
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting a conserved structural element from the SARS-CoV-2 genome using L-DNA aptamers†
Jing
Li
and
Jonathan T.
Sczepanski
 *
*
Department of Chemistry, Texas A&M University, College Station, TX, USA. E-mail: jon.sczepanski@chem.tamu.edu
First published on 20th October 2021
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes COVID-19, has resulted in an ongoing global pandemic that has already claimed more than 4 million lives. While most antiviral efforts have focused on essential SARS-CoV-2 proteins, RNA structural elements within the viral genome are also compelling targets. In this study, we identified high-affinity L-DNA aptamers against a SARS-CoV-2 stem-loop II-like motif (s2m), a highly conserved RNA structure with promising diagnostic and therapeutic potential. Optimized L-C1t and L-C3t aptamers bind selectively to s2m RNA with Kd values in the nanomolar range, and are capable of differentiating the monomeric s2m stem-loop from the proposed homodimer duplex. This structure-specific mode of recognition also allows L-C1t and L-C3t to discriminate between s2m RNAs from SARS-CoV-2 and SARS-CoV-1, which differ by just two nucleotides. Finally, we show that L-C1t and L-C3t induce dramatic conformational changes in s2m structure upon binding, and thus, have the potential to block protein–s2m interactions. Overall, these results demonstrate the feasibility of targeting SARS-CoV-2 RNA using L-aptamers, which has important implications in the diagnosis and treatment of COVID-19. Moreover, the high affinity and selectivity of L-C1t and L-C3t, coupled with the intrinsic nuclease resistance of L-DNA, present an opportunity for generating new tools and probes for interrogating s2m function in SARS-CoV-2 and related viruses.
Introduction
Coronaviruses (CoVs) are a large family of positive-sense single-stranded RNA (+ssRNA) viruses that commonly cause upper-respiratory illnesses in humans.1 Betacoronaviruses in particular have resulted in several serious and widespread diseases, including the current COVID-19 pandemic, which has already claimed the lives of more than 4.9 million people worldwide.2,3 Despite extensive research and clinical efforts to combat severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes COVID-19, there are a lack of efficacious antiviral therapies to treat infected individuals.4 Remdesivir remains the only FDA-approved drug and provides only modest benefits to COVID-19 patients.5 In order to combat the current COVID-19 pandemic, and to prepare for potential future outbreaks by existing and/or novel coronaviruses, there is an urgent need for additional strategies for targeting SARS-CoV-2.While the majority of recent antiviral efforts have focused on targeting essential SARS-CoV-2 proteins, RNA structural elements within the SARS-CoV-2 viral genome are also compelling targets for clinical and basic research applications.6–12 In particular, the 3′ UTR of SARS-CoV-2 genomic RNA contains a 41-nucleotide stem-loop II-like motif (s2m) that has been identified as having high diagnostic and therapeutic value (Fig. 1).13–15 The s2m is a rigorously conserved RNA sequence element found in 3′ UTRs of astroviruses, some noroviruses, and several coronaviruses, including SARS-CoV-2 and SARS-CoV-1, the causative agent of the 2003 epidemic in China.16–18 Remarkably, only 2 nucleotides differ between s2m elements in SARS-CoV-2 and SARS-CoV-1, contrasting the overall 20% genome-wide sequence difference.11 This high level of sequence conservation is likely due to the requirement of the s2m RNA to maintain an elaborate three-dimensional structure. Indeed, a crystal structure of s2m from SARS-CoV-1 shows that it consists of a long stem-loop structure with several non-helical motifs and a unique 90° kink of the helical axis.15 Both chemical mapping and NMR studies indicate that s2m from SARS-CoV-2 also forms an extended stem-loop structure, but the single G to U mutation (position 32 herein) leads to destabilization of the upper stem-loop region and an altered secondary structure relative to SARS-CoV-1 (Fig. 1).7,8,13,19 Importantly, the broad conservation of the s2m RNA structure implies that it confers a selective advantage, making it an attractive antiviral target with low probability of evolving resistance-inducing mutations.18 Moreover, reagents targeting the s2m RNA structure represent powerful tools to study its function, which remains poorly understood.
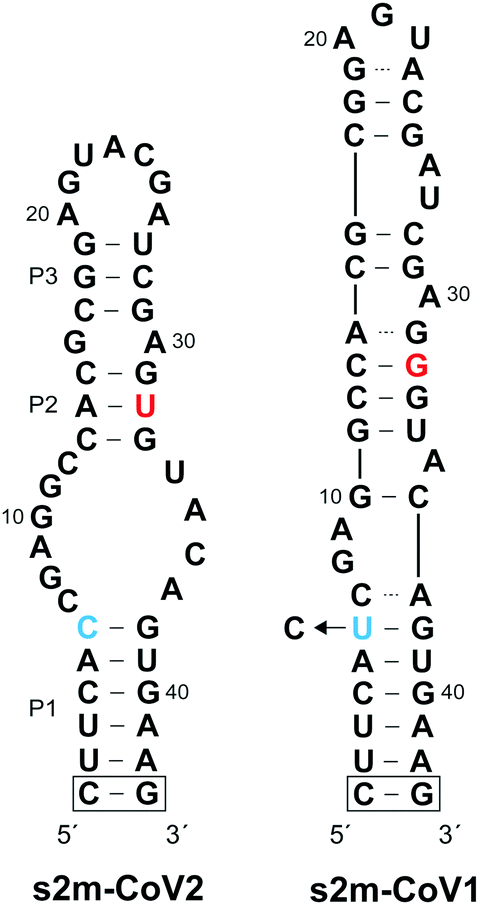 | ||
| Fig. 1 Sequence and secondary structure of s2m RNA elements from SARS-CoV-2 (s2m-CoV2) and SARS-CoV-1 (s2m-CoV1). Sequence differences are highlighted using blue and red text. The boxed G/C base pair was added for structural stability and is not present in the native s2m structure. The s2m-CoV1 sequence used herein contained a U to C transition at position 6. | ||
We and others have shown that L-oligonucleotide aptamers (or Spiegelmers)20 composed of L-DNA and L-RNA, synthetic enantiomers of native D-DNA and D-RNA, respectively, can be evolved to bind structured RNAs.21–25 Because D- and L-oligonucleotides are incapable of forming contiguous WC base pairs with each other,26,27L-aptamers must instead interact with native D-RNA through tertiary interactions. This so called “cross-chiral” mode of nucleic acid recognition results in highly selective binding because it depends on both the sequence and structure of the RNA target.23 These interactions are also high affinity, often achieving low nanomolar dissociation constants under physiological conditions. Importantly, cross-chiral aptamers have been shown to modulate RNA function upon binding their targets, for example, by blocking RNA–protein interactions.21–25 Given these properties, along with the intrinsic nuclease resistance of L-oligonucleotides,28,29 cross-chiral aptamers represent a promising strategy for the development of RNA-targeted therapeutics and probes to study RNA function.
In the current study, we isolated and characterized two L-DNA aptamers that bind tightly to s2m RNA. We show that both L-aptamers can differentiate the monomeric s2m hairpin from the proposed homodimer duplex, highlighting the structure-specific nature of these interactions. Accordingly, both L-aptamers are unable to bind s2m from SARS-CoV-1, which exists almost exclusively as a homodimer in isolation. Finally, footprinting studies show that the structure of s2m RNA changes extensively upon L-aptamer binding, potentially leading to altered function, for example, by preventing recognition of s2m by protein targets. Overall, these results demonstrate the feasibility of targeting SARS-CoV-2 RNA using L-aptamers, providing impetus for future development of L-aptamer-based antiviral therapeutics to treat COVID-19. Moreover, due to their intrinsic nuclease resistance and advantageous binding properties, L-aptamers represent promising biochemical tools for interrogating s2m RNA function.
Results and discussion
Isolation of cross-chiral aptamers for s2m RNA
In order to isolate an L-DNA aptamer capable of binding SARS-CoV-2 s2m RNA (D-s2m-CoV2), we carried out a mirror image in vitro selection experiment against the L-RNA version of s2m (L-s2m-CoV2).21 Here, L-DNA aptamers are initially selected as D-DNA aptamers against the enantiomer of the intended RNA target, which enables enzymatic amplification of the D-DNA library during the selection process. L-s2m-CoV2 was prepared by solid-phase synthesis using L-nucleoside phosphoramidites. The 5′ end was conjugated with a biotin moiety for downstream immobilization and the P1 stem was further stabilized through the addition of a terminal G/C base pair (Fig. 1 and Table S1, ESI†). The selection experiment was initiated by mixing a single-stranded D-DNA pool consisting of a 45-nucleotide random domain (∼1014 unique molecules) together with biotinylated L-s2m-CoV2. D-DNA molecules that bound to L-s2m-CoV2 were captured using streptavidin-coated magnetic beads, and unbound DNA was subsequently washed away. D-Aptamers that remained bound to the beads were eluted by denaturation using NaOH (25 mM) and amplified by PCR. The resulting double-stranded DNA was used to generate the corresponding pool of single-stranded DNAs for the next selection round.The selection pressure was gradually increased during successive rounds by decreasing the concentration of L-s2m-CoV2 and by increasing the bead washing time (see Table S2, ESI† for details). Importantly, a negative selection step was introduced during later rounds of the selection experiment, wherein the single-strand DNA library was first incubated with an L-RNA version of s2m from SARS-CoV-1 (L-s2m-CoV1). In this case, only DNA molecules that did not bind to L-s2m-CoV1 were advanced into the positive selection step against L-s2m-CoV2. The s2m-CoV1 RNA used during the negative selection contains a U → C transition at position 6 such that the only difference between s2m-CoV1 and s2m-CoV2 is at position 32 (Fig. 1). Because these two RNA targets differ by only a single nucleotide, we expected this approach to yield aptamers with high selectivity for the sequence and structure of L-s2m-CoV2.
After a total of eight rounds of in vitro selection, the enriched pool of DNA was cloned and sequenced (Table S3, ESI†). Of the 18 clones obtained, one sequence, D-C1, appeared 8 times, indicating a strong selective advantage. This clone along with three others were tested for their ability to bind L-s2m-CoV2 by electrophoretic mobility shift assay (EMSA) (Fig. S1, ESI†). Clones D-C1 and D-C3 emerged as the tightest binders, with respective Kd values of 87 ± 3 nM and 161 ± 8 nM (Fig. 2 and Fig. S2, ESI†). Therefore, all further studies were based on these two sequences.
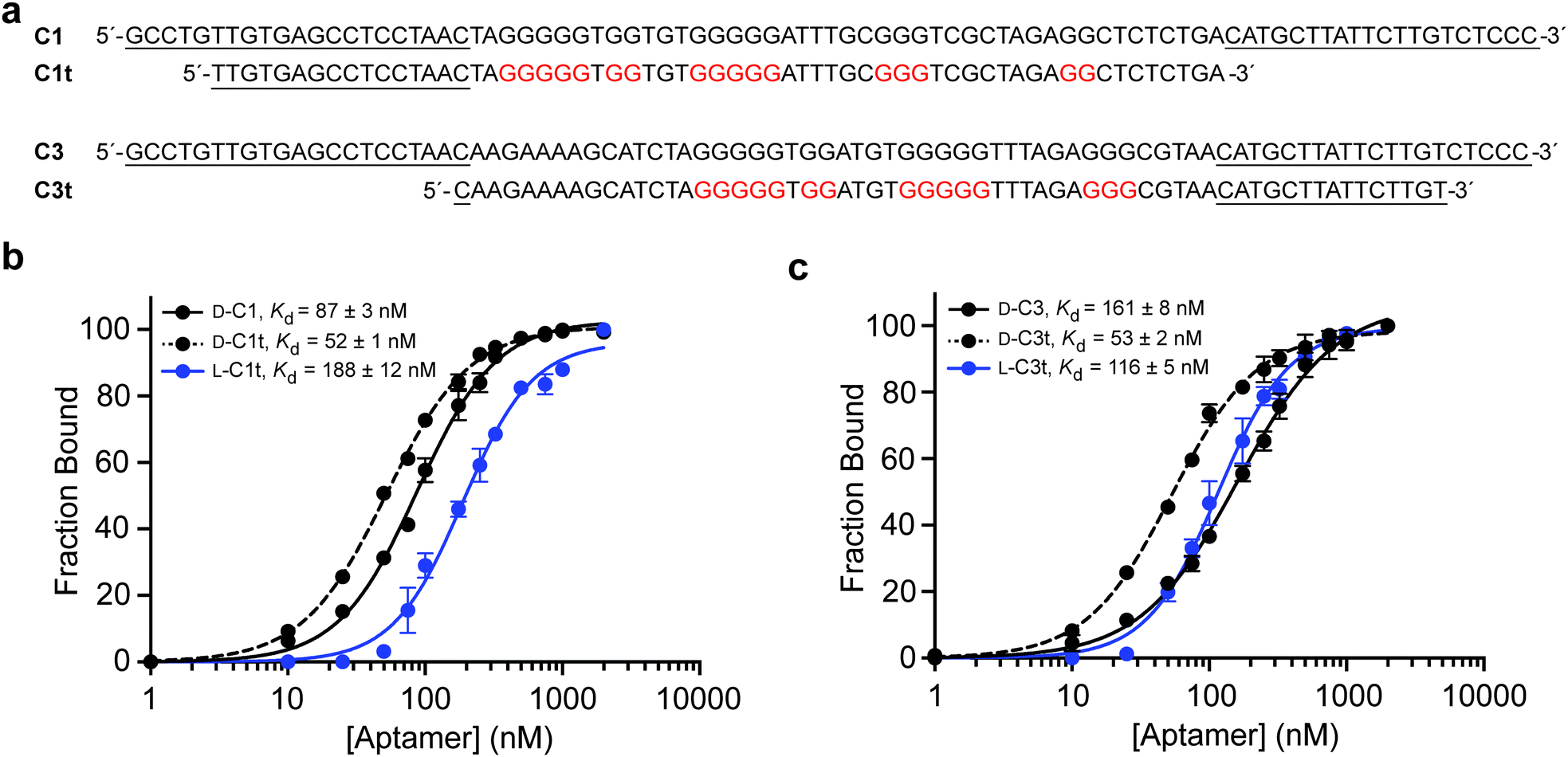 | ||
| Fig. 2 Sequence and binding affinity of L-DNA aptamers for s2m RNA. (a) Sequences of full-length aptamers C1 and C3 and their truncated variants C1t and C3t, respectively. Guanosine residues believed to be involved in formation of G-quadruplexes are colored red. (b) Saturation plots for binding of C1 and C1t to s2m-CoV2 RNA of the opposite stereochemistry. (c) Saturation plots for binding of C3 and C3t to s2m-CoV2 RNA of the opposite stereochemistry. Kd values reported as mean ± S.D. (n = 3). | ||
Preparation of truncated L-DNA aptamers
To help improve the efficiency of downstream L-aptamer synthesis, we sought to remove extraneous nucleotides from D-C1 and D-C3, both of which were 86-nucleotides in length. Unfortunately, initial attempts to rationally truncate these sequences based on predicted secondary structures failed (Fig. S3, ESI†), suggesting that these predictions were not representative of the true aptamer structures. Instead, we generated a series of aptamer variants having a succession of 5′ and 3′ truncations and screened them individually for binding to L-s2m-CoV2 by EMSA (Tables S4 and S5, ESI†). This iterative process resulted in the truncation of both D-C1 and D-C3 by 25 nt, yielding minimal aptamers D-C1t and D-C3t, respectively (Fig. 2a). D-C1t bound L-s2m-CoV2 with a Kd of 52 ± 1 nM, while D-C3t bound L-s2m-CoV2 with a Kd of 53 ± 2 nM, when measured under in vitro selection conditions. It is not uncommon for Kd values for truncated aptamers to be lower than the parent sequences due to reduced interference from steric effects.30,31 As a control, we showed that versions of both D-aptamers having scrambled sequences failed to bind L-s2m-CoV2 (Fig. S4, ESI†).We then prepared L-DNA versions of both truncated aptamers via solid-phase synthesis using L-nucleoside phosphoramidites (Fig. S5, ESI†). Aptamers L-C1t and L-C3t bound D-s2m-CoV2 RNA with Kd values of 188 ± 12 nM and 116 ± 5 nM, respectively, which is somewhat reduced (∼3-fold) relative to their D-DNA counterparts (Fig. 2b and c). The principle of reciprocal chiral substrate specificity dictates that the affinity of the L-DNA aptamers for native D-s2m-CoV2 should be the same as the D-DNA aptamers for L-s2m-CoV2 RNA.32 The discrepancy observed here likely reflects differences in the quality of the synthetic DNAs and/or RNA targets, as well as potential human error in the measurements. Nevertheless, L-C1t and L-C3t aptamers represent the first high-affinity, non-hybridization-based reagents reported to bind this important viral RNA target.
C1t and C3t contain G-quadruplex structures
We observed that both truncated L-DNA aptamers contain at least four tracks of consecutive guanine residues that are diagnostic of G-quadruplexes (Fig. 2a). Indeed, the probability that C1t and C3t form G-quadruplex structures, based on G-score calculations,33 is similar to previously reported L-DNA aptamers with experimentally validated G-quadruplexes (Table S6, ESI†). This is consistent with our inability to truncate parent aptamers C1 and C3 based on secondary structure predictions that do not account for the formation of G-quadruplexes. To verify that aptamers L-C1t and L-C3t form G-quadruplexes, we tested their dependence on K+, which is known to stabilize G-quadruplexes more effectively than other monovalent ions.34 In the absence of K+, both L-DNA aptamers failed to bind D-s2m-CoV2 RNA, even at concentrations that greatly exceeded the observed Kd (Fig. S6, ESI†). In the presence of K+, the circular dichroism (CD) spectrum of D-C1t and D-C3t showed an absorption minimum near 240 nm and maximum near 265 nm, which are characteristic of a parallel-stranded G-quadruplex structure (Fig. 3).35,36 Replacing K+ in the buffer with Li+, which disfavors G-quadruplex formation,34 resulted in a CD spectrum that more closely resembled the unfolded aptamers in the absence of any ions (TE buffer). Taken together, these data strongly suggest that aptamers C1t and C3t contain potassium-dependent, parallel-stranded G-quadruplex structures and that these structures are essential for binding to s2m-CoV2 RNA. Interestingly, we previously showed that L-DNA aptamers isolated against an unrelated RNA hairpin, precursor microRNA-155, also formed an essential G-quadruplex motif.21 Thus, potassium-dependent G-quadruplexes may be a common structural motif for recognition between DNA and RNA molecules of the opposite chirality.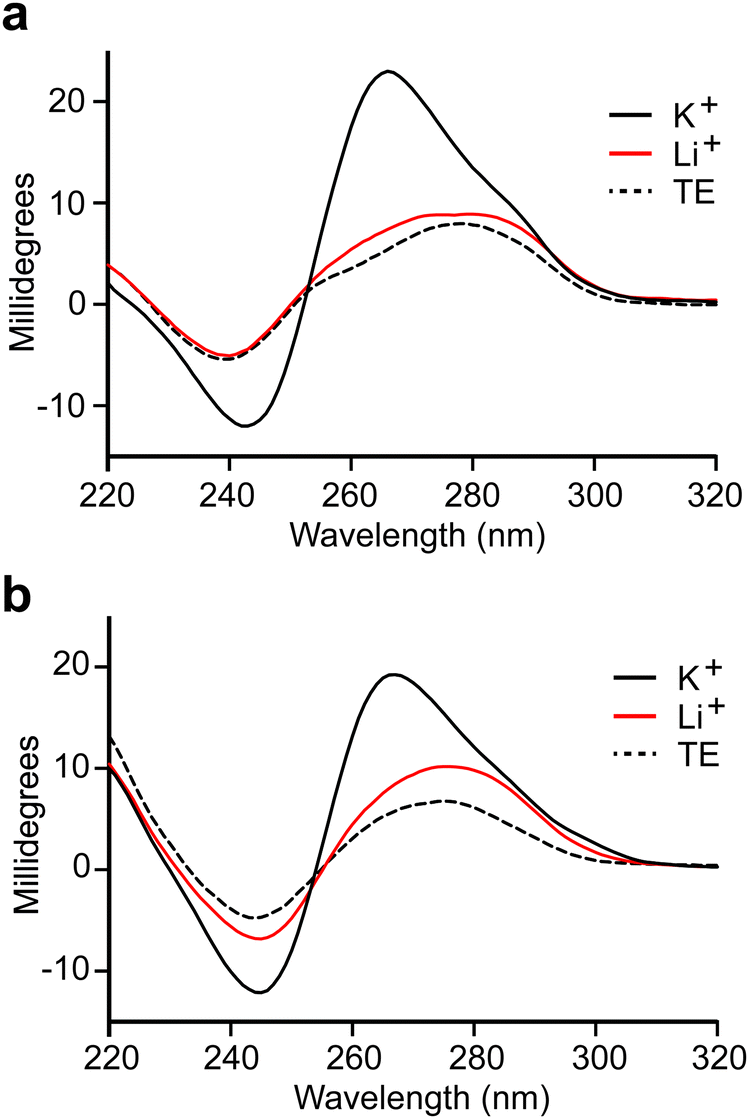 | ||
| Fig. 3 CD spectra of aptamers D-C1t (a) and D-C3t (b). All spectra were obtained in buffer consisting of 20 mM NaCl, 25 mM Tris (pH 7.6), and either 50 mM KCl (black line) or 50 mM LiCl (red line) at room temperature. TE indicates a buffer containing 25 mM Tris (pH 7.6) and 1 mM EDTA (dashed line). | ||
L-C1t and L-C3t bind the upper stem-loop of s2m RNA
To gain insight into the region of binding, we performed an in-line hydrolysis analysis of D-s2m-CoV2 in the presence and absence of saturating concentrations of either L-C1t or L-C3t (Fig. 4 and Fig. S7, ESI†).21 In-line hydrolysis (i.e. cleavage) of a phosphodiester linkage in RNA is dependent on the overall conformational freedom of the corresponding nucleotide, with low conformational freedom being associated with slower cleavage rates (and vice versa).37 Accordingly, conformational changes within D-s2m-CoV2 RNA upon L-aptamer binding are expected to alter hydrolytic cleavage patterns in proximity to the binding site. As seen in Fig. 4, the presence of either L-aptamer resulted in extensive changes to the hydrolytic cleavage pattern within the upper portion of the D-s2m-CoV2 stem-loop comprising residues 13–33, whereas very little change occurred within the strong P1 stem and adjoining bulge. These data strongly suggest that both L-C1t and L-C3t bind the upper portion of the D-s2m-CoV2 stem-loop. Consistently, the strongest protection of D-s2m-CoV2 from hydrolytic cleavage occurred within the loop that closes the P3 stem. In particular, cleavage of A23 and U22 was reduced by more than 3-fold in the presence of L-C1t or L-C3t, respectively, possibly indicating a direct interaction of these otherwise unpaired nucleotides with the L-aptamer. Interestingly, the two L-aptamers had very different effects on the conformation of the P2 and P3 stems. In the presence of L-C1t, the extent of in-line cleavage of several residues within the P2 and P3 stems increased significantly, suggesting that binding of L-C1t to D-s2m-CoV2 destabilizes these secondary structures. In contrast, many of these same residues were either unaffected or had reduced cleavage in the presence of L-C3t, indicating that binding of L-C3t to D-s2m-CoV2 may further stabilize the P2 and P3 stems. Thus, L-C1t and L-C3t bind D-s2m-CoV2 using unique heterochiral interfaces that alter the structure of D-s2m-CoV2 in different ways.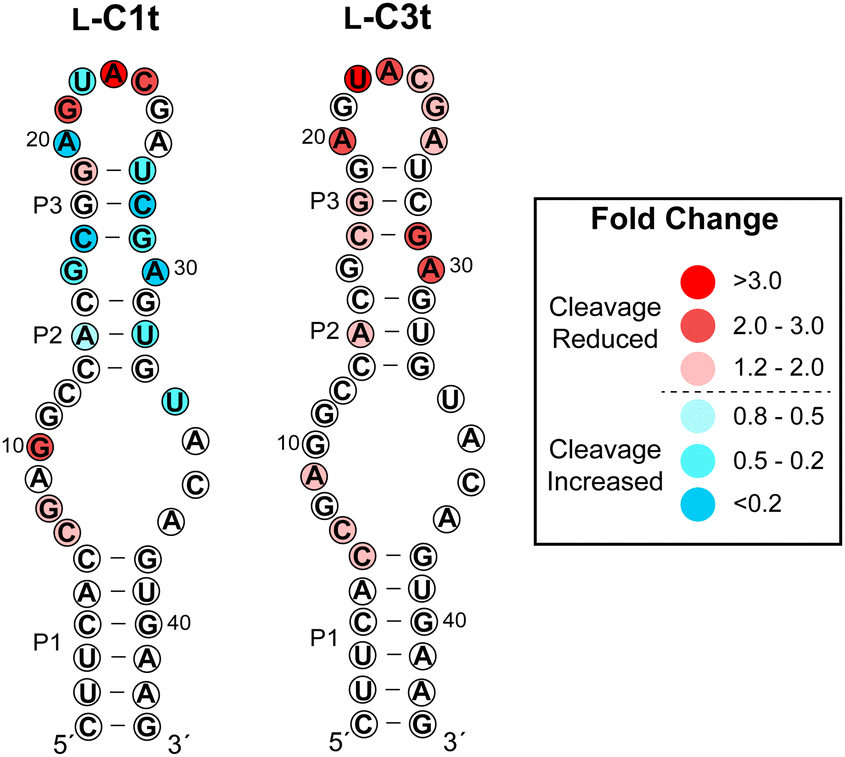 | ||
| Fig. 4 Influence of L-C1t and L-C3t binding on hydrolytic cleavage of D-s2m-CoV2 RNA. Fold change is indicated by colors, and was determined by dividing the fraction cleaved in the absence of the aptamer by the fraction cleaved in the presence of the aptamer. Values greater than one indicate reduced hydrolytic cleavage (i.e. protection) and values less than one indicated increased hydrolytic cleavage in the presence of the L-aptamer. Gel images are shown in Fig. S7 (ESI†). | ||
L-DNA aptamers bind selectively to s2m RNA and do not bind the proposed s2m homodimer
Finally, we sought to demonstrate the binding specificity of L-C1t and L-C3t. We first examined whether these two L-aptamers could differentiate the intended target D-s2m-CoV2 from D-s2m-CoV1, whose sequences differ by just two nucleotides (Fig. 1). As seen in Fig. 5a, D-s2m-CoV1 migrated more slowly than D-s2m-CoV2 when analysed by native gel electrophoresis. This is consistent with previous observations that s2m RNA is capable of forming a homodimer duplex structure, with dimerization of D-s2m-CoV1 being far more favorable than D-s2m-CoV2 in the absence of other cellular factors (Fig. 5b and Fig. S8, ESI†).38 Not unexpectedly, both aptamers bound the monomeric D-s2m-CoV2 hairpin but failed to bind the D-s2m-CoV1 homodimer duplex. Thus, the minor sequence differences between D-s2m-CoV2 and D-s2m-CoV1 result in dramatic conformational differences in vitro, that can be discriminated by L-aptamers. It is likely that the negative selection step against L-SARS-CoV1, which would have existed as a homodimer during the selection experiment, contributed to the structure-specific nature of these aptamers. These data also suggest that L-C1t and L-C3t could differentiate monomeric D-s2m-CoV2 RNA from the proposed homodimer when formed. To test this, we designed a pair of variant D-s2m-CoV2 stem-loops (D-s2m-A and D-s2m-B) having cross-complementary lower stems that greatly favored formation of the heterodimeric complex over the monomeric stem-loops (Fig. 5b and Fig. S8, ESI†). The sequence changes made to the s2m variants fall outside of the aptamer binding site proposed above (Fig. 4), and the structure of the corresponding heterodimer formed between D-s2m-A and D-s2m-B (D-s2m-A/B) is predicted to be identical to the homodimer formed by D-s2m-CoV2 (Fig. S8, ESI†). Accordingly, mixing of D-s2m-A and D-s2m-B resulted in the formation of a slower moving band that co-migrated with the D-s2m-CoV1 homodimer, suggesting formation of a heterodimer between the two s2m variants (Fig. 5c). Importantly, while both L-C1t and L-C3t bound D-s2m-B alone, neither aptamer bound the dimer D-s2m-A/B, based on the absence of a gel shift. As an additional control for specificity, we showed that both L-DNA aptamers also failed to bind several precursor microRNAs (pre-miRs), which also form extended stem-loop structures (Fig. 5a and Table S7, ESI†). Together, these data demonstrate that L-C1t and L-C3t have high selectivity for binding D-s2m-CoV2 RNA hairpin and are unable to bind the corresponding homodimer duplex structure.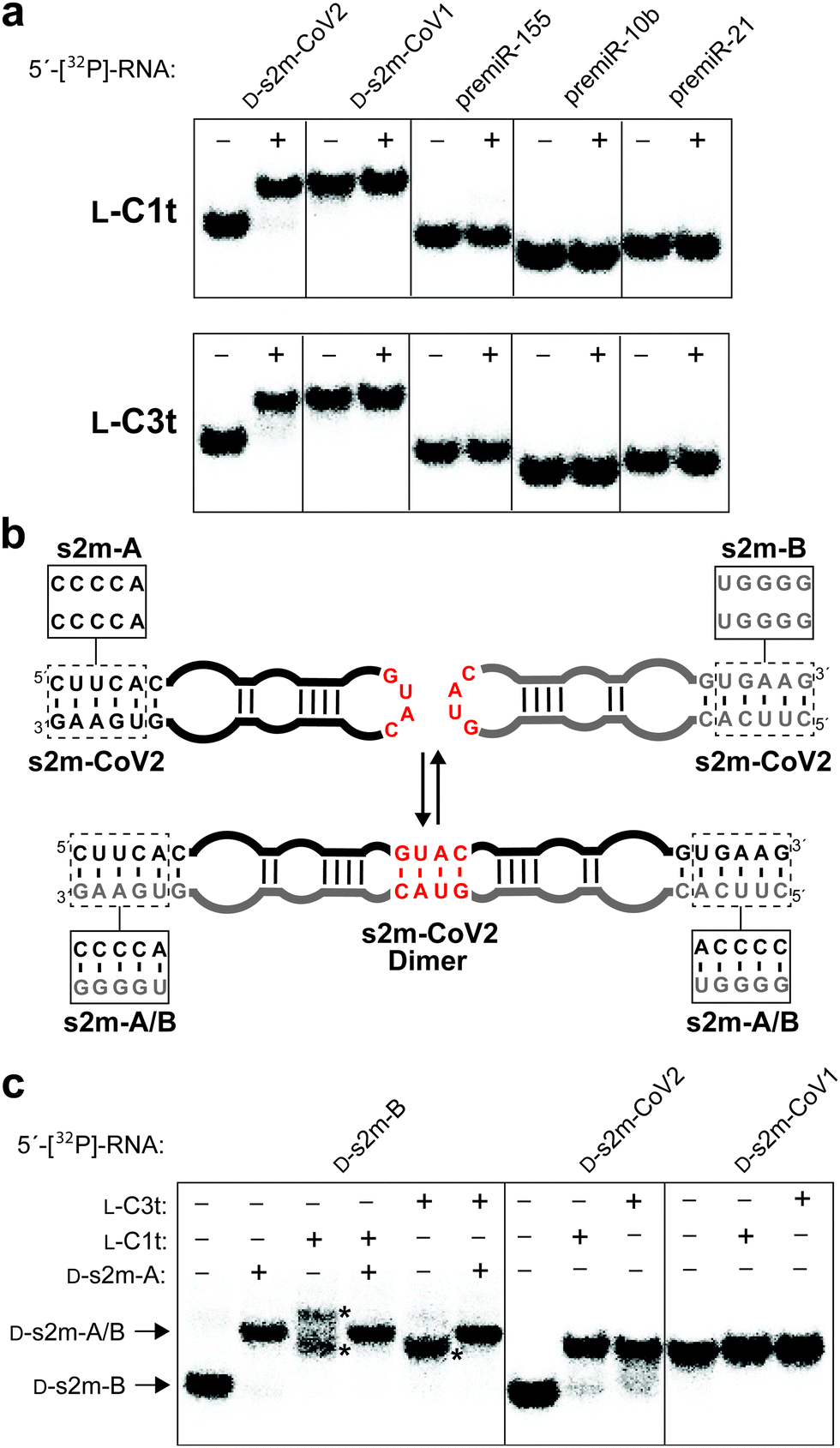 | ||
| Fig. 5 L-DNA aptamers bind selectively to D-s2m-CoV2. (a) Schematic illustration of the proposed s2m dimerization. Boxes indicate sequence differences between s2m-CoV2 and the two variants s2m-A and s2m-B, which favour formation of the s2m-A/B heterodimer (Fig. S8, ESI†). (b) Binding of L-C1t (top) and L-C3t (bottom) to different 5′-[32P]-labelled RNAs. Plus (+) and minus (−) symbols indicate with and without 2 μM L-aptamer, respectively. (c) Binding of L-aptamers to 5′-[32P]-s2m-B or the s2m-A/B heterodimer. Asterisk indicates L-aptamer–D-s2m-B complexes. Binding to D-s2m-CoV2 and D-s2m-CoV1 is shown as a reference. | ||
Conclusions
In summary, we identified and characterized two L-DNA aptamers (L-C1t and L-C3t) against the conserved s2m RNA element from the SARS-CoV-2 genome, thereby demonstrating the feasibility of targeting SARS-CoV-2 and other coronaviruses using L-aptamers. In addition to their nanomolar affinity (<200 nM), both L-aptamers could discriminate the s2m stem-loop target from the proposed dimeric duplex structure, despite the lack of sequence changes at the proposed L-aptamer–s2m interface. Accordingly, L-C1t and L-C3t were unable to bind s2m RNA from SARS-CoV-1, which existed exclusively as the homodimer in our hands. This result further highlights the profound specificity that can be obtained using L-aptamers, which are dependent on the sequence and structure of the RNA target. To the best of our knowledge, this is the first example of an aptamer (of any stereochemistry) that can differentiate between two different structural states of the same RNA sequence. Structure-specific binding of RNA is challenging to achieve with other classes of reagents, such as antisense oligonucleotides and small molecules, and probes capable of discriminating between two different structures of the same RNA are rare. Therefore, the L-aptamers reported herein may provide a unique opportunity to develop molecular switches, sensors, and other devices based on programmable, structure-dependent RNA interactions. Most importantly, due to their intrinsic nuclease resistance and advantageous binding properties, L-C1t and L-C3t represent promising biochemical tools for interrogating s2m function, and can potentially be utilized as affinity reagents for clinical applications related to COVID-19. Indeed, the programmability of WC base pairing allows aptamers to be readily engineered into biosensors for imaging and diagnostic application.39 Finally, although several challenges remain, L-C1t and L-C3t could serve as lead compounds to develop potential RNA-targeted antiviral agents. Thus, the application of these L-aptamers to cellular models of SARS-CoV-2 is a future priority.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institute of General Medical Sciences (R35GM124974) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was provided by the Welch Foundation (A-1909-20190330) and the Cancer Prevention and Research Institute of Texas (RR150038).Notes and references
- P. V’kovski, A. Kratzel, S. Steiner, H. Stalder and V. Thiel, Nat. Rev. Microbiol., 2021, 19, 155–170 CrossRef.
- B. Hu, H. Guo, P. Zhou and Z.-L. Shi, Nat. Rev. Microbiol., 2021, 19, 141–154 CrossRef CAS PubMed.
- WHO Coronavirus (COVID-19) Dashboard, https://covid19.who.int (accessed 10/22/2021).
- T. Asselah, D. Durantel, E. Pasmant, G. Lau and R. F. Schinazi, J. Hepatol., 2021, 74, 168–184 CrossRef CAS PubMed.
- J. H. Beigel, K. M. Tomashek, L. E. Dodd, A. K. Mehta, B. S. Zingman, A. C. Kalil, E. Hohmann, H. Y. Chu, A. Luetkemeyer, S. Kline, D. Lopez de Castilla, R. W. Finberg, K. Dierberg, V. Tapson, L. Hsieh, T. F. Patterson, R. Paredes, D. A. Sweeney, W. R. Short, G. Touloumi, D. C. Lye, N. Ohmagari, M.-D. Oh, G. M. Ruiz-Palacios, T. Benfield, G. Fätkenheuer, M. G. Kortepeter, R. L. Atmar, C. B. Creech, J. Lundgren, A. G. Babiker, S. Pett, J. D. Neaton, T. H. Burgess, T. Bonnett, M. Green, M. Makowski, A. Osinusi, S. Nayak and H. C. Lane, N. Engl. J. Med., 2020, 383, 1813–1826 CrossRef CAS.
- M. G. Costales, J. L. Childs-Disney, H. S. Haniff and M. D. Disney, J. Med. Chem., 2020, 63, 8880–8900 CrossRef CAS.
- N. C. Huston, H. Wan, M. S. Strine, R. de Cesaris Araujo Tavares, C. B. Wilen and A. M. Pyle, Mol. Cell, 2021, 81, 584–598 CrossRef CAS.
- A. Wacker, J. E. Weigand, S. R. Akabayov, N. Altincekic, J. K. Bains, E. Banijamali, O. Binas, J. Castillo-Martinez, E. Cetiner, B. Ceylan, L.-Y. Chiu, J. Davila-Calderon, K. Dhamotharan, E. Duchardt-Ferner, J. Ferner, L. Frydman, B. Fürtig, J. Gallego, J. T. Grün, C. Hacker, C. Haddad, M. Hähnke, M. Hengesbach, F. Hiller, K. F. Hohmann, D. Hymon, V. de Jesus, H. Jonker, H. Keller, B. Knezic, T. Landgraf, F. Löhr, L. Luo, K. R. Mertinkus, C. Muhs, M. Novakovic, A. Oxenfarth, M. Palomino-Schätzlein, K. Petzold, S. A. Peter, D. J. Pyper, N. S. Qureshi, M. Riad, C. Richter, K. Saxena, T. Schamber, T. Scherf, J. Schlagnitweit, A. Schlundt, R. Schnieders, H. Schwalbe, A. Simba-Lahuasi, S. Sreeramulu, E. Stirnal, A. Sudakov, J.-N. Tants, B. S. Tolbert, J. Vögele, L. Weiß, J. Wirmer-Bartoschek, M. A. Wirtz Martin, J. Wöhnert and H. Zetzsche, Nucleic Acids Res., 2020, 48, 12415–12435 CrossRef CAS.
- T. C. T. Lan, M. F. Allan, L. E. Malsick, S. Khandwala, S. S. Y. Nyeo, Y. Sun, J. U. Guo, M. Bathe, A. Griffiths and S. Rouskin, bioRxiv, 2021 DOI:10.1101/2020.06.29.178343.
- R. Rangan, I. N. Zheludev, R. J. Hagey, E. A. Pham, H. K. Wayment-Steele, J. S. Glenn and R. Das, RNA, 2020, 26, 937–959 CrossRef CAS.
- D. Kim, J.-Y. Lee, J.-S. Yang, J. W. Kim, V. N. Kim and H. Chang, Cell, 2020, 181, 914–921 CrossRef CAS.
- H. S. Haniff, Y. Tong, X. Liu, J. L. Chen, B. M. Suresh, R. J. Andrews, J. M. Peterson, C. A. O’Leary, R. I. Benhamou, W. N. Moss and M. D. Disney, ACS Cent. Sci., 2020, 6, 1713–1721 CrossRef CAS PubMed.
- I. Manfredonia, C. Nithin, A. Ponce-Salvatierra, P. Ghosh, T. K. Wirecki, T. Marinus, N. S. Ogando, E. J. Snijder, M. J. van Hemert, J. M. Bujnicki and D. Incarnato, Nucleic Acids Res., 2020, 48, 12436–12452 CrossRef CAS PubMed.
- V. Lulla, M. P. Wandel, K. J. Bandyra, R. Ulferts, M. Wu, T. Dendooven, X. Yang, N. Doyle, S. Oerum, R. Beale, S. M. O’Rourke, F. Randow, H. J. Maier, W. Scott, Y. Ding, A. E. Firth, K. Bloznelyte and B. F. Luisi, J. Virol., 2021, 95, e00663 CrossRef CAS.
- M. P. Robertson, H. Igel, R. Baertsch, D. Haussler, M. Ares Jr and W. G. Scott, PLoS Biol., 2005, 3, e5 CrossRef.
- E. de Wit, N. van Doremalen, D. Falzarano and V. J. Munster, Nat. Rev. Microbiol., 2016, 14, 523–534 CrossRef CAS.
- T. Tengs, A. B. Kristoffersen, T. R. Bachvaroff and C. M. Jonassen, Virol. J., 2013, 10, 132 CrossRef CAS PubMed.
- C. Gilbert and T. Tengs, Sci. Rep., 2021, 11, 16145 CrossRef CAS.
- J. Zhao, J. Qiu, S. Aryal, J. L. Hackett and J. Wang, Viruses, 2020, 12, 1473 CrossRef CAS.
- S. Klussmann, A. Nolte, R. Bald, V. A. Erdmann and J. P. Fürste, Nat. Biotechnol., 1996, 14, 1112–1115 CrossRef CAS.
- S. Dey and J. T. Sczepanski, Nucleic Acids Res., 2020, 48, 1669–1680 CrossRef CAS PubMed.
- A. M. Kabza and J. T. Sczepanski, ChemBioChem, 2017, 18, 1824–1827 CrossRef CAS.
- J. T. Sczepanski and G. F. Joyce, J. Am. Chem. Soc., 2013, 135, 13290–13293 CrossRef CAS.
- M. I. Umar and C. K. Kwok, Nucleic Acids Res., 2020, 48, 10125–10141 CrossRef CAS.
- J. T. Sczepanski and G. F. Joyce, J. Am. Chem. Soc., 2015, 137, 16032–16037 CrossRef CAS PubMed.
- N. C. Hauser, R. Martinez, A. Jacob, S. Rupp, J. D. Hoheisel and S. Matysiak, Nucleic Acids Res., 2006, 34, 5101–5111 CrossRef CAS.
- K. Hoehlig, L. Bethge and S. Klussmann, PLoS One, 2015, 10, e0115328 CrossRef.
- G. W. Ashley, J. Am. Chem. Soc., 1992, 114, 9732–9736 CrossRef.
- H. Urata, K. Shinohara, E. Ogura, Y. Ueda and M. Akagi, J. Am. Chem. Soc., 1991, 113, 8174–8175 CrossRef CAS.
- H. Kaur and L.-Y. L. Yung, PLoS One, 2012, 7, e31196 CrossRef CAS.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- R. C. Milton, S. C. Milton and S. B. Kent, Science, 1992, 256, 1445–1448 CrossRef CAS.
- O. Kikin, L. D'Antonio and P. S. Bagga, Nucleic Acids Res., 2006, 34, W676–W682 CrossRef CAS PubMed.
- D. Bhattacharyya, G. Mirihana Arachchilage and S. Basu, Front. Chem., 2016, 4, 38 Search PubMed.
- M. Vorlíčková, I. Kejnovská, J. Sagi, D. Renčiuk, K. Bednářová, J. Motlová and J. Kypr, Methods, 2012, 57, 64–75 CrossRef PubMed.
- A. I. Karsisiotis, N. M. a. Hessari, E. Novellino, G. P. Spada, A. Randazzo and M. Webba da Silva, Angew. Chem., Int. Ed., 2011, 50, 10645–10648 CrossRef CAS.
- G. A. Soukup and R. R. Breaker, RNA, 1999, 5, 1308–1325 CrossRef CAS.
- J. A. Imperatore, C. L. Cunningham, K. A. Pellegrene, R. G. Brinson, J. P. Marino, J. D. Evanseck and M. R. Mihailescu, bioRxiv, 2020 DOI:10.1101/2020.12.29.424733.
- N. K. Navani and Y. Li, Curr. Opin. Chem. Biol., 2006, 10, 272–281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, Fig. S1–S22 and Tables S1–S7. See DOI: 10.1039/d1cb00172h |
| This journal is © The Royal Society of Chemistry 2022 |