
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe CSY-protecting group in the microwave-assisted synthesis of aggregation-prone peptides†
Truc Lam
Pham
 ab,
Jennifer
Zilke
ab,
Christine Charlotte
Müller
c and
Franziska
Thomas
*ab
ab,
Jennifer
Zilke
ab,
Christine Charlotte
Müller
c and
Franziska
Thomas
*ab
aInstitute of Organic Chemistry, Heidelberg University, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: franziska.thomas@oci.uni-heidelberg.de
bCentre for Advanced Materials, Heidelberg University, Im Neuenheimer Feld 225, 69120 Heidelberg, Germany
cInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1a, 52074 Aachen, Germany
First published on 18th February 2022
Abstract
This report describes the application of cyanosulfurylide (CSY)-protected aspartatic acid building blocks in microwave-assisted synthesis of aggregation-prone protein domains. We present a synthesis of Fmoc-Asp(CSY)-OH on a multigram scale, as well as procedures for the microwave-assisted synthesis of CSY-protected peptides, and CSY cleavage in partially folded or aggregation-prone peptides.
 Franziska Thomas | Franziska Thomas studied chemistry at Humboldt University Berlin, where she received her PhD in 2010. After a postdoctoral stay at the University of Bristol, UK, where she worked with Dek Woolfson, she became an independent research group leader at the University of Göttingen in 2015. In 2019, she became a junior professor of organic chemistry at the University of Heidelberg. Her research interests include protein design, de novo design of functional peptide entities, and identification of small molecules and peptides as drug candidates for the potential treatment of amyotrophic lateral sclerosis. |
Due to recent advances in solid-phase peptide synthesis (SPPS) such as the development of new solid supports1 and the microwave-assisted SPPS,2 peptide sequences with more than fifty amino acid residues can be readily prepared.3 However, a long-standing problem in Fmoc-based peptide synthesis is the aspartimide formation due to side-chain backbone cyclization of aspartic acid (Asp) (Fig. 1).4 This side reaction is particularly pronounced when the downstream amino acid is glycine (Gly).5 Attempts to reduce aspartimide formation include the addition of acids to the Fmoc deprotection solution,6 the use of sterically hindered Asp side-chain protecting groups,7 or backbone amide protection.8 Unfortunately, these methods either cannot completely suppress aspartimide formation or significantly reduce the coupling efficiency.4 It has been shown that a recently developed Asp protecting group, the cyanosulfurylide (CSY) group (Fig. 1(IV)), can prevent undesired aspartimide formation.9 Instead of an ester bond susceptible to nucleophilic cleavage by the backbone amide, the CSY group masks the carboxylate by a carbon–carbon bond that is stable under all relevant conditions of SPPS. After final cleavage of the peptide, CSY is removed under oxidative conditions with N-chlorosuccinimide (NCS). As a pleasant side effect, the polar CSY group increases the solubility of the synthesized peptide and counteracts aggregation on a solid support, an effect also observed when using methionine sulfoxide building blocks in peptide synthesis.10
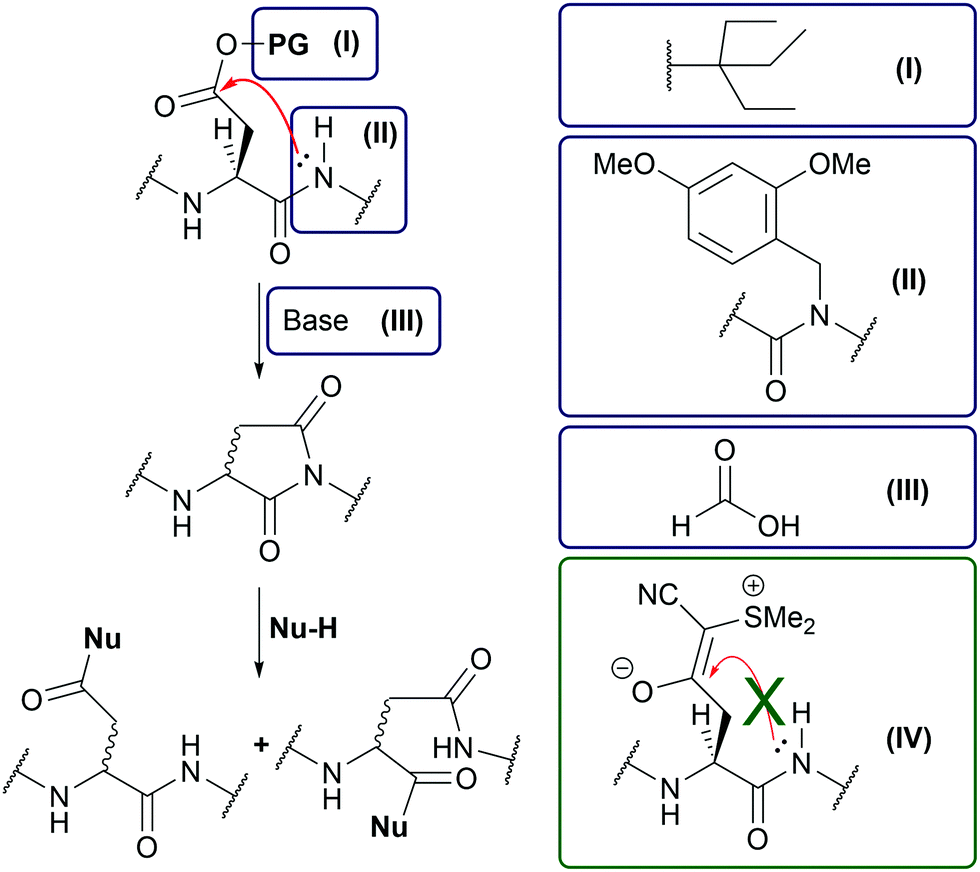 | ||
| Fig. 1 Mechanism of aspartimide formation and strategies to prevent it. Classical attempts: (I) Bulky β-carboxyl protection group, (II) backbone protection group, (III) addition of acid to the deprotection solution. (IV) CSY protection group. | ||
Encouraged by this report, we intended to apply this new Asp building block to the synthesis of β-sheet peptides (sequences Table 1) that contain Asp in their sequences and that we could not previously access using standard peptide synthesis protocols. Peptides 1 and 2 are derived from WW domains that are involved in neuronal signal transduction and are linked to Alzheimer-dementia11 and Golabi-Ito-Hall syndrome,12 respectively. SH3 domain 3 is a part of a proto-oncogenic protein.13 Peptides 1 and 2 have only been expressed but never chemically synthesized,12,14 whereas chemical synthesis of peptide 3 was achieved by chemical ligation of two fragments.15
| Peptide | Description | Sequence |
|---|---|---|
| 1 | hPin1 WW domain (6–39), Met15Nle, Arg17_Ser19delinAlaAsp |

|
| 2 | PQBP1 WW domain (47–80), Cys60Ser |
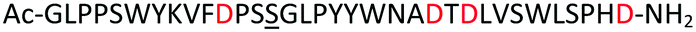
|
| 3 | c-Crk N-terminal SH3 domain (134–190), Met181Nle |

|
| 4 | Test peptide |
|
As our target peptides contain many Asp residues, we first optimized the synthesis of Fmoc-Asp(CSY)-OH to be feasible on a multigram scale with inexpensive reagents (see Fig. 2A). The CSY salt was generated in situ and coupled to Fmoc-Asp-OtBu using HBTU. The resulting Fmoc-Asp(CSY)-OtBu was isolated in 95% yield. Removal of the tert-butyl group was carried out with formic acid. After recrystallization from ethyl acetate, the desired product Fmoc-Asp(CSY)-OH was isolated in 88% yield (overview of costs compared to the original procedure, see ESI†). With the building block in hand, we performed the automated microwave-assisted solid-phase synthesis of the Asp-Gly-containing WW domain 1. After acidic cleavage and deprotection, the crude peptide was analyzed by HPLC and MALDI-TOF-MS. Surprisingly, a significant amount of aspartimide- and piperidine amide-containing peptides were identified (Fig. 2B and C). The main peak of the chromatogram contains a mixture of the desired CSY-protected peptide and the aspartimide-containing side product (Fig. 2D). Peptides completely lacking the CSY group were not found.
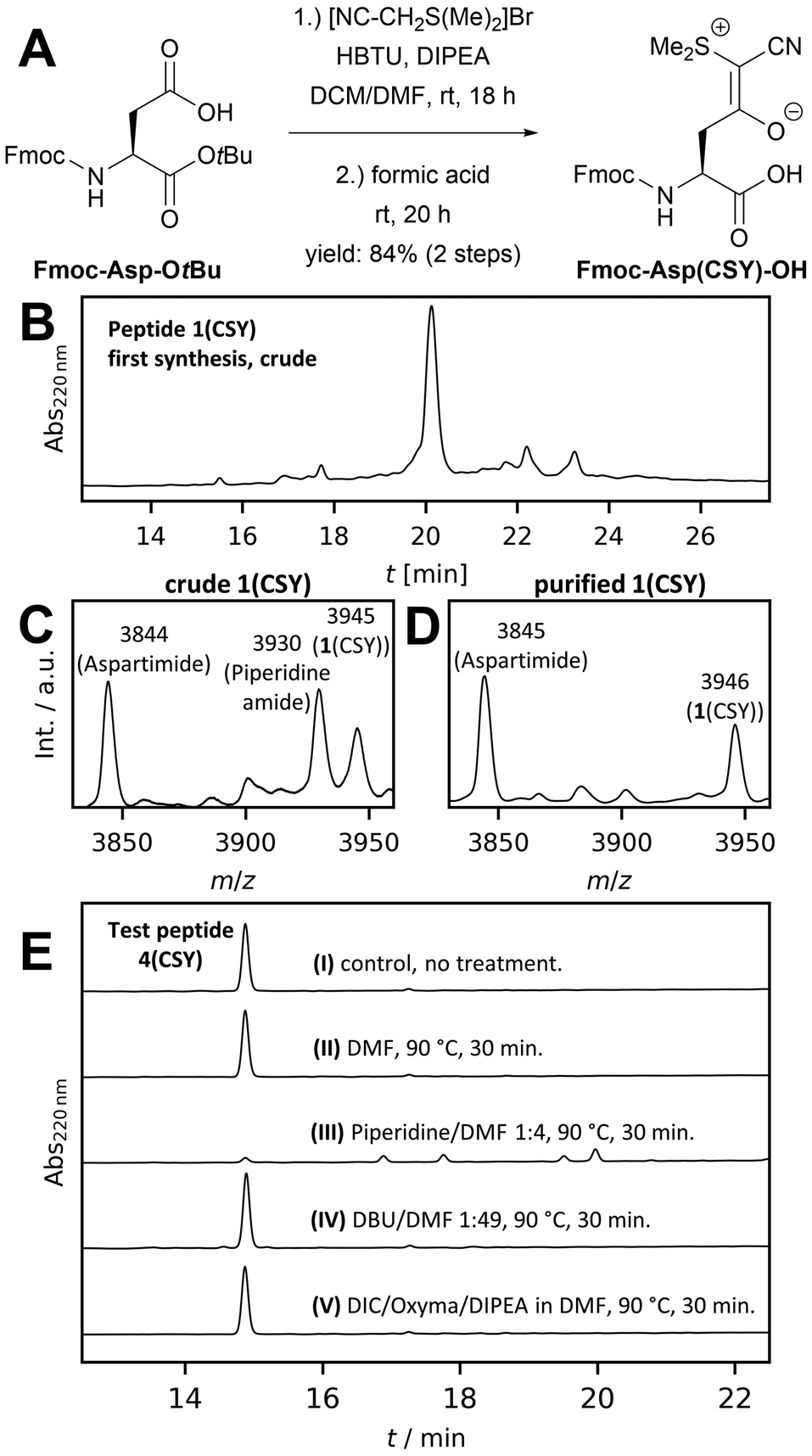 | ||
| Fig. 2 Optimization of the building block and microwave-assisted solid-phase synthesis. (A) Synthesis of Fmoc-Asp(CSY)-OH (DCM: dichloromethane). (B) HPLC of crude WW domain 1 syntesized with standard procedure (coupling and deprotection at 90 °C). (C) MALDI-TOF-MS of crude 1. (D) MALDI-TOF-MS of main HPLC peak. (E) Treatment of test peptide 4 on resin under different conditions. HPLC traces of crude peptide after cleavage. | ||
A closer look at the original work showed that the authors synthesized their peptides only at room temperature.9 Therefore, we hypothesized that microwave-heating would lead to undesired aspartimide formation. To investigate this issue in depth, a short test peptide (4) was synthesized at room temperature to avoid aspartimide formation (crude 4(CSY) see Fig. 2E(I)). The fully protected, resin-bound peptide was then subjected to the various conditions used in microwave-assisted SPPS, cleaved with trifluoroacetic acid (TFA), and analyzed by HPLC and mass spectrometry. Microwave heating in N,N-dimethylformamide (DMF) (Fig. 2E(II)) did not result in decomposition of 4(CSY), demonstrating that CSY in principle is stable at elevated temperatures. However, when 4(CSY) was subjected to microwave heating and Fmoc deprotection solution, decomposition was observed (Fig. 2E(III)). Short-term heating resulted in aspartimide formation and longer heating in the opening of the aspartimide and the formation of piperidine amide (Fig. S1, ESI†). To test whether the basicity and/or nucleophilicity of piperidine is of relevance of the partial CSY cleavage, we treated 4(CSY) with 2% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in DMF under microwave heating (Fig. 2E(IV)). DBU, although a stronger base than piperidine, is not nucleophilic and a well-known reagent for Fmoc deprotection, which induces aspartimide formation even stronger than piperidine.4 Since no aspartimide formation was observed in this case, we assumed that CSY is nucleophilically decomposed by piperidine at elevated temperatures (for the proposed mechanism, see Fig. S2, ESI†) and that DBU would be a good alternative. Exposure of 4(CSY) to N,N-diisopropylcarbodiimide (DIC), oxyma, diispropylethlyamine (DIPEA) and DMF under microwave heating showed that the CSY group is stable to our standard coupling conditions (Fig. 2E(V)), thus, allowing microwave-assisted amino acid coupling.
Based on these results we used a solid-phase synthesis protocol with microwave heating only during the coupling step. The Fmoc deprotection was carried out at room temperature (details see ESI†). After standard microwave-assisted amino acid coupling the resin was washed with DMF to allow it to cool down to room temperature before new deprotection solution was added. This method allowed excellent quality synthesis of test peptide 4 (Fig. S3, ESI†). Next, we synthesized WW domain 1 and obtained a crude product of significantly improved quality than when synthesized using our standard microwave-assisted synthesis protocol (Fig. 3A). Furthermore, the synthesis of even more complex WW domain 2 that contains four aspartate residues and is aggregation-prone succeeded with excellent crude product quality (Fig. 3B). These results encouraged us to chemically synthesize the N-terminal SH3 domain of c-Crk (3), which consists of 57 amino acids and contains six aspartate residues. Again, crude product 3 was obtained in high quality (Fig. 3C). Purification of all CSY-protected peptides by semipreparative HPLC proceeded smoothly.
 | ||
| Fig. 3 Final CSY deprotection and characterization. Chromatograms of the crude and purified peptides of (A) 1(CSY) and 1, (B) 2(CSY) and 2, (C) 3(CSY) and 3. Deprotection conditions: NCS (2.2 eq. regarding amount of CSY, added in four portions), used solvent: (a) ACN, acetate buffer pH 4.5 (according to Neumann et al.).9 (b) HFIP, ACN + 0.1% TFA, water + 0.1% TFA. (c) DMF, HFIP, water + 0.1% TFA. HPLC gradients were identical, but different solvent systems of the sample led to shifts in retention time. CD spectra of (D) 1(CSY) and 1, (E) 2(CSY) and 2, (F) 3(CSY) and 3. | ||
The final step of peptide synthesis with CSY-protected aspartic acid residues involved removal of the CSY group under oxidative conditions. First, we followed the described protocol of Neumann et al. (NCS in acetonitrile (ACN) and buffered aqueous solution).9 However, in the case of peptides 1 to 3, the CSY groups were not completely removed and a significant amount of by-products were formed (Fig. 3). Therefore, we took a step back and tried different oxidants and different solvent systems on Fmoc-Asp(CSY)-OH as a test substrate with NCS proving to be most effective (Fig. S4, ESI†). We then tested different solvent systems for deprotection of peptide 1 (Fig. S5, ESI†). The best procedure included dissolving 1 in water with 10% 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and 0.1% TFA. NCS was dissolved in ACN with 0.1% TFA and four times 0.55 equivalents were added to 1 over 20 min. The reaction was then quenched with sodium ascorbate, diluted with water containing 0.1% TFA, and lyophilized to remove HFIP. The lyophilized peptide was successfully purified by HPLC (see ESI,† peptide characterization section). This procedure was subsequently applied to peptides 2 and 3, but successful deprotection was achieved only in the case of 3 (Fig. 3C). Peptide 2 aggregated, requiring slight modifications to the solvent mixture. This mainly included replacing ACN with DMF as solvent for NCS, while the other conditions remained unchanged. In this case, however, freeze-drying of the mixture was not possible due to the high amount of DMF, and the mixture was purified directly by HPLC giving unprotected 2 in high purity (see ESI†). To test the universality, we applied this procedure to 1(CSY) and 3(CSY) on an analytical scale, which was also successful (Fig. S5, ESI†).
We measured circular dichroism (CD) spectra and thermal denaturation curves of CSY-protected and unprotected peptides 1 to 3 to investigate the influence of the CSY group on peptide folding and structural integrity of 1 to 3 after oxidative deprotection. Interestingly, CSY-protected peptide 1, which contained only one CSY group, was still folded (Fig. 3D), and residual folding was also observed in CSY-protected peptides 2 and 3, which also gave sigmoidal thermal denaturation profiles, albeit with broad folding-to-unfolding transitions (Fig. S6, ESI†). After removal of the CSY groups, folding of peptides 1 to 3 was restored. The CD spectra of the WW domains (1 and 2) showed the typical maximum at 227 nm, which was due to the exciton coupling of the aromatic residues of the hydrophobic core.16 Thermal denaturation analysis also revealed a cooperative folding-to-unfolding transition with melting temperatures of 78.5 °C (1) and 36.3 °C (2), which were significantly higher than those of CSY-protected peptides. The CD spectrum of peptide 3 was characteristic of an SH3 domain structure17 and thermal denaturation revealed steep folding-to-unfolding transition with a melting temperature of 44.5 °C.
The residual folding of CSY-protected peptides explains the need for fluorinated solvents such as HFIP in the CSY cleavage cocktail. We assumed that CSY-protected Asp side chains would be buried and less accessible to the oxidizing agent when the reaction was carried out in pure buffer. HFIP and other fluorinated solvents, however, are known to induce structural change.18 To prove this, we recorded the CD spectra of peptides 1(CSY) and 3(CSY) in both water containing 10% HFIP and 0.1% TFA and in acetate buffer, the solvent used for the deprotection procedure originally reported by Neumann et al.9 Peptide 2(CSY) could not be measured because its solubility was too low under these conditions. While 1(CSY) and 3(CSY) had a similar secondary structure in acetate buffer as in phosphate buffer (Fig. 3D, F and Fig. S7, ESI†), the structure in the water-HFIP mixture was strongly α-helical, as indicated by the characteristic minima at 208 and 222 nm in the respective CD spectra (Fig. S7, ESI†).19 Thus, the CSY-protected aspartates are no longer buried in the folded peptide and can be readily accessed by NCS.
Conclusions
The two most common problems in SPPS are peptide aggregation during synthesis and aspartimide formation, which can naturally coincide.20 Approaches such as the use of bulky protecting groups for the Asp side chain or acidic additions to the Fmoc deprotection solution usually do not solve both problems and can only reduce aspartimide formation to some extent.4,6,7 Protection of the amide backbone addresses both problems, but significantly reduces coupling efficiency, thus requiring the coupling of dipeptide fragments.4,8 The CSY protecting group developed by Neumann et al. completely suppresses aspartimide formation and, due to its polarity, should prevent peptide aggregation during synthesis, representing a breakthrough in this field. However, one should keep in mind that this approach is not suitable for peptides with oxidation-sensitive methionine residues.9We used the CSY-Asp building block for the synthesis of difficult β-sheet peptides, which are prone not only to aspartimide formation but also, in some cases, to aggregation. This required modification of the reported synthesis protocol: First, we established a microwave-assisted synthesis adapted to the latent base sensitivity of the CSY group. Second, we optimized oxidative CSY cleavage on aggregation-prone and partially folded CSY peptides using HFIP as a solvent additive. These adjustments enabled the synthesis of three complex peptides, two WW domains of 33 and 34 amino acids and one SH3 domain of 57 amino acids that contained 1 to 6 aspartic acid residues. The PQBP1 WW domain (2) had never been chemically synthesized before and presented a particular challenge because of its high aggregation propensity. Due to the CSY group, the peptides were obtained in excellent quality of the crude products. The optimized cleavage of the CSY group proceeded quantitatively and without the formation of by-products. We are therefore convinced that the optimized synthesis protocols will greatly expand the applications of the CSY-protected Asp in the synthesis of difficult peptides.
Author contributions
F. T. and T. L. P. designed the project. T. L. P., J. Z. and C. C. M. optimized the synthesis of Fmoc-Asp(CSY)-OH. T. L. P. developed the microwave assisted method for incorporation of Fmoc-Asp(CSY)-OH. T. L. P. and J. Z. synthesised the peptides and optimized the deprotection condition. T. L. P. and J. Z. performed CD spectroscopy. All authors contribute to the analysis of the data. T. L. P. and F. T. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. L. P is grateful to the Fonds der Chemischen Industrie for financial support through a Kekulé Fellowship. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) 414261058 and by the DFG under Germany's Excellence Strategy 2082/1 390761711. We are grateful to Prof. Dr. Christian Klein (Heidelberg University) for providing access to a CD spectrometer.References
- F. García-Martín, M. Quintanar-Audelo, Y. García-Ramos, L. J. Cruz, C. Gravel, R. Furic, S. Côté, J. Tulla-Puche and F. Albericio, J. Comb. Chem., 2006, 8, 213–220 CrossRef PubMed.
- J. M. Collins, K. A. Porter, S. K. Singh and G. S. Vanier, Org. Lett., 2014, 16, 940–943 CrossRef CAS PubMed.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
- R. Subirós-Funosas, A. El-Faham and F. Albericio, Tetrahedron, 2011, 67, 8595–8606 CrossRef.
- J. L. Lauer, C. G. Fields and G. B. Fields, Lett. Pept. Sci., 1995, 1, 197–205 CrossRef CAS.
- T. Michels, R. Dölling, U. Haberkorn and W. Mier, Org. Lett., 2012, 14, 5218–5221 CrossRef CAS PubMed.
- R. Behrendt, S. Huber, R. Martí and P. White, J. Pept. Sci., 2015, 21, 680–687 CrossRef CAS PubMed.
- A.-B. M. Abdel-Aal, G. Papageorgiou, R. Raz, M. Quibell, F. Burlina and J. Offer, J. Pept. Sci., 2016, 22, 360–367 CrossRef CAS PubMed.
- K. Neumann, J. Farnung, S. Baldauf and J. W. Bode, Nat. Commun., 2020, 11, 982 CrossRef CAS PubMed.
- V. Reusche and F. Thomas, ChemBioChem, 2021, 22, 1779–1783 CrossRef CAS PubMed.
- K. P. Lu and X. Z. Zhou, Nat. Rev. Mol. Cell Biol., 2007, 8, 904–916 CrossRef CAS PubMed.
- E. Pucheta-Martinez, N. D’Amelio, M. Lelli, J. L. Martinez-Torrecuadrada, M. Sudol, G. Saladino and F. L. Gervasio, Sci. Rep., 2016, 6, 30293 CrossRef CAS PubMed.
- R. B. Birge, C. Kalodimos, F. Inagaki and S. Tanaka, Cell Commun. Signaling, 2009, 7, 13 CrossRef PubMed.
- M. Jäger, Y. Zhang, J. Bieschke, H. Nguyen, M. Dendle, M. E. Bowman, J. P. Noel, M. Gruebele and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10648–10653 CrossRef PubMed.
- F. Mende, M. Beisswenger and O. Seitz, J. Am. Chem. Soc., 2010, 132, 11110–11118 CrossRef CAS PubMed.
- M. Jäger, M. Dendle and J. W. Kelly, Protein Sci., 2009, 18, 1806–1813 CrossRef PubMed.
- J. A. Camarero, D. Fushman, S. Sato, I. Giriat, D. Cowburn, D. P. Raleigh and T. W. Muir, J. Mol. Biol., 2001, 308, 1045–1062 CrossRef CAS PubMed.
- N. Hirota, Y. Goto and K. Mizuno, Protein Sci., 1997, 6, 416–421 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional spectroscopic data (CD and NMR spectra), characterization of synthesized peptides (HPLC chromatograms and MALDI-TOF mass spectra). See DOI: 10.1039/d1cb00252j |
| This journal is © The Royal Society of Chemistry 2022 |