
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStapling proteins in the RELA complex inhibits TNFα-induced nuclear translocation of RELA†
Smit
Kour‡
a,
Sandeep
Rana‡§
a,
Smitha
Kizhake
a,
Dragana
Lagundžin
ab,
David
Klinkebiel
c,
Jayapal Reddy
Mallareddy
a,
Tom
Huxford
 d,
Nicholas T.
Woods
ae and
Amarnath
Natarajan
*aef
d,
Nicholas T.
Woods
ae and
Amarnath
Natarajan
*aef
aEppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE 68198, USA. E-mail: anatarajan@unmc.edu
bMass Spectrometry and Proteomics Core Facility, University of Nebraska Medical Center, Omaha, NE 68198, USA
cDepartment of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, NE 68198, USA
dStructural Biochemistry Laboratory, Department of Chemistry & Biochemistry, San Diego State University, San Diego, CA 92182, USA
eFred & Pamela Buffett Cancer Center, University of Nebraska Medical Center, Omaha, NE 68198, USA
fDepartment of Pharmaceutical Sciences and Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE 68198, USA
First published on 28th October 2021
Abstract
Tumor necrosis factor (TNF) α-induced nuclear translocation of the NF-κB subunit RELA has been implicated in several pathological conditions. Here we report the discovery of a spirocyclic dimer (SpiD7) that covalently modifies RELA to inhibit TNFα-induced nuclear translocation. This is a previously unexplored strategy to inhibit TNFα-induced NF-κB activation.
Induction of the canonical NF-κB pathway activated by tumour necrosis factor (TNF) α, results in the nuclear translocation of RELA.1 The presence of TNFα in the tumour microenvironment promotes constitutive activation of this pathway in ovarian and pancreatic tumours as well as other cancers.2–4 Several studies have shown that activation/inhibition of the pathway via genetic manipulation of the pathway proteins leads to either decreased/increased survival in mouse models of experimental cancer.5–10 Several strategies have been employed to develop therapeutic agents that block nuclear translocation of RELA. Therapeutics currently in clinical use include antibodies against TNFα, such as Humira®, and small molecule inhibitors, such as Velcade®. In addition, we and others have reported an array of reversible and irreversible inhibitors that block the nuclear translocation of RELA and thus inhibit activation of RELA mediated transcription.11–19
The human RELA protein is 551 amino acids (aa) in length with a Rel homology region (RHR) containing an amino-terminal DNA recognition domain and a dimerization domain followed by a transcription activation domain (TAD) at the carboxy-terminal end.20 In resting cells, RELA is retained in the cytoplasm by virtue of its binding to endogenous IκB inhibitor proteins that mask the RELA nuclear localization signal (NLS).21 TNFα-induced activation of the pathway leads to the phosphorylation of IκBα by the IκB kinase (IKK) β subunit, resulting in the ubiquitination-dependent degradation of IκBα thus allowing the nuclear translocation of RELA (Fig. S1, ESI†).22
There are 9 cysteine (Cys) residues in RELA and 6 (38, 95, 105, 109, 120, and 195) of the 9 Cys residues are solvent exposed (pdb id: 1IKN).23 Surface exposed Cys residues that are not associated with catalytic or structural functions are less conserved and less abundant, thus making them attractive targets for precise modulation by covalent binders.24 Recent reports show α-methylene-γ-butyrolactone containing natural product Helenalin analogues covalently target solvent exposed Cys residues 38 and 120 of RELA.25,26 Another α-methylene-γ-butyrolactone natural product, Parthenolide, was previously shown to covalently bind solvent exposed Cys 179 on IKKβ.27 Additionally, we previously showed that an isatin-derived α-methylene-γ-butyrolactone analogue 19, covalently binds to both RELA and IKKβ.16
The α-methylene-γ-butyrolactone moiety in the sesquiterpene natural products is fused to an alicyclic ring system. However, in analogue 19 the α-methylene-γ-butyrolactone moiety and the oxindole core form a spirocyclic system. Therefore, we hypothesized that the spirocyclic disposition of the α-methylene-γ-butyrolactone in analogue 19 could target different solvent exposed Cys residues in RELA and IKKβ. To test this, we incubated recombinant RELA and IKKβ with 19 or DMSO for 1 h on ice and the trypsinized samples were subjected to LC-MS/MS analyses (Fig. 1). Analyses of the tryptic digests revealed excellent coverage (>85%) in both treated and control samples. We identified 2 and 24 spectra in the 19-treated RELA and IKKβ digests, respectively, that had a +215 Da adduct indicating covalent addition of 19 (Fig. S2, ESI†). Analyses of the MS2 spectra revealed that 19 covalently binds to Cys105 on RELA and Cys46, 299, 412 and 524 on IKKβ. Docking studies using Schrödinger GLIDE identified basic amino acids proximal to each Cys residue that potentially increased the basicity of the sulfhydryl groups, facilitating covalent C–S bond formation (Fig. S3, ESI†). Based on the docking score and assuming a two-step process (i.e., binding followed by C–S bond formation) the pocket adjacent to Cys105 on RELA was deemed the most accessible, followed by the pocket adjacent to Cys412 on IKKβ (Table S1, ESI†). Covalent modification of Cys105 and not Cys 38 on RELA by 19 is a novel finding, as Cys38 is the primary target of well characterized NF-κB inhibitors, such as parthenolide and dehydroxymethylepoxyquinomicin (DHMEQ).28
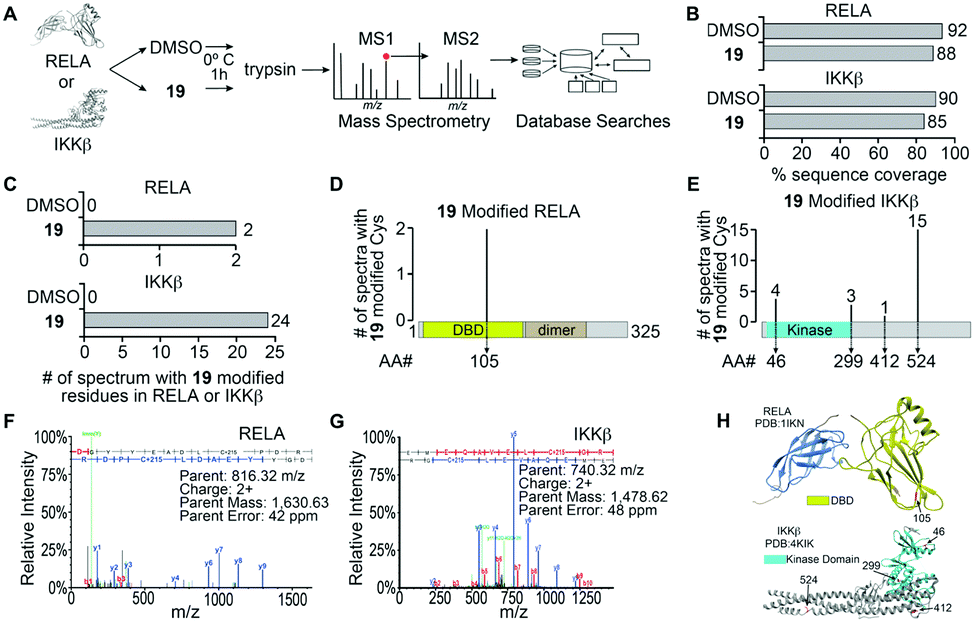 | ||
| Fig. 1 Identification of 19 modified RELA and IKKβ peptides. (A) Purified recombinant RELA or IKKβ was incubated with 19 or DMSO and tandem mass spectrometry was performed on both trypsin and chymotrypsin digests of the protein to identify all RELA and IKKβ peptides. Database searches with variable modification of cysteine residues that correspond to conjugation of 19 (+215 Da) were performed to identify modified peptides. (B) Graph of the total sequence coverage of RELA and IKKβ accomplished by peptide mapping with mass spectrometry. (C) Analysis of the number of significant MS2 spectra with a Peptide Prophet probability ≥95% mapping to RELA and IKKβ with 19 modified sites. (D and E) Illustration of the mapping and abundance of 19 modified cysteine residues in RELA and IKKβ. (F and G) Representative MS2 spectrum of a 19 modified peptide in RELA and IKKβ. H. Protein Data Bank (PDB) structure of RELA (1IKN) and IKKβ (4KIK) with 19-modified cysteine residues indicated in red and the kinase domain of IKKβ coloured cyan. | ||
Next, we assessed if 19 will covalently bind RELA and IKKβ in cells using an alkyne-tagged 19. Ovarian cancer cells (A2780) were subjected to alkyne-tagged 19. Following a 1 h incubation the cells were pelleted, thoroughly washed and lysed. The lysates were subjected to click chemistry using biotin-azide and 19-bound-biotin-tagged proteins in the lysates were pulled down using avidin beads following previously reported methods (Fig. 2).15 Western blot analyses revealed the presence of RELA and IKKβ in the pull down but not IKKγ or IκBα despite the presence of solvent exposed Cys residues in the latter two proteins. Although limited to the proteins in the IKK complex this suggests that 19 selectively and covalently binds to RELA and IKKβ in cells.
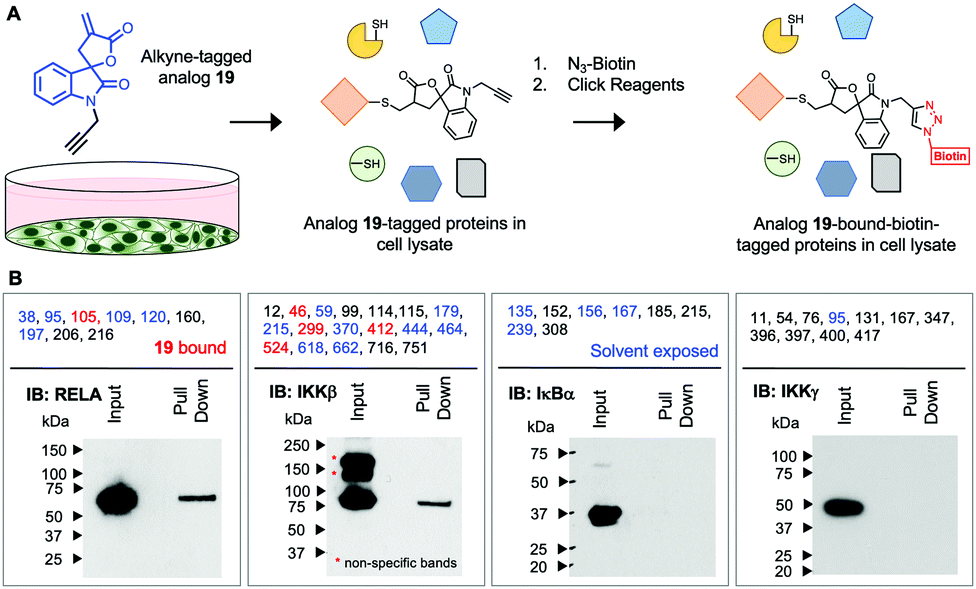 | ||
| Fig. 2 Covalent binding of 19 to RELA and IKKβ in cells. (A) Cancer cells (A2780) were incubated with 10 μM of alkyne-tagged 19 for 1 h. The cells were thoroughly washed to remove unreacted 19 and lysed. The lysate was subjected to click reagents (TCEP, TBTA and CuSO4) and azido-biotin. Biotin-tagged-19-bound proteins were subjected to mono-avidin column. Biotin-tagged proteins were captured, the column was washed to removed untagged proteins and the biotin-tagged-19-bound proteins were eluted with regeneration buffer (6 M urea/PBS). (B) The eluted lysates were subjected to western blot analyses and probed for proteins (IKKβ, RELA, IκBα and IKKγ) in the IKK complex. Cys residues for the protein in the IKK complex that bound to 19in vitro are shown in red and the solvent exposed Cys residues are shown in blue. | ||
Since RELA and IKKβ are part of an ∼900 kDa protein complex,29,30 we rather naively speculated that a dimer of 19 could crosslink RELA and/or IKKβ to their binding partners, which is a previously unexplored modality for targeting the IKK complex. We synthesized a dimer of isatin (1), by alkylating the ends of 1,7-dibromoheptane in the presence of NaH. The isatin-dimer 1 was subjected to an indium catalysed Barbier-type reaction to set up the carbon framework for the spirocyclic core structure.31 An acid catalysed ring closure of the acyclic compound 2 yielded the desired spirocyclic dimer 7 (SpiD7) (Scheme 1). Iodoacetamide is commonly used in mass spectrometry to alkylate Cys residues. To examine the role of the isatin-derived spirocyclic core, we also synthesized an iodoacetamide dimer analogue SpiD7-C (Scheme 1). The distance between the active methylene groups in SpiD7-C is ∼16 Å, which is similar to the distance between the Michael acceptors in SpiD7 (blue arrow, Scheme 1).
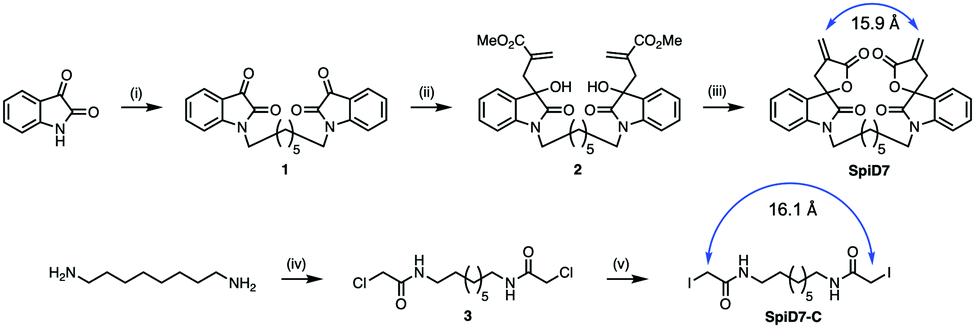 | ||
| Scheme 1 Synthesis of spirocyclic dimer 7 (SpiD7) and a control iodoacetamide dimer compound (SpiD7-C) where in the electrophilic carbon atoms (blue arrows) are ∼16 Å apart. Reaction conditions: (i). 1,7-Dibromoalkane, NaH, DMF, RT, 16 h; 80%; (ii). Methyl 2-(bromomethyl)acrylate, In, THF: water, RT, 24 h, 75%; (iii). TsOH, DCM, RT, 24 h, 80%; (iv) 2-chloroacetyl chloride, K2CO3, DCM; (v) NaI, acetone, reflux, 24 h. | ||
Next, to determine if the dimers crosslink proteins we set up in vitro reactions with recombinant RELA and IKKβ in the presence and absence of 19, SpiD7 or SpiD7-C (Fig. 3A and B). The reactions were analysed via SDS-PAGE and visualised either by silver staining or by membrane transfer and immunodetection by western blot (WB) with anti-His antibody to detect his-tagged recombinant IKKβ and anti-RELA antibody. When compared to RELA and IKKβ alone lanes, we observed additional high molecular weight (MW) bands present only in the SpiD7-treated lane but not in 19- or SpiD7-C-treated lanes. Also, we observed a modest reduction of RELA and IKKβ levels in the SpiD7-treated lane, but not in the controls. Moreover, the banding pattern in 19- and SpiD7-C-treated lanes was similar to that of RELA and IKKβ alone lanes.
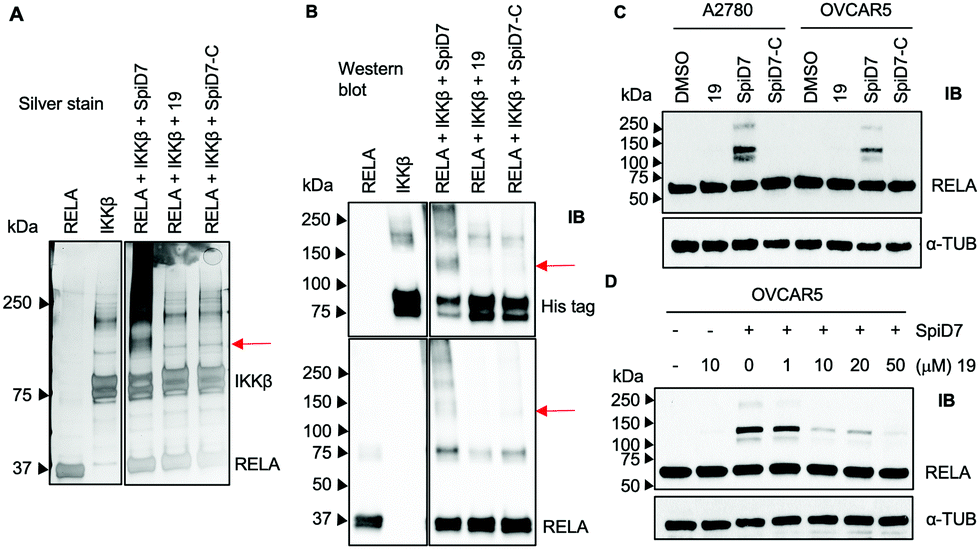 | ||
| Fig. 3 In vitro and cellular crosslinking studies with SpiD7, 19 and SpiD7-C. (A) Equimolar concentrations (7.72 μM) of the reactants were mixed and incubated for 24 h at 4 °C. Following incubation, the mixture was subjected to SDS-PAGE and the gel subjected to silver staining. (B) WB analysis of the in vitro reactions. Red arrows indicate the HMW protein cross-link bands. (C) Cancer cells (A2780 and OVCAR5) were treated with and without 19, SpiD7 and SpiD7-C (10 μM) for 2 h. The lysates were subjected to WB analyses with antibodies against the indicated proteins. (D) OVCAR5 cells were incubated with 10 μM of either 19 or SpiD7 for 2 h (lanes 1–3). In parallel, cells were treated with increasing concentration of 19 (1–50 μM) followed by 10 μM of SpiD7 (lanes 4–7). Following a 2 h incubation lysates from the above, treatments were probed with RELA and α-tubulin antibodies. | ||
To determine if the in vitro observation translates to a cellular system, we subjected cancer cells to either DMSO, 19, SpiD7 or SpiD7-C and probed the lysates for RELA. In both cell lines tested, SpiD7 showed three distinct high MW bands (∼105, 130 and 230 kDa) while no such high MW bands were observed in 19 or SpiD7-C-treated cells indicating that dimerization and the spiroisatin core are required for crosslinking RELA to its binding partners (Fig. 3C). We did not observe any high MW bands when we probed for IKKβ (data not shown). To determine if dimer (SpiD7) and monomer 19 bind to the same proteins, we conducted a competition assay. Cells were treated with increasing concentrations (1–50 μM) of 19, followed by 10 μM of SpiD7, and incubated for 2 h. Western blot analyses of the lysates showed a dose-dependent decrease in the high MW bands (lanes 4–7, Fig. 3D). This demonstrates that monomer 19 and dimer SpiD7 bind to the same sites on RELA and the high MW bands (∼105, 130 and 230 kDa) are a direct result of the dimerization of 19.
To determine the functional consequence of SpiD7-induced RELA crosslinking (stapling), we subjected two sets of cancer cells to either DMSO, 19 or SpiD7. One set of cells were then stimulated with TNFα while the other served as the control. Following a 15 min incubation, the lysates were fractionated and the cytoplasmic and nuclear fractions of all the treated and control samples were subjected to WB analyses (Fig. 4). Consistent with the above studies we observed high MW RELA bands only in the cytoplasmic fraction of the SpiD7-treated lanes. Consistent with in vitro studies we observed a reduction in the IKKβ levels in the SpiD7-treated cells. As expected in the control lanes (1 and 4) of the cytoplasmic fraction, TNFα stimulation resulted in reduced IκBα levels due to ubiquitination-dependent degradation. This reduction was modestly inhibited in 19-treated cells, while it was completely blocked in the SpiD7-treated cells (lanes 4–6, Fig. 4A). Conversely, as expected, in the control lanes (7 and 10) of the nuclear fraction TNFα stimulation resulted in increased levels of RELA. This increase was modestly diminished in 19-treated cells and completely lost (compare unstimulated control lane 7 and lane 12) in the SpiD7-treated cells (lanes 10–12, Fig. 4A). Together these data show that stapling RELA to its binding partners by SpiD7 resulted in potent inhibition of TNFα-induced RELA translocation. Consistent with this observation, SpiD7 inhibited TNFα-induced RELA activity in a luciferase reporter assay in a dose-dependent manner (Fig. 4B).
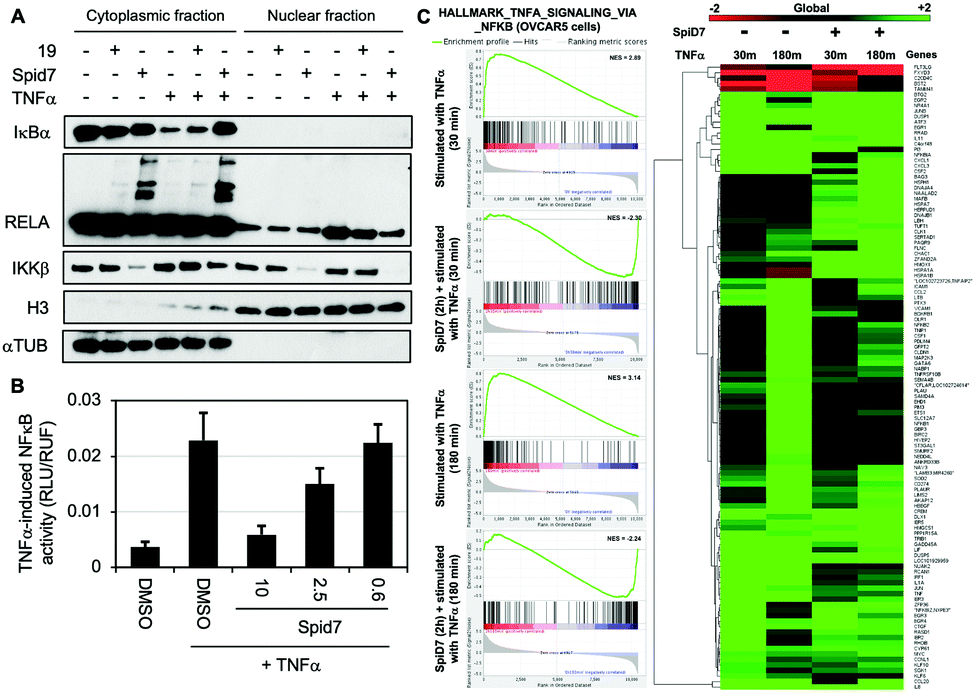 | ||
| Fig. 4 Functional consequence of RELA stapling by SpiD7 (A) Cancer cells (OVCAR5) were treated with and without 19 or SpiD7 (10 μM) and either stimulated with TNFα (20 ng mL−1) for 15 min or held as control. Cytoplasmic and nuclear cell fractions were subjected to WB analyses using antibodies for the indicated proteins. (B) Dose–response study with SpiD7 to assess inhibition of TNFα-induced RELA activation in a dual luciferase reporter assay. (C) OVCAR5 cells treated with either DMSO or SpiD7 (10 μM) and stimulated by TNFα (20 ng mL−1) for 30 min or 180 min. Gene Set Enrichment Analysis (GSEA) for the Hallmark TNFα_signaling_via_NFκB (200) gene set. Euclidean hierarchical clustering of the log2-fold change of genes, where at least one of the treatment comparisons was significantly different (FDR adjusted p-value <0.05). | ||
We conducted RNA-seq studies to assess the changes in RNA levels upon TNFα stimulation in the presence and absence of SpiD7. OVCAR5 cells were treated with SpiD7 for 2 h and stimulated with TNFα for 30 min or 180 min. Isolated RNA from these samples were subjected to RNA-seq analysis (Fig. 4C). Gene set enrichment analysis (GSEA) for Hallmark_TNFα_signaling_via_NFκB yielded normalized enrichment scores (NES) of +2.89 and +3.14 for the TNFα stimulated samples, while the SpiD7 treatment followed by TNFα stimulation yielded −2.30 and −2.24 NES. The heat map provides an unbiased view of changes wherein one or more of the treatments resulted in a significant difference. Representative genes that are known to be regulated by TNFα stimulation at 30 min and 180 min, are NFKBIA (IκBα) and NFKB1 (p105), respectively.32 As expected, TNFα induced NFKBIA (IκBα) expression is inhibited by SpiD7 at the 30 min time point (compare lanes 1 and 3 of the heat map) and NFKB1 which is elevated at the 180 min timepoint, is inhibited by SpiD7 (compare lanes 2 and 4 of the heatmap). Together these data strongly suggest that SpiD7 inhibits TNFα-induced NF-κB transcription. We conducted a growth inhibition assay with OVCAR8 cells to determine the relative potency of SpiD7 and Analog 19 (Fig. S4, ESI†). The results showed that SpiD7 was ∼15-fold more potent than 19. We also show that SpiD7 selectively induced Caspase-7 and PARP cleavage in OVCAR5 but not in FTE282C11 cells (Fig. S5, ESI†).
In conclusion, here we report the Cys residues on RELA and IKKβ targeted by the α-methylene-γ-butyrolactone-containing spirocyclic compound 19. The spirocyclic architecture allows targeting Cys residues that are not accessible to the fused α-methylene-γ-butyrolactone-containing sesquiterpene natural products or their analogues. We also show that SpiD7, a dimer of 19, staples RELA to its binding partners to yield stable high MW bands. Characterization of the stable high MW bands are currently underway and will be reported in due course. More importantly, SpiD7 inhibits TNFα-induced nuclear translocation of RELA resulting in the blockade of NF-κB gene transcription, through a previously unexplored modality.
Conflicts of interest
A. N. and S. R. are listed as inventors on US patent 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 104684.
104684.
Acknowledgements
This work was supported in part by NIH grants CA197999, CA127297 and CA036727. S. Ko was supported by a predoctoral UNMC fellowship. Research resources at UNMC are supported in part by the Nebraska Research Initiative. Biochemistry research at SDSU is supported in part by the California Metabolic Research Foundation. We would like to thank the Natarajan lab members for helpful discussions.References
- M. S. Hayden and S. Ghosh, Cell, 2008, 132, 344–362 CrossRef CAS PubMed.
- D. F. Lee, H. P. Kuo, C. T. Chen, J. M. Hsu, C. K. Chou, Y. Wei, H. L. Sun, L. Y. Li, B. Ping, W. C. Huang, X. He, J. Y. Hung, C. C. Lai, Q. Ding, J. L. Su, J. Y. Yang, A. A. Sahin, G. N. Hortobagyi, F. J. Tsai, C. H. Tsai and M. C. Hung, Cell, 2007, 130, 440–455 CrossRef CAS.
- B. S. Harrington and C. M. Annunziata, Cancers, 2019, 11, 1182 CrossRef CAS.
- W. Wang, J. L. Abbruzzese, D. B. Evans, L. Larry, K. R. Cleary and P. J. Chiao, Clin. Cancer Res., 1999, 5, 119–127 CAS.
- K. Vlantis, A. Wullaert, Y. Sasaki, M. Schmidt-Supprian, K. Rajewsky, T. Roskams and M. Pasparakis, J. Clin. Invest., 2011, 121, 2781–2793 CrossRef CAS PubMed.
- E. Maniati, M. Bossard, N. Cook, J. B. Candido, N. Emami-Shahri, S. A. Nedospasov, F. R. Balkwill, D. A. Tuveson and T. Hagemann, J. Clin. Invest., 2011, 121, 4685–4699 CrossRef CAS PubMed.
- H. Shaked, L. J. Hofseth, A. Chumanevich, A. A. Chumanevich, J. Wang, Y. Wang, K. Taniguchi, M. Guma, S. Shenouda, H. Clevers, C. C. Harris and M. Karin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14007–14012 CrossRef CAS PubMed.
- J. Ling, Y. Kang, R. Zhao, Q. Xia, D. F. Lee, Z. Chang, J. Li, B. Peng, J. B. Fleming, H. Wang, J. Liu, I. R. Lemischka, M. C. Hung and P. J. Chiao, Cancer Cell, 2012, 21, 105–120 CrossRef CAS PubMed.
- Y. Xia, N. Yeddula, M. Leblanc, E. Ke, Y. Zhang, E. Oldfield, R. J. Shaw and I. M. Verma, Nat. Cell Biol., 2012, 14, 257–265 CrossRef CAS.
- M. Naramura and A. Natarajan, Pancreas, 2018, 47, e27–e29 CrossRef.
- J. A. Prescott and S. J. Cook, Cells, 2018, 7, 115 CrossRef CAS PubMed.
- M. Suhail, M. Tarique, N. Muhammad, H. Naz, A. Hafeez, T. A. Zughaibi, M. A. Kamal and M. Rehan, Curr. Med. Chem., 2021, 28, 4117–4132 CrossRef CAS.
- T. D. Gilmore and M. Herscovitch, Oncogene, 2006, 25, 6887–6899 CrossRef CAS.
- S. Sagar, S. Singh, J. R. Mallareddy, Y. A. Sonawane, J. V. Napoleon, S. Rana, J. I. Contreras, C. Rajesh, E. L. Ezell, S. Kizhake, J. C. Garrison, P. Radhakrishnan and A. Natarajan, Eur. J. Med. Chem., 2021, 222, 113579 CrossRef CAS PubMed.
- S. Rana, S. Kour, Y. A. Sonawane, C. M. Robb, J. I. Contreras, S. Kizhake, M. Zahid, A. R. Karpf and A. Natarajan, Chem. Biol. Drug Des., 2020, 96, 773–784 CrossRef CAS.
- S. Rana, E. C. Blowers, C. Tebbe, J. I. Contreras, P. Radhakrishnan, S. Kizhake, T. Zhou, R. N. Rajule, J. L. Arnst, A. R. Munkarah, R. Rattan and A. Natarajan, J. Med. Chem., 2016, 59, 5121–5127 CrossRef CAS PubMed.
- P. Radhakrishnan, V. C. Bryant, E. C. Blowers, R. N. Rajule, N. Gautam, M. M. Anwar, A. M. Mohr, P. M. Grandgenett, S. K. Bunt, J. L. Arnst, S. M. Lele, Y. Alnouti, M. A. Hollingsworth and A. Natarajan, Clin. Cancer Res., 2013, 19, 2025–2035 CrossRef CAS PubMed.
- V. C. Bryant, G. D. Kishore Kumar, A. M. Nyong and A. Natarajan, Bioorg. Med. Chem. Lett., 2012, 22, 245–248 CrossRef CAS.
- J. V. Napoleon, S. Singh, S. Rana, M. Bendjennat, V. Kumar, S. Kizhake, N. Y. Palermo, M. M. Ouellette, T. Huxford and A. Natarajan, Chem. Commun., 2021, 57, 4678–4681 RSC.
- T. Huxford and G. Ghosh, Cold Spring Harbor Perspect. Biol., 2009, 1, a000075 Search PubMed.
- S. Malek, Y. Chen, T. Huxford and G. Ghosh, J. Biol. Chem., 2001, 276, 45225–45235 CrossRef CAS.
- F. Liu, Y. Xia, A. S. Parker and I. M. Verma, Immunol. Rev., 2012, 246, 239–253 CrossRef PubMed.
- T. Huxford, D. B. Huang, S. Malek and G. Ghosh, Cell, 1998, 95, 759–770 CrossRef CAS PubMed.
- S. M. Marino and V. N. Gladyshev, J. Mol. Biol., 2010, 404, 902–916 CrossRef CAS.
- J. C. Widen, A. M. Kempema, J. W. Baur, H. M. Skopec, J. T. Edwards, T. J. Brown, D. A. Brown, F. A. Meece and D. A. Harki, ChemMedChem, 2018, 13, 303–311 CrossRef CAS.
- J. C. Widen, A. M. Kempema, P. W. Villalta and D. A. Harki, ACS Chem. Biol., 2017, 12, 102–113 CrossRef CAS.
- B. H. Kwok, B. Koh, M. I. Ndubuisi, M. Elofsson and C. M. Crews, Chem. Biol., 2001, 8, 759–766 CrossRef CAS PubMed.
- M. Yamamoto, R. Horie, M. Takeiri, I. Kozawa and K. Umezawa, J. Med. Chem., 2008, 51, 5780–5788 CrossRef CAS.
- E. Zandi, D. M. Rothwarf, M. Delhase, M. Hayakawa and M. Karin, Cell, 1997, 91, 243–252 CrossRef CAS PubMed.
- G. Chen, P. Cao and D. V. Goeddel, Mol. Cell, 2002, 9, 401–410 CrossRef CAS.
- S. Rana and A. Natarajan, Org. Biomol. Chem., 2013, 11, 244–247 RSC.
- B. Tian, D. E. Nowak and A. R. Brasier, BMC Genomics, 2005, 6, 137 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00183c |
| ‡ Equal contribution. |
| § Current address: SR (NIH/NCATS, MD). |
| This journal is © The Royal Society of Chemistry 2022 |