. Manjili, CD44+/CD24−/Low Cancer Stem/Progenitor Cells Are More Abundant in Triple-Negative Invasive Breast Carcinoma Phenotype and Are Associated with Poor Outcome, Hum. Pathol., 2012, 43(3), 364–373, DOI:10.1016/j.humpath.2011.05.005
- V. Orian-Rousseau, CD44 Acts as a Signaling Platform Controlling Tumor Progression and Metastasis, Front. Immunol., 2015, 6, 154, DOI:10.3389/fimmu.2015.00154
- M. E. Prince, R. Sivanandan, A. Kaczorowski, G. T. Wolf, M. J. Kaplan, P. Dalerba, I. L. Weissman, M. F. Clarke and L. E. Ailles, Identification of a Subpopulation of Cells with Cancer Stem Cell Properties in Head and Neck Squamous Cell Carcinoma, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(3), 973–978, DOI:10.1073/pnas.0610117104
- M. Najafi, B. Farhood and K. Mortezaee, Cancer Stem Cells (CSCs) in Cancer Progression and Therapy, J. Cell. Physiol., 2019, 234(6), 8381–8395, DOI:10.1002/jcp.27740
- A. Aruffo, I. Stamenkovic, M. Melnick, C. B. Underhill and B. Seed, CD44 Is the Principal Cell Surface Receptor for Hyaluronate, Cell, 1990, 61(7), 1303–1313, DOI:10.1016/0092-8674(90)90694-A
- F. Dosio, S. Arpicco, B. Stella and E. Fattal, Hyaluronic Acid for Anticancer Drug and Nucleic Acid Delivery, Adv. Drug Delivery Rev., 2016, 97, 204–236, DOI:10.1016/j.addr.2015.11.011
- S. Y. Lee, M. S. Kang, W. Y. Jeong, D.-W. Han and K. S. Kim, Hyaluronic Acid-Based Theranostic Nanomedicines for Targeted Cancer Therapy, Cancers, 2020, 12(4), 940, DOI:10.3390/cancers12040940
- L. H. Robert, a Truly “Youthful” Polysaccharide. Its Medical Applications, Pathol. Biol., 2015, 63(1), 32–34, DOI:10.1016/j.patbio.2014.05.019
- R. Stern and H. I. Maibach, Hyaluronan in Skin: Aspects of Aging and Its Pharmacologic Modulation, Clin. Dermatol., 2008, 26(2), 106–122, DOI:10.1016/j.clindermatol.2007.09.013
- Pharmacopée Hyaluronate de Sodium.
- M. Dai, E. Georgilis, G. Goudounet, B. Garbay, J. Pille, J. C. M. van Hest, X. Schultze, E. Garanger and S. Lecommandoux, Refining the Design of Diblock Elastin-Like Polypeptides for Self-Assembly into Nanoparticles, Polymers, 2021, 13(9), 1470, DOI:10.3390/polym13091470
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, A Strain-Promoted [3+2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems, J. Am. Chem. Soc., 2004, 126(46), 15046–15047, DOI:10.1021/ja044996f
- G. Seitz, L. Pohl and R. Pohlke, 5,6-Didehydro-11,12-Dihydrodibenzo[a,e]-Cyclooctene, Angew. Chem., Int. Ed. Engl., 1969, 8(6), 447–448, DOI:10.1002/anie.196904471
- A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Surface Functionalization Using Catalyst-Free Azide−Alkyne Cycloaddition, Bioconjugate Chem., 2010, 21(11), 2076–2085, DOI:10.1021/bc100306u
- S. Munneke, J. R. C. Prevost, G. F. Painter, B. L. Stocker and M. S. M. Timmer, The Rapid and Facile Synthesis of Oxyamine Linkers for the Preparation of Hydrolytically Stable Glycoconjugates, Org. Lett., 2015, 17(3), 624–627, DOI:10.1021/ol503634j
- H. Duan, M. Donovan, A. Foucher, X. Schultze and S. Lecommandoux, Multivalent and Multifunctional Polysaccharide-Based Particles for Controlled Receptor Recognition, Sci. Rep., 2018, 8(1), 14730, DOI:10.1038/s41598-018-32994-y
- P. Greenspan and S. D. Fowler, Spectrofluorometric Studies of the Lipid Probe, Nile Red, J. Lipid Res, 1985, 26(7), 781–789, DOI:10.1016/S0022-2275(20)34307-8
- I. N. Kurniasih, H. Liang, P. C. Mohr, G. Khot, J. P. Rabe and A. Mohr, Nile Red Dye in Aqueous Surfactant and Micellar Solution, Langmuir, 2015, 31(9), 2639–2648, DOI:10.1021/la504378m
- N. Sarkar, K. Das, D. N. Nath and K. Bhattacharyya, Twisted Charge Transfer Processes of Nile Red in Homogeneous Solutions and in Faujasite Zeolite, Langmuir, 1994, 10(1), 326–329, DOI:10.1021/la00013a048
- P. Greenspan, E. P. Mayer and S. D. Fowler, Nile Red: A Selective Fluorescent Stain for Intracellular Lipid Droplets, J. Cell Biol., 1985, 100(3), 965–973, DOI:10.1083/jcb.100.3.965
- A. P. Goodwin, J. L. Mynar, Y. Ma, G. R. Fleming and J. M. J. Fréchet, Synthetic Micelle Sensitive to IR Light via a Two-Photon Process, J. Am. Chem. Soc., 2005, 127(28), 9952–9953, DOI:10.1021/ja0523035
- M. C. A. Stuart, J. C. van de Pas and J. B. F. N. Engberts, The Use of Nile Red to Monitor the Aggregation Behavior in Ternary Surfactant–Water–Organic Solvent Systems, J. Phys. Org. Chem., 2005, 18(9), 929–934, DOI:10.1002/poc.919
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of ELPs, DBCO-ELP and HA-N3, and additional characterization of the self-assembly of all HA-b-ELP bioconjugates. See DOI: https://doi.org/10.1039/d2bm01149b |
| This journal is © The Royal Society of Chemistry 2022 |