
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeveloping crosslinkers specific for epimerization domain in NRPS initiation modules to evaluate mechanism†
Woojoo E.
Kim‡
 a,
Fumihiro
Ishikawa‡
*b,
Rebecca N.
Re
a,
Takehiro
Suzuki
c,
Naoshi
Dohmae
c,
Hideaki
Kakeya
d,
Genzoh
Tanabe
*b and
Michael D.
Burkart
*a
a,
Fumihiro
Ishikawa‡
*b,
Rebecca N.
Re
a,
Takehiro
Suzuki
c,
Naoshi
Dohmae
c,
Hideaki
Kakeya
d,
Genzoh
Tanabe
*b and
Michael D.
Burkart
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla CA 92093-0358, USA. E-mail: mburkart@ucsd.edu
bFaculty of Pharmacy, Kindai University, 3-4-1 Kowakae, Higashi-osaka, Osaka 577-8502, Japan. E-mail: ishikawa@phar.kindai.ac.jp; g-tanabe@phar.kindai.ac.jp
cBiomolecular Characterization Unit, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
dGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo, Kyoto, 606-8501, Japan
First published on 27th January 2022
Abstract
Nonribosomal peptide synthetases (NRPSs) are complex multi-modular enzymes containing catalytic domains responsible for the loading and incorporation of amino acids into natural products. These unique molecular factories can produce peptides with nonproteinogenic D-amino acids in which the epimerization (E) domain catalyzes the conversion of L-amino acids to D-amino acids, but its mechanism remains not fully understood. Here, we describe the development of pantetheine crosslinking probes that mimic the natural substrate L-Phe of the initiation module of tyrocidine synthetase, TycA, to elucidate and study the catalytic residues of the E domain. Mechanism-based crosslinking assays and MALDI-TOF MS were used to identify both H743 and E882 as the crosslinking site residues, demonstrating their roles as catalytic bases. Mutagenesis studies further validated these results and allowed the comparison of reactivity between the catalytic residues, concluding that glutamate acts as the dominant nucleophile in the crosslinking reaction, resembling the deprotonation of the Cα-H of amino acids in the epimerization reaction. The crosslinking probes employed in these studies provide new tools for studying the molecular details of E domains, as well as the potential to study C domains. In particular, they would elucidate key information for how these domains function and interact with their substrates in nature, further enhancing the knowledge needed to assist combinatorial biosynthetic efforts of NRPS systems to produce novel compounds.
Introduction
In nature, microorganisms and plants produce secondary metabolites as a function of survival and protection against other organisms.1,2 Included among these natural products are peptide secondary metabolites known as nonribosomal peptides, which are synthesized by large, multi-modular enzyme machineries called nonribosomal peptide synthetases (NRPSs). These compounds exhibit a wide variety of potent clinical bioactivities for human health, functioning as antibiotics, immunosuppressants, and anticancer reagents, among others.1 NRPSs are commonly very large proteins composed of several modules, each of which contains multiple catalytic domains responsible for recognizing, activating, and incorporating one amino acid at a time, constructing a final peptide in assembly line fashion. Recent evidence indicates that protein–protein interactions between a peptidyl carrier protein domain (PCP), which loads substrates and covalently chaperones intermediates within the pathway, and the enzymatic domains within each module are crucial for monomer fidelity and biosynthetic organization.3,4 In the last decade, there has been a lot of interest in engineering these NRPS pathways to yield redesigned peptides but it has proved challenging for many groups.5,6 Continuing to gain a full understanding for how the individual domains function and interact within these systems will therefore be instrumental in their manipulation.Among the unique abilities of NRPSs, these pathways can incorporate nonproteinogenic D-amino acids into final natural products, creating unique conformations that enable bioactivity and avoid protease degradation of peptide natural products.7 While D-amino acids are also present in ribosomally synthesized and post-translationally modified peptides (RiPPs) through an epimerization reaction catalyzed by radical-S-adenosylmethionine (SAM) enzymes, their incorporation in NRPSs can follow various routes.8D-amino acid monomers can be directly recognized and activated by an adenylation (A) domain to serve as a building block, as in the case of cyclosporine A biosynthesis.9 More recently, crystal structures of two thioesterases, NocTE and Skyxy-TE, revealed the key residues involved in their unusual ability to also catalyze an epimerization reaction to include D-amino acids into their linear and cyclic peptides, specifically in norcardicin and skyllamycin biosynthesis, respectively.10,11 However, the majority of NRPS-incorporated D-amino acids are converted from L-amino acids subsequent to covalent loading on PCPs through the use of auxiliary epimerization (E) domains. Unlike amino acid racemases, which utilize the PLP cofactor, E domains catalyze epimerization without the use of cofactors.12 Despite multiple biochemical, structural, and computational studies, the fundamental mechanism of the E domains remains hypothetical and has yet to be fully characterized.
Mutational studies of the E domain within the initiation module of the gramicidin S synthetase (GrsA) reported that His753 and Glu892 act as a base/acid to deprotonate and re-protonate Cα-H.13 While the H753A mutant completely abrogates activity, the mutant E892A only diminishes its activity by ∼6-fold relative to wild-type GrsA, suggesting that His753 functions as a base catalyst.13 This study also identified that epimerization occurs in both the forward and reverse directions until an equilibrium of 1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 D- to L-amino acid is reached.13
:1 D- to L-amino acid is reached.13
Crystal structures of the excised E domain of the initiation module of the tyrocidine synthetase, TycA (A-PCPPhe-E), and the PCP-E didomain of GrsA have been reported.14,15 Both support the homology prediction that E domains are similar in structure to condensation (C) domains.16,17 The PCP-E didomain structure of GrsA was solved in apo- and holo- form, identifying the protein interface and how PCP can be oriented to deliver the substrate-loaded 4′-phosphopantetheine arm towards the active site of the E domain.15 Based on the active-site geometry of the TycA E domain, Samel et al. proposed that Glu882 is acting as an acid/base catalyst, whereas His743 stabilizes a transient enolate intermediate during the L- to D- and D- to L-amino acid isomerization.14 From structure-based calculations of protonation states of the E domain of GrsA, catalytic residue H753 was calculated to have a local pKa of 7.8–9.0. This pKa prediction shows that histidine remains in its protonated state, suggesting that Glu882 may act as a base catalyst (Fig. 1).18 Despite these previous biochemical, structural, and computational studies, several questions related to E domain substrate recognition, binding, and detailed chemical mechanism remain to be addressed.
 | ||
| Fig. 1 The proposed mechanism of the E domain in TycA. Glu882 is hypothesized to deprotonate Cα-H of L-Phe, which can then be reprotonated by His743 to form D-Phe. In the reverse process, it is plausible that H743 acts as the base in the reaction. | ||
Mechanism-based crosslinking probes have been used to probe protein–protein interactions in NRPSs.19,20 Recently, Aldrich and co-workers utilized crosslinking probes to study the interactions between the PCP and C domains19 and Eguchi and co-workers applied crosslinking probes to obtain structural information of PCP-A domain complexes.20 Here we introduce the development of new crosslinking probes and their application with mutagenesis studies to investigate the NRPS E domain mechanism. We had previously identified chlorovinylglycine, a mechanism-based inhibitor of alanine racemase, as a PCP and E domain crosslinker.18 However, it revealed only modest crosslinking activity and proved difficult to evaluate due to rapid hydrolysis. In this study, we describe the design, synthesis, and evaluation of sulfonate crosslinking probes that, when tethered to the PCP in an initiation module, allow capture of the PCP-E bound complex and allow us to further elucidate the biochemical mechanism of the E domain.
Probe development and activity
Weerapana et al. recently reported proteome reactivity profiles for a phenylsulfonate-based probe using a mass spectrometry platform referred to as tandem orthogonal proteolysis-activity-based protein profiling (ABPP) for simultaneous identification of protein targets and sites of probe modification.21 This study demonstrated that the probe displays unique reactivity with several amino acids, including Asp, Cys, Glu, His, and Tyr.21 Tsukiji et al. developed this ABPP probe further and published ligand-directed tosyl chemistry as a way to introduce synthetic probes nongenetically onto a protein in vivo.22 This method utilizes a protein ligand and synthetic probe that are connected by a phenylsulfonate group. The ligand binds tightly to the protein, where it situates the electrophilic phenylsulfonate moiety in close proximity to a nucleophilic amino acid (e.g. His) for an SN2-type chemical reaction to occur. Inspired by these findings, we sought to utilize a sulfonate warhead to target the active site residues, specifically Glu and/or His, in the E domain. We reasoned that preparing a crosslinker with a pantetheinamide sulfonate warhead would allow it to be loaded onto a PCP, which will situate the sulfonate warhead towards the active site residues (Glu and/or His) of an E domain (Fig. 2A).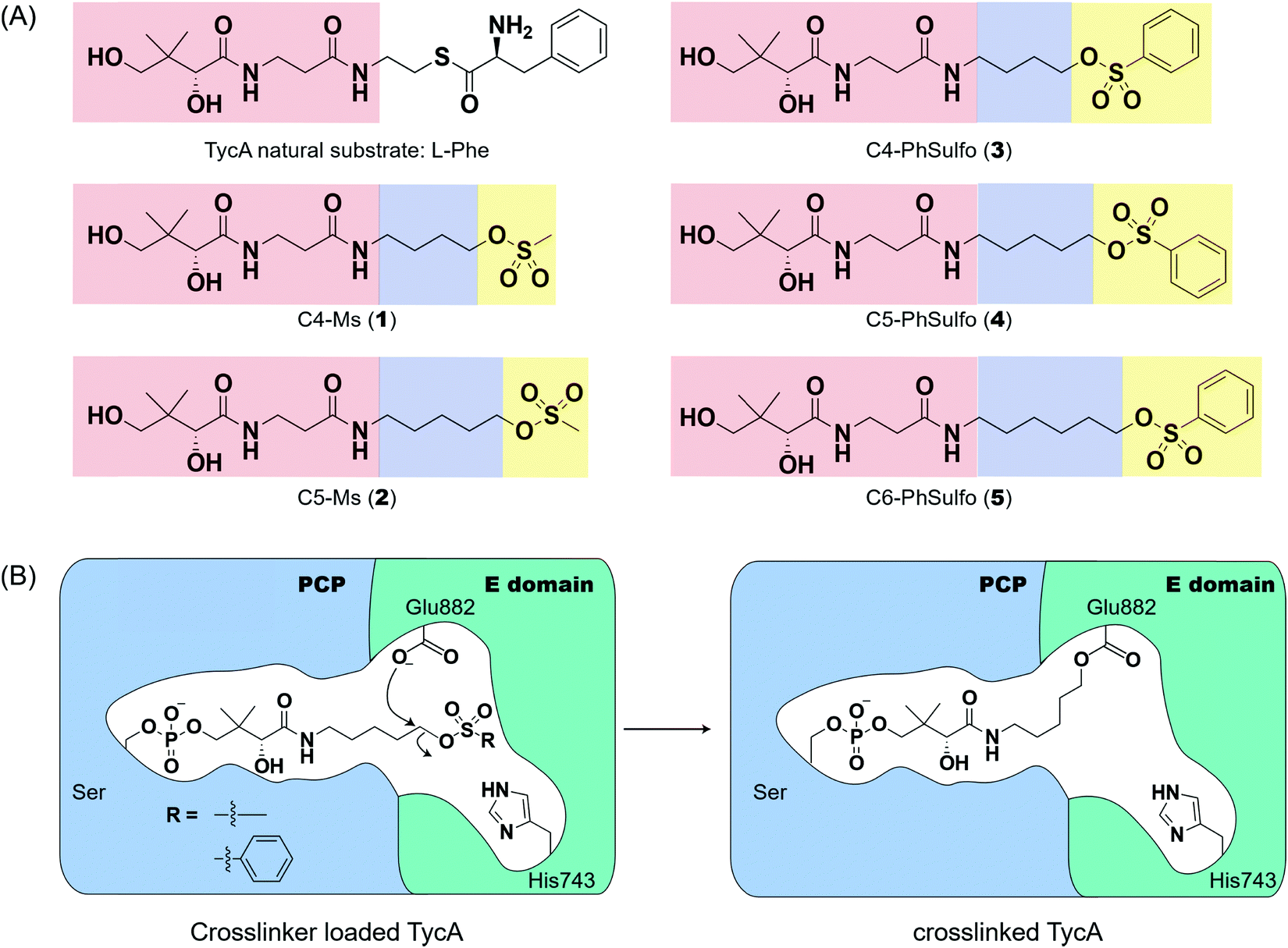 | ||
| Fig. 2 Crosslinkers designed to target the catalytic residues of the E domain to study its mechanism. (A) The panel of pantetheine analog crosslinkers with different linker lengths and warheads. Red: pantetheine portion, blue: linker, yellow: warhead. (B) Proposed mechanism of the crosslinker targeting the catalytic residue Glu882 of the TycA E domain. | ||
Our biochemical studies began by examining the ability of probes 1 and 2 to form a covalent linkage between the PCP and the E domain of TycA (Fig. 2B). Probes 1 and 2 (500 μM) were chemoenzymatically loaded onto the PCP of TycA using CoA biosynthetic enzymes CoaA, CoaD, and CoaE and the promiscuous 4′-phosphopantetheinyl transferase (PPTase), Sfp,23 followed by intramolecular crosslinking. Sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis demonstrated the clear crosslinking of TycA after 12 h at 25 °C. These crosslinked products resulted in observed gel shifts of apo-TycA band from ∼116 kDa to an apparent ∼160 kDa for the intramolecular crosslinked complex. We have previously demonstrated this gel shift phenomenon with crosslinking of other large synthases.24 We speculate that this phenomenon is due to the large size of the protein (124 kDa), such that when it is intramolecularly crosslinked, it does not completely denature and therefore runs differently on an SDS-PAGE gel. The observation of a TycA gel shift under these conditions, and only when incubated with Sfp, indicated that probes 1 and 2 facilitated intramolecular crosslinking between the PCP and the E domain (Fig. 3A).
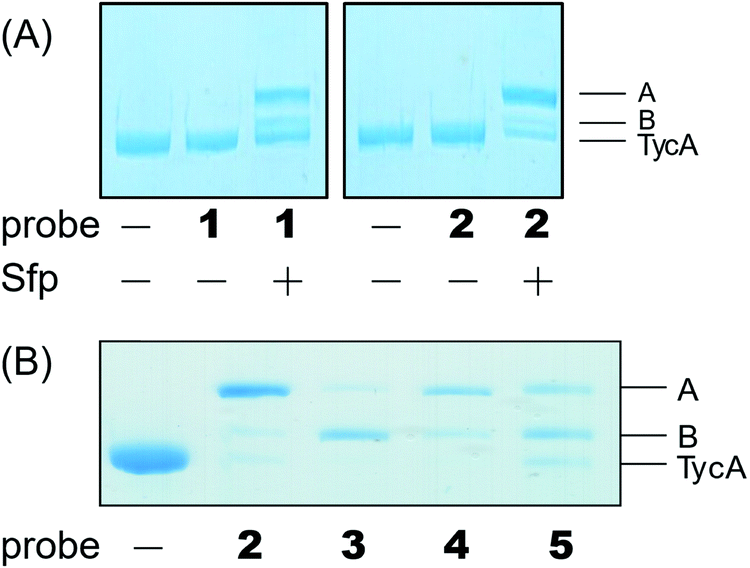 | ||
| Fig. 3 Crosslinking reactions confirmed by running SDS-PAGE gel. (A) Different crosslinked bands (A and B) were shown as gel shifts on SDS-PAGE above apo-TycA with mesylate warhead probes 1 and 2. Crosslinking reactions occurred only when Sfp was present. (B) Crosslinking reaction showed different populations of crosslinking bands depending on probe identity. | ||
The crosslinking experiment resulted in three different bands, two that we presumed to be different crosslinked, gel-shifted TycA complexes (bands A and B) and a third band running similar to that of apo-TycA in a 1:0.27:1.20 ratio for probe 1 and 4.14:0.41:1 ratio for probe 2. To explain these findings, we found that the mesylate moiety has been shown to be degraded by deprotonation to form the sulfene in the presence of a non-nucleophilic base.25 Given that Sfp loading conditions in an excess of probe do not run to completion, we reasoned that the mesylate warhead degraded while tethered to the PCP, accounting for the presence of non-crosslinked TycA along with the crosslinked products.
Second generation probes
Due to the instability of the mesylate warhead, we chose to design additional probes containing a phenylsulfonate moiety that would better mimic the natural substrate L-Phe and also avoid sulfone degradation (Fig. 2A). Interestingly, the crosslinking results showed the two crosslinked band populations (A and B) present above apo-TycA similar to the mesylate probes but in different ratios depending on the linker lengths. This further confirms our hypothesis that we are observing two crosslinked complexes that run differently on a gel (Fig. 3B). Probes 2 and 4, which mimic the length of the natural substrate, displayed similar crosslinking profiles, depicting the majority of the crosslinking bands at the A position. Conversely, probe 3 showed its majority of crosslinking bands at the B position, while probe 5 displayed similar crosslinking populations at both A and B positions.Crosslinking site validation – protein digestion followed by mass spectrometry
In order to locate the actual crosslinking site(s), protease digestion followed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) was performed. When compared with apo-TycA, we were able to identify two fragments that contained covalent crosslinking sites. One of the fragments was located on PCP (DSIQAIQVVAR) and the other on the E domain (DLLLAALGLAFAEWSKLAQIVIHLEGHGRE), both of which were only found in the apo-TycA digestion results and not visible in the crosslinked protein digestion (Fig. S2, ESI†). The fragment from the PCP contained the phosphopantetheinylation site S563 of TycA where the crosslinking probe is covalently loaded onto by Sfp. The other fragment from the E domain contained several nucleophilic residues along with the catalytic residue Glu882, all of which were potential candidates for crosslinking sites that could represent either of the crosslinking bands. We generated six alanine mutants out of the eight nucleophilic residues within the E domain fragment as well as the catalytic residue alanine mutants of the E domain, H743A and E882A. In addition, the double alanine mutant of the catalytic residues, H743A/E882A (HE), was generated to examine if the crosslinking was occurring on both residues.Crosslinking site validation – alanine mutant library crosslinking reaction
We saw an opportunity to use differential site reactivity to probe the active site mechanism and uncover the identity of bands A and B. Crosslinking experiments on the alanine mutant library were performed with 4 and run on an 8% SDS-PAGE gel (Fig. 4A). Interestingly, only the catalytic residue mutants showed differences from wild-type TycA crosslinking. In the case of the wild-type TycA crosslinking reaction with 4, band A showed a larger population than band B (2.6:1). To our surprise, crosslinking reactions performed on the catalytic residue mutants revealed that each led to only one crosslinking band (A or B) seen with the wild-type crosslinking reaction. The crosslinking band for H743A exhibited only band A, while the crosslinking band for E882A displayed only band B but in much higher yield than the wild-type crosslinking. When both catalytic residues were mutated to alanine residues (HE), no crosslinking occurred. This indicated that crosslinking solely occurred with the catalytic residues H743 and E882. It can be speculated that for the crosslinking reaction on the H743A mutant, E882 acts as a nucleophile (indicated by the blue box in Fig. 4B) and for the crosslinking reaction on the E882A mutant, H743 acts as a nucleophile (indicated by the green box in Fig. 4B). We deduced that the differential gel shifts correlate with different internal crosslinking loop sizes between the S563-H743 and S563-E882 crosslinking complexes, which is further corroborated by Dehling et al where they observed different migration behaviors between L- and T-branched NRPS crosslink isomers on SDS-PAGE gels.26
 | ||
| Fig. 4 Pinpointing the actual crosslinking site on the catalytic residues of the E domain of TycA. (A) Crosslinking reaction on alanine mutant library with probe 4. HE refers to H743A/E882A mutant. (B) Schematic depiction of the crosslinking reaction for wild-type TycA (red box), H743A mutant (blue box), and E882A mutant (green box). | ||
E domain activity assay – DKP assay
To further understand the roles of these active site residues, we decided to perform a diketopiperazine (DKP) assay, an in vitro condensation assay that allows us to indirectly study the activity of the E domain.18,27 This assay allows us to quantify the dipeptide formed between the initiation module (TycA) and the subsequent downstream module TycB1 (C-A-PCPPro), which is responsible for the selective uptake of epimerized D-Phe substrate in its condensation reaction. First, the A domains of TycA and TycB1 catalyze the adenylation of L-Phe and L-Pro using ATP, and the activated amino acids are loaded onto their respective PCPs producing L-Phe-TycA and L-Pro-TycB1. If the TycA E domain is functional and able to catalyze the epimerization reaction, it will then convert the L-Phe-TycA to the desired D-Phe-TycA that the TycB1 module can then recognize, mediated by the communication (COM) domain between the two modules.26,28 The acceptor module's C domain will catalyze the peptide bond formation generating D-Phe-L-Pro-TycB1, which undergoes an intramolecular cyclization to yield D-Phe-L-Pro DKP in a stereopreference of 50:1 over its L,L-diastereomer counterpart.13
Using the three key constructs employed in our studies, H743A, E882A, and the double mutant HE, their ability to perform the DKP reaction was tested and compared to that of the wild-type (Fig. 5). NMR analysis of the detected DKP product confirmed the formation of the D-Phe-L-Pro-DKP diastereomer in accordance with published literature.27 As quantified by HPLC, a significant decrease in DKP formation was observed for all the TycA mutants. The H743A mutant exhibited a 99% decrease in DKP product, the E882A mutant had a 99.5% decrease in product, and the double HE mutant showed no DKP formation. This can be explained by the considerable decrease in D-Phe-L-Pro dipeptide formation as a result of mutating the key catalytic residues involved in the epimerization reaction. When each residue is mutated, they individually inhibit the E domain's activity to produce the preferred Phe stereoisomer through this assay. This demonstrates how both active site residues are critical for the epimerization reaction to occur in which both residues are needed to shuttle the proton involved in the reaction as an acceptor or donor, dependent on the direction of equilibrium. However, the DKP assay only indirectly represents E domain activity where the catalytic residue mutants, H743A and E882A, exclusively show the deprotonation half-reaction, which does not fully mirror the wild-type E domain activity. While further structural information is needed to further elucidate the exact roles that each of these residues play, these results support our hypothesis that both residues must function together in a base/acid catalysis in order to perform the L-to-D isomerization. Studies are currently underway to obtain structural data of these crosslinked complexes to better understand their roles.
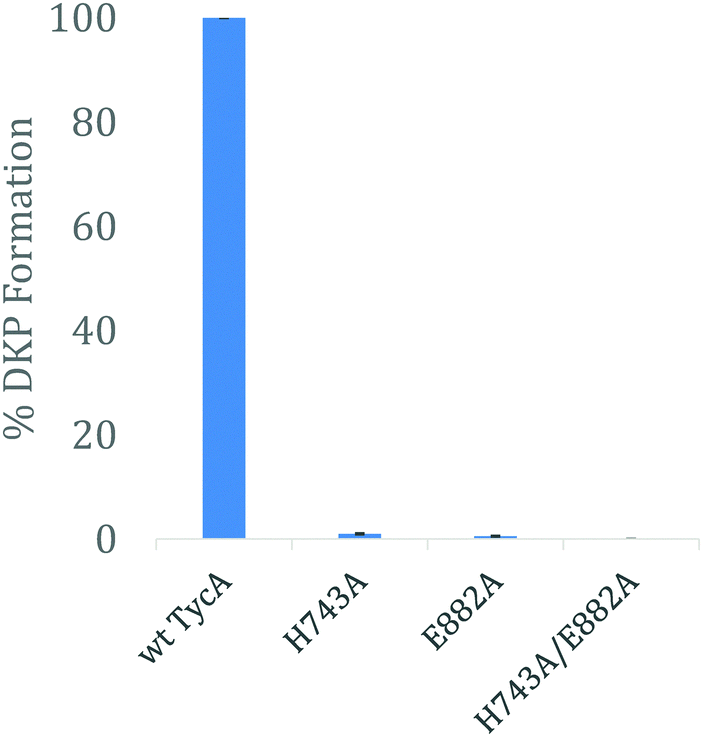 | ||
| Fig. 5 HPLC analysis of DKP formation comparing the effects of the E domain active site mutants on its activity. The error bars represent standard deviation of % DKP formation observed from the assay, performed in triplicate. | ||
To gain insight into the crosslinking domains/sites and the probe's selectivity toward the E domain, we examined the selective labeling of crosslinked TycA (Fig. 6). We have developed ABPP probes toward NRPSs, which enable the selective labeling of the A domains of NRPSs in purified proteins and proteomes (Fig. 6A).29,30L-Phe-AMS-Bpyne, an A domain labelling reagent, (1 μM) was treated with the crosslinking reaction mixtures of TycA and probe 2 for 10 min at room temperature. The sample was then photoactivated with UV light (365 nm) for 30 min at 0 °C, reacted with rhodamine-azide using copper(I)-catalyzed Huisgen's azide–alkyne cycloaddition, and visualized by SDS-PAGE coupled with in-gel fluorescence imaging (Fig. 6B and C, left lanes). To further confirm which domain was being labeled, a secondary study was performed in which the crosslinking reaction mixtures were pre-incubated with L-Phe-AMS, a known A domain inhibitor, (100 μM) for 10 min at room temperature to block the A domain pocket.4 Then, following addition of L-Phe-AMS-BPyne, SDS-PAGE analysis showed how the labeling process of crosslinked TycA was completely inhibited by pre-treatment with L-Phe-AMS (Fig. 6C, right lanes). This demonstrates the presence of an active A domain in the crosslinked TycA. Since the crosslinking band(s) of TycA were unaffected in this process, we can confirm the crosslinking probes to be selective for the substrate binding pocket of the TycA's E domain.
 | ||
| Fig. 6 Crosslinking domain site validation using A domain labeling reagent. (A) The structure of the A domain inhibitor, L-Phe-AMS, and A domain labeling reagent, L-Phe-AMS-BPyne. (B) Schematic depiction showing how A domain labeling reagent functions on a crosslinked TycA construct. (C) SDS-PAGE gel showing the effect of A domain labeling reagent on crosslinked TycA construct with or without A domain inhibitor present. The gel was visualized with fluorescent imager (FL) and Coomassie Brilliant Blue stain (CBB). TycA crosslinking was performed with probe 2. | ||
We next performed additional crosslinking reactions with another initiation module, GrsA, to fully understand the sulfonyl probes’ selectivity (Fig. S8, ESI†). Here, we observed similar crosslinking patterns, which further support the use of these probes to specifically crosslink E domains in these types of modules. Additionally, we tested crosslinking within TycB1, which lacks an E domain, yet still observed a gel-shifted, crosslinking band (Fig. S8, ESI†). Due to the high similarity between E and C domains, we hypothesize that the PCP-loaded probe intramolecularly crosslinked with the C domain. While additional studies are still underway, these results broaden the applicability of the sulfonyl probes in each type of domain, though also limit their use in modules that include both C and E domains.
Conclusions
We have developed pantetheine analog crosslinking probes containing sulfonyl warheads that target catalytic residues (His and Glu) of the E domain in initiation modules of NRPSs. The mechanism of the epimerization reaction in NRPSs is proposed to occur with both catalytic histidine and glutamate residues by deprotonating/reprotonating Cα-H to racemize the tethered amino acid. However, limited assays, such as radiolabeling amino acids, exist to directly measure TycA epimerization, and despite structural evidence, the mechanism remains unclear. Previously, pKa calculations indicated that the catalytic histidine residue was in a protonated state, suggesting that glutamate would act as a base in the epimerization mechanism.14,18 This correlates with our data that in the wild-type TycA crosslinking, band A indicates 2.6 times more crosslinking than band B. For the H743A mutant crosslinking, where the catalytic residue E882 acts as a nucleophile, band A only shows 73% crosslinking compared to that of the wild-type crosslinking band A. However, the E882A mutant crosslinking, where the catalytic residue H743 acts as a nucleophile, shows 2.5 times more crosslinking for band B in comparison to the wild-type crosslinking band B. With the higher crosslinking observed in band A over band B for the wild-type TycA paired with the mutagenesis studies, this indicates that the catalytic glutamate acts as the dominant nucleophile toward the sulfonyl probe compared to histidine, in agreement with recent E domain mechanisms where glutamate serves as the base. Given the hydrophobic nature of the sulfonyl probes, similar to phenylalanine, the pKa of the active site residues would likely be affected through differential acidity and shift toward more normative pKa's.31 This would challenge previous hypotheses of histidine remaining in its protonated state, thereby allowing itself to serve as a base in the epimerization reaction, supporting our results that both residues participate in the crosslinking reaction. The catalytic role of the active site histidine residue can also be seen in the epimerization activity of NocTE and Skyxy-TE.10,11 Since the E domain epimerization is a reversible process as shown in Fig. 1, both catalytic residues must function as both acid and base with water in the active site assisting in proton shuttling between the residues to re-establish their roles in the epimerization for continuous activity. The DKP assay also corroborates that both histidine and glutamate serve as acid and base in the epimerization reaction indicated by the reduced DKP formation when tested with each of the catalytic residue mutants. Our results validate the previous hypothesis on the E domain mechanism where two active site residues, glutamate and histidine, serve as catalytic bases. Additionally, we were able to qualitatively compare the reactivity between those catalytic residues through mechanism-based crosslinking gel assays. For GrsA, the epimerization equilibrium ratio lies at 1.9:1 of D-Phe to L-Phe. This asymmetric equilibrium cannot be easily explained without having actual substrate-bound structural information. With the differing rates of crosslinking we observed for the wild-type and probe 4 in Fig. 4A, we hypothesize that each residue is responsible for acting as the base in one direction of epimerization and that the ratio we see between band A and B (2.6:1) is proportional to the equilibrium constant of D-Phe to L-Phe, 1.9:1. Furthermore, by comparing the mutant crosslinking with the wild-type crosslinking, we concluded that glutamate acts as a dominant nucleophile/base over histidine. This may explain why there is a kinetic preference for one amino acid over the other.
From this study, we were able to directly target the catalytic residues of the E domain by the intramolecular crosslinking of TycA. We revealed that both catalytic residues act as a base/nucleophile towards the probe, where glutamate acts as a dominant nucleophile over histidine, which is consistent with previous work and validates the use of our probes to study the E domain mechanism.14,18 When glutamate is mutated to alanine, histidine by itself can function as a base/nucleophile towards the probe. In addition to the crosslinking observed in two initiation modules of NRPSs as well as C domain crosslinking in TycB1, the sulfonyl probes can be used to study downstream modules containing E domains where the C domain is lacking or deleted. Thus, the development of this crosslinker sets the stage for the next step in studying the molecular basis of the epimerization domain in its modular settings, and this knowledge will ultimately help further combinatorial biosynthetic endeavors to incorporate D-amino acids into novel compounds.
Author contributions
W. E. K., F. I., and M. D. B. designed research; W. E. K., F. I., R. N. R., T. S., and N. D. performed research; W. E. K., F. I., R. N. R., T. S., and N. D. analyzed data; and W. E. K., F. I., R. N. R., H. K., G. T., and M. D. B. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH grant R01 GM095970 (to M. D. B.), a Grants-in-Aid for Scientific Research on Innovative Areas (17H05438 and 19H04664 to F. I.), a Grants-in-Aid for Research (C) (19K05722 to F. I.) from the Japan Society of the Promotion of Science (JSPS), and a grant from the Takeda Science Foundation (F. I.). This work was partly supported by NIH grant F31 GM139327 (to R. N. R.), a Grants-in-Aid for Research (C) (17K08377 to G. T.) from JSPS and by grants from the Noda Institute for Scientific Research (F. I.) and the Hoansha Foundation (G. T.). We are also thankful for the financial support extended by the Antiaging Project for Private Universities. We thank Dr Anthony Mrse and Dr Yongxuan Su for assistance with acquisition of NMR and MS data.References
- G. H. Hur, C. R. Vickery and M. D. Burkart, Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology, Nat. Prod. Rep., 2012, 29, 1074–1098 RSC.
- T. Izoré and M. J. Cryle, The many faces and important roles of protein–protein interactions during non-ribosomal peptide synthesis, Nat. Prod. Rep., 2018, 35, 1120–1139 RSC.
- M. A. Fischbach and C. T. Walsh, Assembly-Line Enzymology for Polyketide and Nonribosomal Peptide Antibiotics: Logic, Machinery, and Mechanisms, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- A. M. Gulick and C. C. Aldrich, Trapping interactions between catalytic domains and carrier proteins of modular biosynthetic enzymes with chemical probes, Nat. Prod. Rep., 2018, 35, 1156–1184 RSC.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Structural, functional and evolutionary perspectives on effective re-engineering of non-ribosomal peptide synthetase assembly lines, Nat. Prod. Rep., 2018, 35, 1210–1228 RSC.
- K. A. Bozhüyük, J. Micklefield and B. Wilkinson, Engineering enzymatic assembly lines to produce new antibiotics, Curr. Opin. Microbiol., 2019, 51, 88–96 CrossRef PubMed.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Comparison of the proteolytic susceptibilities of homologous L-amino acid, D-amino acid, and N-substituted glycine peptide and peptoid oligomers, Drug Dev. Res., 1995, 35, 20–32 CrossRef CAS.
- M. A. Funk and W. A. van der Donk, Ribosomal natural products, tailored to fit, Acc. Chem. Res., 2017, 50, 1577–1586 CrossRef CAS PubMed.
- J. Dittmann, R. M. Wenger, H. Kleinkauf and A. Lawen, Mechanism of cyclosporin A biosynthesis. Evidence for synthesis via a single linear undecapeptide precursor, J. Biol. Chem., 1994, 269, 2841–2846 CrossRef CAS.
- K. D. Patel, F. B. d’Andrea, N. M. Gaudelli, A. R. Buller, C. A. Townsend and A. M. Gulick, Structure of a bound peptide phosphonate reveals the mechanism of nocardicin bifunctional thioesterase epimerase-hydrolase half-reactions, Nat. Commun., 2019, 10, 1–12 CrossRef CAS PubMed.
- J. Yu, J. Song, C. Chi, T. Liu, T. Geng and Z. Cai, et al., Functional Characterization and Crystal Structure of the Bifunctional Thioesterase Catalyzing Epimerization and Cyclization in Skyllamycin Biosynthesis, ACS Catal., 2021, 11, 11733–11741 CrossRef CAS.
- C. T. Walsh, R. V. O’Brien and C. Khosla, Nonproteinogenic Amino Acid Building Blocks for Nonribosomal Peptide and Hybrid Polyketide Scaffolds, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS PubMed.
- T. Stachelhaus and C. T. Walsh, Mutational Analysis of the Epimerization Domain in the Initiation Module PheATE of Gramicidin S Synthetase, Biochemistry, 2000, 39, 5775–5787 CrossRef CAS PubMed.
- S. A. Samel, P. Czodrowski and L.-O. Essen, Structure of the epimerization domain of tyrocidine synthetase A, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1442–1452 CrossRef CAS PubMed.
- W.-H. Chen, K. Li, N. S. Guntaka and S. D. Brunauer, Interdomain and Intermodule Organization in Epimerization Domain Containing Nonribosomal Peptide Synthetases, ACS Chem. Biol., 2016, 11, 2293–2303 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Nonribosomal Peptide Synthesis—Principles and Prospects, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- K. Bloudoff and T. M. Schmeing, Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: discovery, dissection and diversity, Biochim. Biophys. Acta., Proteins Proteomics, 1865, 2017, 1587–1604 Search PubMed.
- W. E. Kim, A. Patel, G. H. Hur, P. Tufar, M. G. Wuo, J. A. McCammon and M. D. Burkart, Mechanistic Probes for the Epimerization Domain of Nonribosomal Peptide Synthetases, ChemBioChem, 2018, 20, 147–152 CrossRef PubMed.
- C. Shi, B. R. Miller, E. M. Alexander, A. M. Gulick and C. C. Aldrich, Design, Synthesis, and Biophysical Evaluation of Mechanism-Based Probes for Condensation Domains of Nonribosomal Peptide Synthetases, ACS Chem. Biol., 2020, 15, 1813–1819 CrossRef CAS PubMed.
- A. Miyanaga, S. Kurihara, T. Chisuga, F. Kudo and T. Eguchi, Structural characterization of complex of adenylation domain and carrier protein by using pantetheine cross-linking probe, ACS Chem. Biol., 2020, 15, 1808–1812 CrossRef CAS PubMed.
- E. Weerapana, G. M. Simon and B. F. Cravatt, Disparate proteome reactivity profiles of carbon electrophiles, Nat. Chem. Biol., 2008, 4, 405–407 CrossRef CAS PubMed.
- S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Ligand-directed tosyl chemistry for protein labeling in vivo, Nat. Chem. Biol., 2009, 5, 341–343 CrossRef CAS PubMed.
- A. S. Worthington and M. D. Burkart, One-pot chemo-enzymatic synthesis of reporter-modified proteins, Org. Biomol. Chem., 2006, 4, 44–46 RSC.
- S. Konno, J. J. La Clair and M. D. Burkart, Trapping the Complex Molecular Machinery of Polyketide and Fatty Acid Synthases with Tunable Silylcyanohydrin Crosslinkers, Angew. Chem., Int. Ed., 2018, 57, 17009–17013 CrossRef CAS PubMed.
- J. F. King, J. Y. L. Lam and S. Skonieczny, Mechanisms of hydrolysis and related nucleophilic displacement reactions of alkanesulfonyl chlorides: pH dependence and the mechanism of hydration of sulfenes, J. Am. Chem. Soc., 1992, 114, 1743–1749 CrossRef CAS.
- E. Dehling, J. Rüschenbaum, J. Diecker, W. Dörner and H. D. Mootz, Photo-crosslink analysis in nonribosomal peptide synthetases reveals aberrant gel migration of branched crosslink isomers and spatial proximity between non-neighboring domains, Chem. Sci., 2020, 33, 8945–8954 RSC.
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, Peptide bond formation in nonribosomal peptide biosynthesis. Catalytic role of the condensation domain, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef CAS PubMed.
- C. D. Fage, S. Kosol, M. Jenner, C. Öster, A. Gallo and M. Kaniusaite, et al., Communication Breakdown: Dissecting the COM Interfaces between the Subunits of Nonribosomal Peptide Synthetases, ACS Catal., 2021, 11, 10802–10813 CrossRef CAS.
- S. Konno, F. Ishikawa, T. Suzuki, N. Dohmae, M. D. Burkart and H. Kakeya, Active site-directed proteomic probes for adenylation domains in nonribosomal peptide synthetases, Chem. Commun., 2015, 51, 2262–2265 RSC.
- F. Ishikawa, S. Konno, T. Suzuki, N. Dohmae and H. Kakeya, Profiling Nonribosomal Peptide Synthetase Activities Using Chemical Proteomic Probes for Adenylation Domains, ACS Chem. Biol., 2015, 10, 1989–1997 CrossRef CAS PubMed.
- S. D. Fried and S. G. Boxer, Thermodynamic framework for identifying free energy inventories of enzyme catalytic cycles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12271–12276 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cb00005a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |