
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRT-qPCR as a screening platform for mutational and small molecule impacts on structural stability of RNA tertiary structures†
Martina
Zafferani
 ,
Dhanasheel
Muralidharan
,
Nadeska I.
Montalvan
and
Amanda E.
Hargrove
*
,
Dhanasheel
Muralidharan
,
Nadeska I.
Montalvan
and
Amanda E.
Hargrove
*
Department of Chemistry, Duke University, 124 Science Drive, Durham, NC 27705, USA. E-mail: amanda.hargrove@duke.edu
First published on 6th June 2022
Abstract
The exponential increase in the discovery and characterization of RNA tertiary structures has highlighted their active role in a variety of human diseases, yet often their interactome and specific function remain unknown. Small molecules offer opportunities to both decode these cellular roles and develop therapeutics, however there are few examples of small molecules that target biologically relevant RNA tertiary structures. While RNA triple helices are a particularly attractive target, discovery of triple helix modulators has been hindered by the lack of correlation between small molecule affinity and effect on structural modulation, thereby limiting the utility of affinity-based screening as a primary filtering method. To address this challenge, we developed a high-throughput RT-qPCR screening platform that reports on the effect of mutations and additives, such as small molecules, on the stability of triple helices. Using the 3′-end of the oncogenic long non-coding RNA MALAT1 as a proof-of-concept, we demonstrated the applicability of both a two-step and a one-pot method to assess the impact of mutations and small molecules on the stability of the triple helix. We demonstrated the adaptability of the assay to diverse RNA tertiary structures by applying it to the SARS-CoV-2 pseudoknot, a key viral RNA structure recently identified as an attractive therapeutic target for the development of antivirals. Employment of a functional high-throughput assay as a primary screen will significantly expedite the discovery of probes that modulate the structural landscape of RNA structures and, consequently, help gain insight into the roles of these pervasive structures.
Introduction
Advances in biophysical techniques aimed at studying RNA structure have resulted in an exponential increase in discovery and characterization of RNA tertiary structures.1,2 For example, the recent surge in interest and characterization of RNA triple helices across various kingdoms of life has led to coining of the term ‘triplexome’, referring to the diverse and large number of RNA triple helices involved in cellular processes.3 While many of the functions of triplexome members are still being elucidated, the recognized roles of this RNA structural topology include protection of RNA from degradation, protein recruitment, and nuclear localization.4–7 RNA triple helices have been found in a variety of long non-coding RNAs, and the structuredness of the triplex motif has been shown to render the transcript refractory to exonuclease degradation, ultimately promoting its cellular accumulation.3–5,8 Despite the continuous increase in the size of the triplexome, small molecule targeting of these structures has significantly lagged, with only a few examples published.9–11 Notably, recent studies showed that small molecule modulation of RNA structure and function cannot always be predicted by affinity alone, highlighting the need for more cost efficient high-throughput assays that report on the effects of small molecule:RNA interactions in relationship to structural stability.12 Furthermore, affinity-based screening platforms for RNA tertiary structures are limited due to the inherent challenges of optimizing binding assays for larger, more complex structures.12 Function-based assays would help bridge the gap between in vitro screening and biological activity, thereby expediting the discovery of small molecule modulators for the continuously growing number of RNA tertiary structures such as triple helices. In turn, affordable high-throughput screening platforms can enable researchers to screen both mutations and small molecules and assess their impact on RNA structural stability, ultimately providing essential insight into the role of complex RNA structures in disease pathways.Methodologies commonly employed to assess RNA structural stability include circular dichroism (CD), UV absorbance spectroscopy (UV-Vis), differential scanning calorimetry (DSC), and differential scanning fluorimetry (DSF).13–16 While CD and UV-Vis can provide a variety of thermodynamic parameters, they suffer from low throughput and generally require large amounts of RNA, severely limiting their scope and application to sizeable screenings. DSC and DSF can be optimized for high-throughput screening and can provide information on the impact of a variety environmental factors on RNA structure, but the use of heat can lead to results that do not reflect an output relevant to biological environments.16 Furthermore, while DSC and DSF can readily identify molecules with thermal stabilizing effects, the identification of molecules with destabilizing effects is rare. Finally, recent developments and optimization of RNA enzymatic degradation assays have shown them to be uniquely poised to interrogate the effect of additives on RNA structural stability and resistance to enzymatic degradation in a more biologically relevant setting.9,10 However, enzymatic degradation assays are often analyzed through gel electrophoresis and are thus severely limited both in throughput and quantitative potential. It is imperative to develop high-throughput methodologies that assess the effect of small molecules and other additives on RNA structure reliably, quickly, and cost-effectively under biologically relevant conditions.
While the link between structural stability of RNA triplexes and their biological function is a relatively recent discovery, the stability-function relationship is a well-established relationship for frameshifting pseudoknots and RNA G-quadruplexes (rG4s), making them a good reference for our functional assay.17 For example, highly stable rG4s can result in a premature stop in ribosomal scanning and prevent translation of the mRNA transcript downstream.17 Similarly, rG4s stability leads to stalling of reverse transcriptases and premature termination of reverse transcription, an outcome that has been recently used to map the presence of rG4s across the transcriptome.18,19 This finding led to the preliminary investigation of quantitative PCR (qPCR) on the reverse transcribed cDNA template as a potential method to identify G-quadruplex structures.20,21 Recently, Katsuda and co-workers were able to employ this method in a screen to identify small molecules that increased the stability of RNA G-quadruplexes by observing their effect on the length and amount of cDNA formed during reverse transcription of the RNA.22 The small molecule identified as an inhibitor of the elongation reaction of the reverse transcriptase (RT) in the mRNA TERRA was also confirmed as a translation inhibitor in cellulo, corroborating the biological relevance of the in vitro RT assay.22 Based on the success of this method in identifying functional modulators of a G-quadruplex, we sought to ask whether an RT-qPCR reaction could be optimized for application in a general cost-effective high-throughput platform for RNA tertiary structural stability. In addition to the use of a two-step method and high input of RT in the previous experiments, the reported structuredness and thermal stability of RNA G-quadruplexes brings into question whether this assay can be optimized and applied to more complex and/or less stable RNA tertiary structures.22 We also aimed to assess whether an RT-qPCR method could identify both stabilizing and destabilizing mutations and small molecules. We hypothesized that such an assay could both elucidate key interactions necessary for triplex formation and stability via mutant analysis and provide useful modulators to facilitate elucidation of triplex-mediated regulatory pathways to unravel new potential therapeutic avenues.
To ensure the applicability and biological relevance of the newly developed high-throughput RT-qPCR screening platform presented herein, we chose the well-studied MALAT1 triple helix as our initial proof-of-concept. The A-U rich blunt-ended MALAT1 triple helix forms at the 3′-end of a 6.7 kb long non-coding RNA (lncRNA) found to be overexpressed in several types of cancers and implicated in a variety of human diseases, including diabetes.23,24 While the structure and functions of the entire transcript are still under investigation, the triple helix has been reported as essential for protection of the transcript from enzymatic degradation, ultimately leading to increased cellular accumulation of MALAT1.24,25 Indeed, knockdown of MALAT1 in small cell lung adenocarcinoma mouse models led to significant decrease in tumor size and metastasis, confirming the oncogenic role of this lncRNA.26 Biophysical studies by Steitz and co-workers further showed that mutations aimed at destabilizing the triple helix structure led to a significant depletion of MALAT1 in cellulo, establishing the modulation of the MALAT1 triplex as a potential therapeutic avenue.24 To further probe the general applicability of the assay to RNA tertiary structures of biological relevance, we adapted this method to an RNA tertiary structure within the SARS-CoV-2 genome.27 Specifically, the SARS-CoV-2 frameshifting pseudoknot has been highlighted as an essential structure for programmed ribosomal frameshifting to occur.28,29 This process is a common strategy amongst viruses that allows the pathogen to increase the coding potential of its genome by translating two overlapping reading frames (ORFs) and, consequently, control the expression of structural and non-structural viral proteins at different stages of viral lifecycle.30 Consequently, the application of the RT-qPCR assay to this recently discovered RNA structure revealed that two recently found frameshifting inhibitors result in stabilization of the SARS-CoV-2 pseudoknot, thus providing a new stability-centric platform to discover small molecule modulators and expedite RNA-targeted antiviral development.
Results and discussion
Assay and MALAT1 construct design
The 3′-MALAT1 triple helix forms via recruitment of a genomically encoded A-rich tail to the adjacent U-rich region after processing of the full-length transcript.31 Given the critical function and sequestration of the 3′-end, we sought to avoid the use of an A-tail specific reverse primer for the reverse transcription reaction to retain its ability to form a triplex structure. We thus synthesized a construct containing a primer handle commonly used in chemical probing experiments (SHAPE cassette, Fig. 1, orange).32,33 Indeed, SHAPE cassettes have been successfully employed in chemical probing experiments of MALAT1 and MALAT1-like evolutionarily conserved transcripts, and the results were consistent with triple helix formation (Fig. S1, ESI†).34 In the system designed here, small molecules that stabilize the triple helix will result in inhibition of the reverse transcription reaction due to the inability of the chosen RT enzyme (SuperScript IV, ThermoFisher) to unwind structured regions. Inhibition of the elongation reaction will ultimately result in lower levels of cDNA produced, which can be measured quantitatively via qPCR and expressed as cycle threshold (Ct), values (Fig. 1). Analogously, small molecules that destabilize the triple helix conformation will enable for more efficient readthrough of the RT, yielding higher cDNA and a resulting lower Ct value than the control (Fig. 1). Specifically, Ct value is the number of cycles needed during qPCR for the fluorescent signal of the dye (two-step RT-qPCR) or the FRET probe (one-pot RT-qPCR) to exceed a set threshold above background signal. Thus, the Ct value is inversely proportional to the amount of target cDNA present in the sample and, consequently, to the efficiency of reverse transcription.35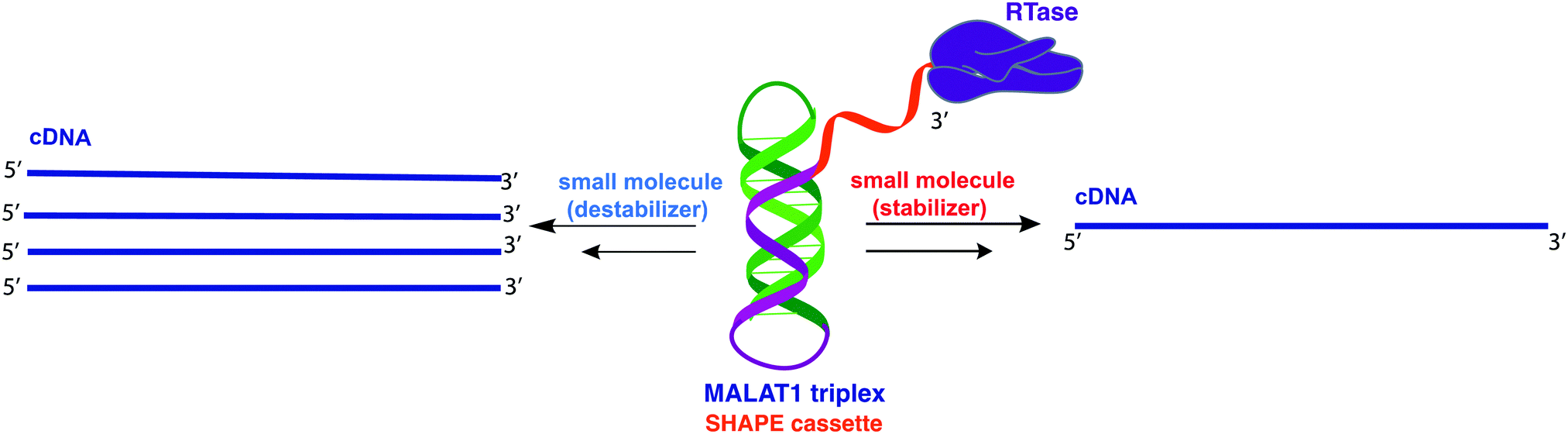 | ||
| Fig. 1 Schematic representation of small molecule-induced structural modulation of the MALAT1 triple helix assessed via RT-qPCR assay. The MALAT1 triple helix (green/purple) is equipped with a structural SHAPE cassette (orange) to prevent competition between primer binding and triplex formation. According to the assay design, small molecules that destabilize the triple helix construct result in lower frequency of RT stalling and, consequently, in more full-length cDNA synthesis (left). Small molecules that stabilize the triple helix structure result in higher occurrence of RT stalling, ultimately resulting in lower amounts of full cDNA synthesis (right). Reverse transcription reactions are then followed by qPCR for quantification (not shown). | ||
Upon successful synthesis of the designed MALAT1 construct (Table S1 and Fig. S1, ESI†), we first optimized the assay for the evaluation of mutational effects on structural stability, particularly destabilizing effects. Accordingly, we synthesized a construct containing a MALAT1 mutant where one of the uracils involved in a base triple within the triplex core is mutated to a cytosine (U13C). This U13C mutant was reported by Steitz and co-workers as a triplex destabilizing mutation that resulted in a decrease of full-length MALAT1 levels in cellulo (Fig. 2).24
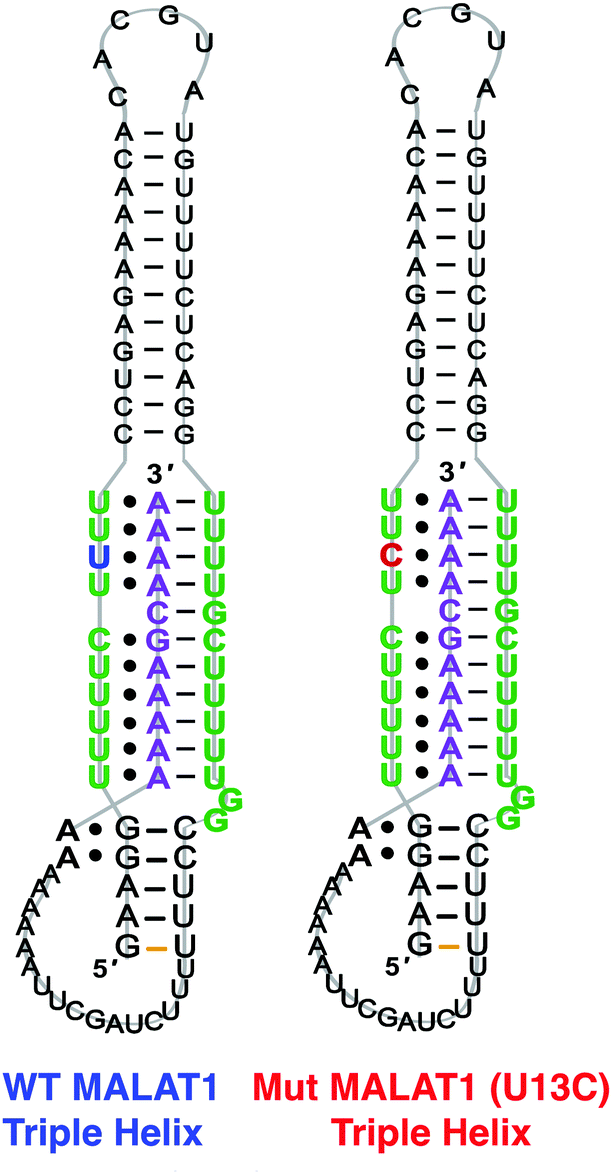 | ||
| Fig. 2 2D structure and sequence of MALAT1 WT and U13C mutant. | ||
Two and one-step RT-qPCR
A two-step RT-qPCR was first utilized to allow for optimization of the reverse transcription and qPCR steps independently. The MALAT1 wild type (WT) and mutant (U13C) were incubated with DMSO control at room temperature after which reverse transcriptase (SSIV), magnesium, primers, and dNTPs were added on ice. The reverse transcription reaction was then carried out at 37 °C, the optimal temperature for the activity of the enzyme, and inactivated by heating to 98 °C after 15 minutes from the start of the reaction. The reaction was then aliquoted in a 96-well light cycler plate and qPCR mix was added to each well and placed in a real-time qPCR machine for cDNA amplification (Fig. 3(A)). Both reverse transcription and qPCR steps were optimized to obtain WT amplification with a Ct that would allow detection of ΔΔCt = ±4 without exceeding the instrument detection limit (<2 Ct) or incurring non-specific amplification (>25 Ct).36 Detection of ΔCt = ±2 corresponds to a 75% change in RT efficiency.22 Initial RNA quantities were in line with manufacturer recommendations. The reverse transcription reaction was optimized by testing whether the reaction needed the presence of “first-strand buffer” for reverse transcription (1 M DTT, 0.5 M tris–HCl, 0.25 M KCl, at pH 8.0). Two reverse transcription reactions were carried out in small scale (20 μL) with first strand buffer or water and the reaction was quenched and cleaned up after 30 minutes at 37 °C. The presence of cDNA was assessed by Agarose gel and by Nanodrop, which qualitatively showed equal amounts of cDNA in both reactions. Given the presence of denaturant and the non-biologically relevant pH of the first strand buffer, the RT reaction was subsequently optimized without first strand buffer. Subsequent optimization included variation of reverse transcriptase amount (Thermo Fisher, 30–150 units), magnesium chloride concentration (1–7.5 nM), reverse primer concentration (150–500 nM), dNTP concentration (100–600 nM), and RNA concentration (10–15 nM). After successful optimization of the reverse transcription reaction (final conditions: 15 SSIV units, 10 nM of RNA, 150 nM of primers, 600 nM of dNTPs, 300 nM of MgCl2), qPCR (KAPA) was optimized by varying the amount of cDNA added to the mix (1–3 μL of a max 200 ng μL−1 RT reaction). Analysis of the qPCR curve yielded a lower Ct value for MALAT1-U13C when compared to the MALAT1-WT, ultimately yielding a ΔCt = 2.1 ± 0.2 (Fig. 3(B) and (C)).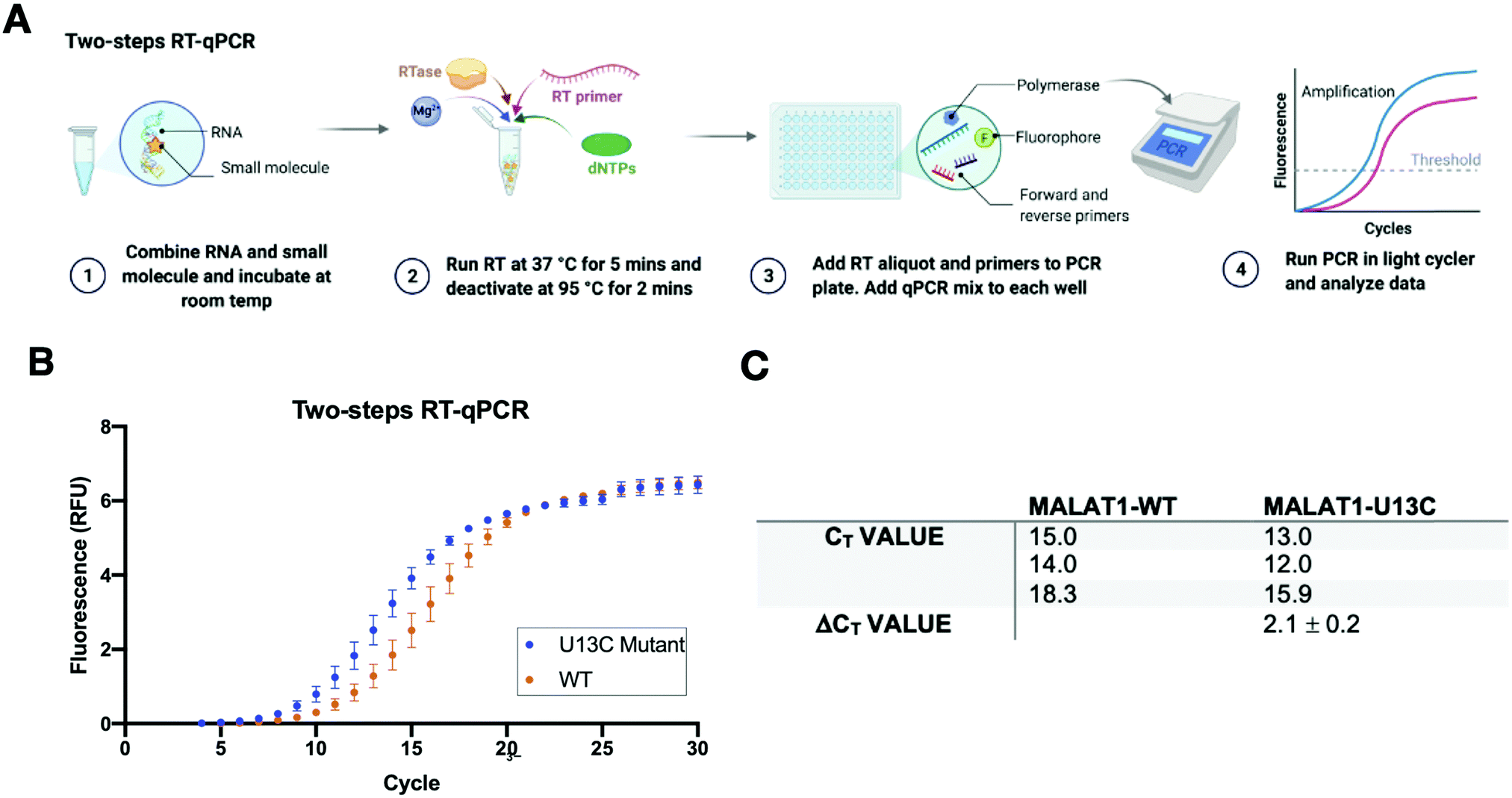 | ||
Fig. 3 Two-step RT-qPCR system. (A) Schematic of the two-step RT-qPCR reaction with the reverse transcription reaction being performed in a thermocycler and then being aliquoted in a 96-well plate for qPCR amplification, which is performed in a light cycler. (B) Amplification curves obtained from 3 independent replicates of RT-qPCR of the U13C MALAT1 mutant and the WT triple helix. Error bars are standard deviation calculated over the three independent experiments. (C) Ct values calculated over three independent experiments for WT and U13C mutant (ΔCt value = Ct![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) WT − CtMut). The mutated destabilized construct amplifies faster than the WT, in agreement with trends reported by Steitz and co-workers. WT − CtMut). The mutated destabilized construct amplifies faster than the WT, in agreement with trends reported by Steitz and co-workers. | ||
Having optimized a two-step reaction, the MALAT1 WT and Mut were analogously employed in a one-pot RT-qPCR kit (QuantaBio), which would enable cost-efficient, high-throughput screening of additives and small molecules. In this system, both the reverse transcription and the qPCR amplification are performed directly in a real-time qPCR instrument (Fig. 4(A)). The one-pot reaction was optimized by varying the RNA concentration in the reaction (1–20 nM) and the forward and reverse primer concentration (200–500 nM) in the same buffer previously used to obtain Ct values in the same range as the two-step RT-qPCR optimized above. Once again, we found that the MALAT1 mutant has lower Ct value than the wild type, yielding a ΔCt = 2.2 ± 0.6 (Fig. 4(B) and (C)). In summary, both methodologies were confirmed to report on mutation-induced changes in MALAT1 triplex structural stability and, most importantly, were consistent with the trends Steitz and co-workers observed in a cellular environment.
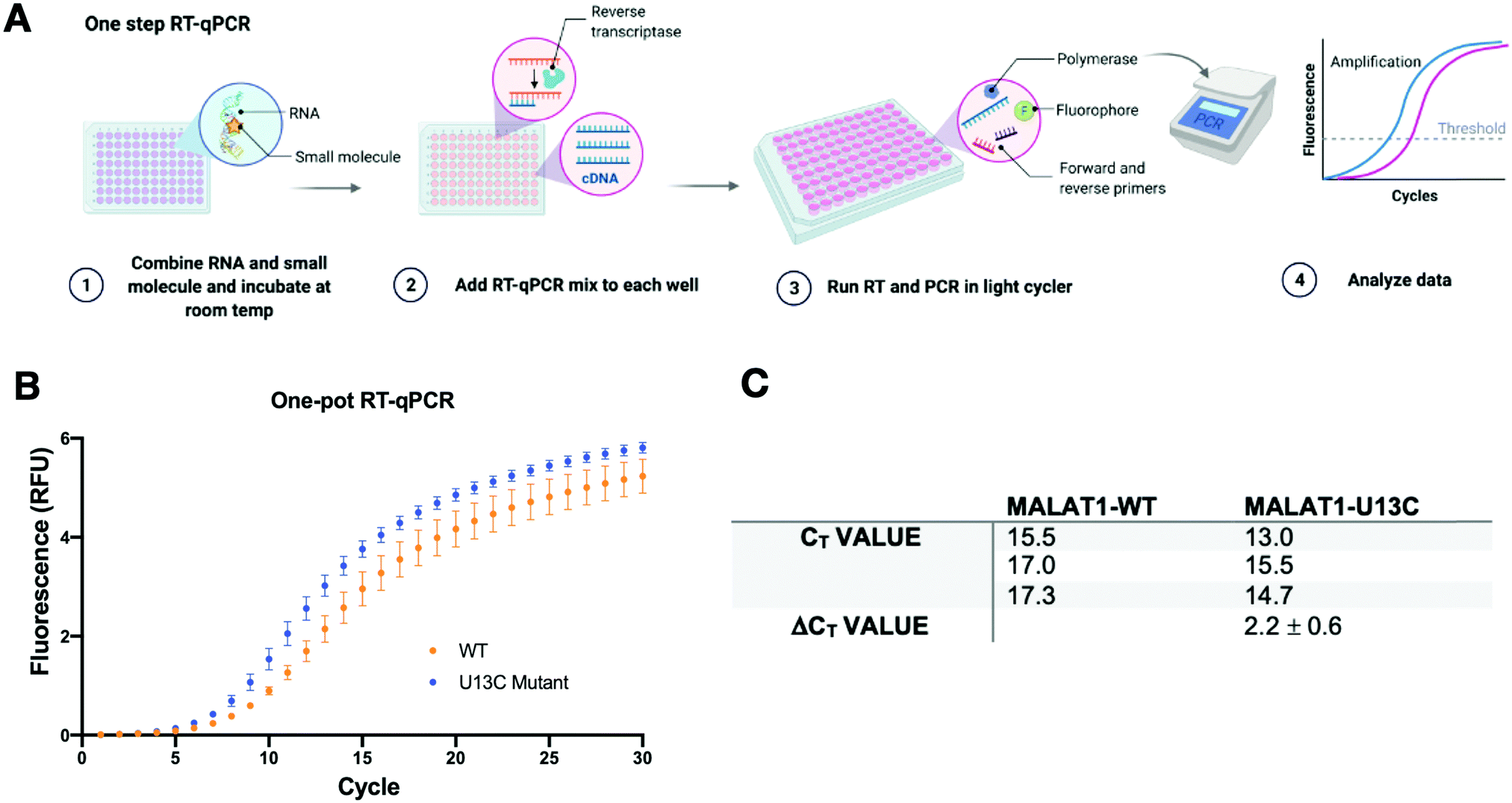 | ||
| Fig. 4 One pot RT-qPCR system. (A) Schematic of the one-pot RT-qPCR reaction with reverse transcription reaction being performed in a 96-well format in a light cycler instrument. (B) Raw data obtained from 3 independent replicates of RT-qPCR of the U13C MALAT1 mutant and the WT triple helix. Error bars are standard deviation calculated over the three independent experiments. (C) Ct values obtained for the Mut and WT constructs (ΔCt value = CtWT − CtMut). The mutated destabilized construct amplifies faster than the WT, in agreement with trends reported by Steitz and co-workers. | ||
Small molecule screening against the MALAT1 triple helix
Next, we tested the applicability of both RT-qPCR routes to evaluate the impacts of small molecules on the MALAT1 WT triple helix. For this purpose, we chose two previously published small molecules that have been classified as stabilizers or destabilizers, respectively (Fig. 5(A)).9,10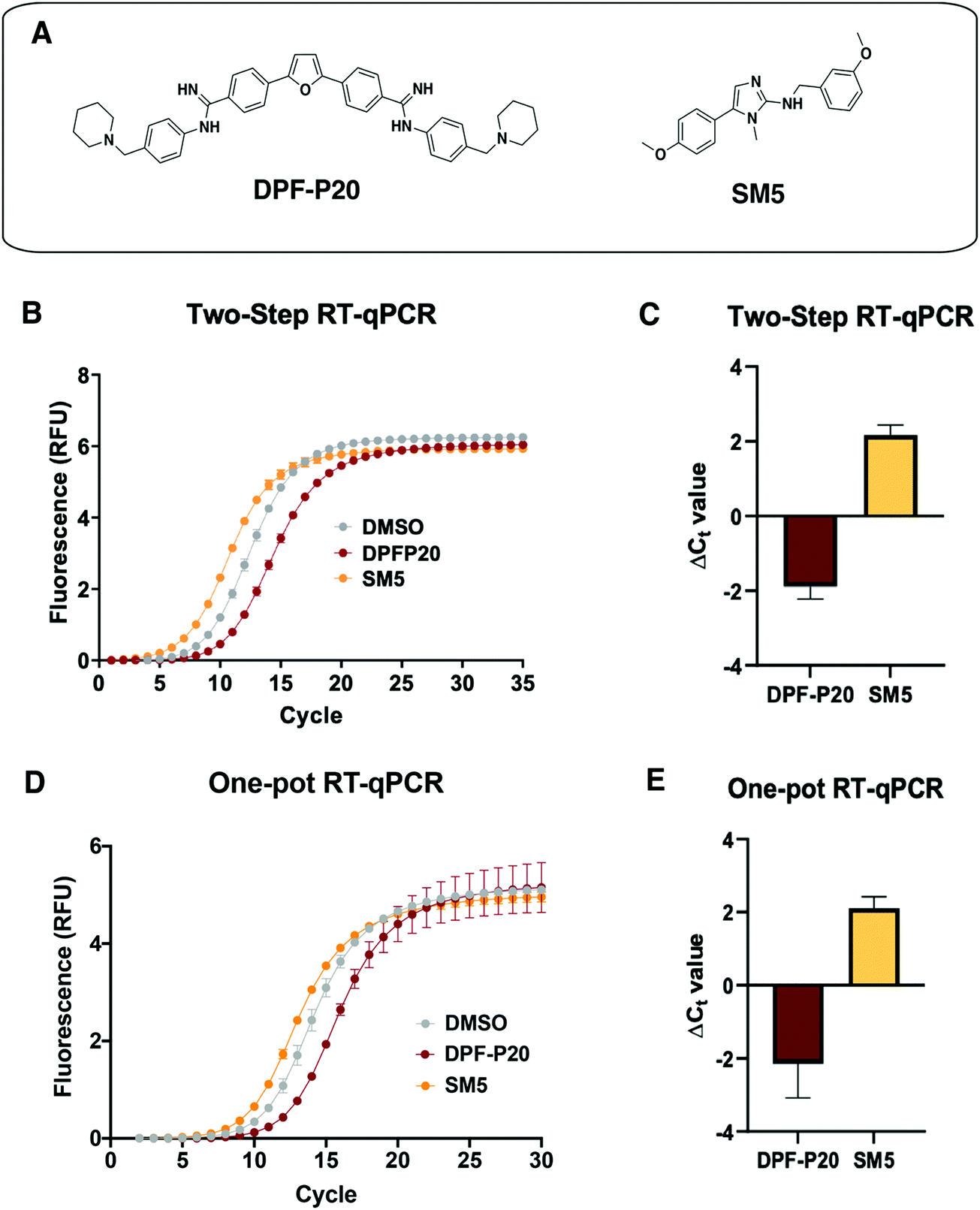 | ||
| Fig. 5 Application of RT-qPCR assays to assess small molecule effect on MALAT1 WT triple helix stability. (A) Structures of two small molecules chosen for assay validation. Both DPF-P20 (a MALAT1 triplex stabilizer) and SM5 (a MALAT1 triplex destabilizer) were previously evaluated in relationship to their effect on triplex enzymatic degradation.9,10 (B) Raw data obtained for the two-step RT-qPCR procedure for both small molecules. (C) Small molecule ΔCt values calculated in reference to DMSO (ΔCt value = CtDMSO − CtSM) from 4 independent replicates are in agreement with their reported effect on MALAT1 triplex enzymatic degradation. (D) Raw data obtained for the one-pot RT-qPCR procedure for both small molecules. (E) Small molecule ΔCt values calculated in reference to DMSO (ΔCt value = CtDMSO – CtSM) from 4 independent replicates in reference to DMSO are in agreement with their reported effect on MALAT1 triplex enzymatic degradation and in line with the values obtained in the two-step RT-qPCR procedure. All error bars represent the standard deviation calculated over the four independent experiments. | ||
DPF-P20 has been recently reported as a MALAT1 triple helix stabilizers as it increases the triplex thermal stability measured via DSF as well as inhibits RNase R-mediated exonucleolytic degradation of the triplex (Fig. 5(A)).9 Both the two-step and the one-pot method identified DPF-P20 as a triplex stabilizer, yielding less cDNA and higher Ct values than the DMSO control (ΔCt(2step) = −1.9 ± 0.3 ΔCt(1pot) = −2.3 ± 0.7) (Fig. 5(C)–(E)). Recently reported by Le Grice and co-workers, SM5 was identified as a destabilizer of the MALAT1 triplex resulting in reduction of MALAT1 accumulation in ex vivo organoid breast cancer models (Fig. 5(A)).10SM5 has also been shown by Donlic and co-workers to increase RNase R-mediated exonucleolytic degradation of the triple helix over time.9 In line with previous studies, SM5 resulted in lower Ct values than the DMSO control, confirming its triplex destabilizing properties (ΔCt(2step) = 2.2 ± 0.3 ΔCt(1pot) = 2.1 ± 0.3) (Fig. 5(B) and (C)). ΔCt values were in the same range in both the two-step and the one-pot RT-qPCR method, showcasing the consistency and applicability of both approaches. (Fig. 5(D) and (E)). As expected, given the sensitivity of RT-qPCR, Z-factor control experiments for the one-pot method were suitable for a high-throughput screening platform (Z-factor = 0.93, Fig. S3, ESI†). The |ΔCt| values observed for the MALAT1 triplex modulators are comparable to the values obtained by Katsuda and co-workers for the best identified leads of G-quadruplex stabilizers. The authors used ΔCt ≥ 2 as a cutoff for lead molecules since a value of 2 corresponded to a 75% decrease in RT elongation.22 Both small molecules, DPF-P20 and SM5, would be classified as hits under these conditions, and we propose the same cutoff for the assay reported here. For promising small molecule stabilizers, it will be important to rule out possible interference from small molecule inhibition of the RT or polymerase enzymes, which can be accomplished in complementary or secondary assays.
Application of RT-qPCR assay to functional RNA elements in SARS-CoV-2 genome
Viral protein expression is finely tuned by a tertiary structure element at the interface of two overlapping open reading frames (ORF) in the SARS-CoV-2 genome.37–39 The structuredness of this RNA element is known to cause mechanochemical tension in ribosomal elongation, ultimately resulting in stalling and re-positioning in a different frame before continuing elongation.30,40–43 This process, also referred to as programmed ribosomal frameshifting, is essential for viral replication and thus has been identified as a potential antiviral target (Fig. 6).38,44,45 Consequently, modulating the stability of the RNA pseudoknot can lead to an increase or decrease in frameshifting events, both of which have been shown to lead to viral inhibition. To date, no high-throughput assays have been developed that report on the structural stability of RNA pseudoknots. Indeed, most RNA pseudoknot-focused studies, though limited, resort to in vitro or in cellulo reporter-based frameshifting assays or biophysical techniques such as Cryo-EM and NMR.28,46 The reporter-based approach can lead to false positive or false negative results as effects on frameshifting may not reflect engagement of the RNA pseudoknot structure. The structural techniques, on the other hand, are both time- and cost-intensive, thereby preventing its application to sizable screenings. Therefore, the application of the RT-qPCR assay developed herein can significantly expedite the discovery of CoV-2 pseudoknot (CoV-2 PK) binders while providing insight on small molecule recognition of this class of underexplored tertiary structures for which very limited examples have been published to date. | ||
| Fig. 6 Application of the RT-qPCR assay to the SARS-CoV-2 pseudoknot. (A) 3D structure of the SARS-CoV-2 frameshifting element pseudoknot structure obtained via NMR (PDB: 7LYJ).28 (B) 2D representation of the pseudoknot and the relative base-pairing according to the structure of D’Amare and co-workers. (C) Chemical structure of two frameshifting inhibitors merafloxacin29,47 and nafamostat.48 (D) Two-step RT-qPCR adapted to the SARS-CoV-2 pseudoknot identifies frameshifting inhibitors as stabilizing small molecules. | ||
We first aimed to optimize the two-step RT-qPCR assay to the pseudoknot construct alone. Equimolar amounts of CoV-2 PK and MALAT1 triplex input RNA led to faster (lower Ct value) amplification by qPCR for the CoV-2 PK. Optimal Ct values were achieved by decreasing the reverse transcription time and RNA concentrations of the CoV-2 PK. This trend is in agreement with recently reported biophysical characterization of the CoV-2 PK constructs, which displays a shorter region of base triples than MALAT1, potentially impacting its stability in the knotted conformer.28 Furthermore, frameshifting pseudoknots are known to utilize significant conformational plasticity to control the balance of in-frame vs. frameshifted translation. These results suggest that the RT-qPCR platform could, potentially, be employed to gain insight into the differential stability between the two structures.
To benchmark this assay for small molecule screening against the CoV-2 pseudoknot we chose two small molecules recently discovered as frameshifting inhibitors in two separate studies, merafloxacin and nafamostat.29,47,48 Discovered by Sun and co-workers, merafloxacin was identified through a cell-based luciferase assay in HEK293T cells and was found to inhibit frameshifting and SARS-CoV-2 replication.49 Nafamostat, on the other hand, was identified in a cell-free luciferase assay as a frameshifting inhibitor of a variety of bat CoVs.48 Indeed, both studies highlighted the potential of frameshifting elements and, specifically, the SARS-CoV-2 pseudoknot as an attractive therapeutic target to develop mutation-resistant and broad-spectrum antivirals. At the same time, these assays did not inform the relationship between pseudoknot structural stability and inhibition of frameshifting. By screening merafloxacin and nafamostat in our assay, we confirmed direct engagement of the CoV-2 PK target as well as stabilization of the CoV-2 PK as a plausible mechanism of action. Both merafloxacin and nafamostat led to similar levels of stabilization (ΔCtMeraflox. = −2.3 ± 0.6 ΔCtNafam. = −2.7 ± 0.4) in our assay, and Munshi and co-workers found similar levels of SARS-CoV-2 frameshifting inhibition between the two molecules, supporting the relevance of this assay for future screening and characterization efforts.48
Conclusion
Here, we report the development and optimization of a new high-throughput screening platform that assesses the effects of mutations and small molecule additives on the structural stability of the MALAT1 3′-end RNA triple helix, one of the best studied disease-relevant RNA triple helices. While recent efforts aimed at small molecule targeting and modulation of this RNA motif have identified small molecule binders via screening, the effect of the reported small molecules on MALAT1 triplex stability has been studied through low throughput and/or non-biologically relevant techniques.9,11In this work, we developed a high-throughput RT-qPCR screening platform, accessible via both two-step and one-pot protocols, utilizing the MALAT1 triple helix and a biologically relevant mutant construct. Robust differences in Ct values of the U13C mutant relative to wild type recapitulated the destabilizing effects of the point mutation, which was previously reported to lead to a decrease of MALAT1 transcript accumulation in cellulo. These findings underscored the applicability of this platform to evaluate the effects of mutations on structural stability as well as the likely biological relevance of the trends observed. We then chose two MALAT1 small molecule ligands previously published as stabilizers or destabilizers and evaluated their impacts on the MALAT1 triple helix in both methods. Once again, both approaches resulted in trends consistent with previously published effects of the small molecules on RNase R-mediated exonucleolytic degradation of the triplex. To further highlight the applicability of the assay to other disease-relevant RNA structures we investigated the SARS-CoV-2 frameshifting pseudoknot. We chose two small molecules recently identified in separate studies as frameshifting inhibitors that resulted in a decrease of SARS-CoV-2 replication. Both small molecules were identified as pseudoknot stabilizers, providing first evidence of direct engagement of the SARS-CoV-2 pseudoknot structure and preliminary insight into the relationship between frameshifting inhibition and small molecules’ effect on pseudoknot structural stability.
The RT-qPCR-based screening method developed herein establishes a high-throughput platform that can identify RNA-targeted small molecules that have both stabilizing and destabilizing effects on RNA tertiary structure. The ability to identify probes with opposite impacts can greatly help elucidate the many biological roles of RNA tertiary structures such as triple helices and pseudoknots in human disease and expedite the discovery of RNA-targeted therapeutics. Having access to a cost-efficient high-throughput structural stability screening platform can significantly increase the ability to evaluate small molecule selectivity for one structure over another in relationship to structural stability. In turn, the data gathered can help address several unanswered questions such as what molecular properties make a small molecule a stabilizer or a destabilizer and whether we can use structural stability data as a guiding principle for future small molecule design and synthesis. We expect that answering these remaining questions will move the scientific community toward the efficient development of RNA-targeted small molecule therapeutics.
Experimental procedures
Synthesis of the MALAT1 RNA constructs
DNA template sequence was purchased from Dharmacon, and forward and reverse primers were purchased from Integrated DNA technologies (IDT) (Table S1, ESI†). For PCR amplification the following reagents were added for a given 50 μL final reaction volume. First, the entire working space was treated with RNase Zap to prevent contamination. Next, in the desired amount of sterile PCR tubes (ThermoFisher) the reaction's component were adding as detailed in Table 1. The DNA template was then amplified for 30 cycles in an Eppendorf Echo thermocycler. A Zymo DNA-clean-up kit was then utilized to clean up the desired DNA sequence. A solution of amplified DNA in water was made to reach 28–35 ng μL−1. The sequence was then in vitro transcribed (IVT) using the protocol detailed in Table 1 per 50 μL reaction.| Component | Stock concentration | Volume added (μL) | |
|---|---|---|---|
| PCR | Nuclease-free water | N/A | 32.5 |
| Q5 reaction buffer (NEB) | 10× | 10 | |
| dNTPs | 10 mM | 1 | |
| Forward primer | 10 mM | 2.5 | |
| Reverse primer | 10 mM | 2.5 | |
| DNA template | 50 ng μL−1 | 1 | |
| Q5 polymerase (NEB) | 2000 U mL−1 | 0.5 | |
| IVT | Nuclease-free water | N/A | 31.75 |
| MgCl2 | 1 M | 1.25 | |
| Tris–HCl, pH 8.0 | 1 M | 2 | |
| rNTP mix | 10 mM | 5 | |
| Spermidine | 0.1 M | 1.25 | |
| Triton-X | 0.1% | 0.5 | |
| DTT | 1 M | 0.5 | |
| Pyrophosphatase (NEB) | 100 U μL−1 | 0.2 | |
| DNA from PCR | 28–35 ng μL−1 | 5 | |
| T7 polymerase (Tolbert lab) | 50000 U mL−1 |
2.5 | |
The reaction was then incubated at 37 °C for 12 hours. Following incubation, the reaction was treated with 2 μL of DNase I (NEB) and 5 μL of DNase I buffer twice in intervals of 30 minutes, followed by addition of 10% of the reaction volume of EDTA. The desired RNA was then extracted using phenol chloroform extraction and further purified via ethanol precipitation. Purity and size of the RNA construct was confirmed by Small RNA chip on Agilent Bioanalyzer and 10% TBE denaturing gel (Fig. S2, ESI†).
Synthesis of the SARS-CoV-2 RNA pseudoknot constructs
DNA template sequence was purchased from Dharmacon, and forward and reverse primers were purchased from Integrated DNA technologies (IDT) (Table S1, ESI†). For PCR amplification the following reagents were added to for a given 50 μL final reaction volume as detailed in Table 2. First, the entire working space was treated with RNase Zap to prevent contamination.| Component | Stock concentration | Volume added (μL) | |
|---|---|---|---|
| PCR | Nuclease-free water | N/A | 30 |
| Q5 reaction buffer (NEB) | 10× | 10 | |
| dNTPs | 10 mM | 1 | |
| Forward primer | 10 mM | 2.5 | |
| Reverse primer | 10 mM | 2.5 | |
| DNA template | 50 ng μL−1 | 1 | |
| DMSO | N/A | 2.5 | |
| Q5 polymerase (NEB) | 2000 U mL−1 | 0.5 | |
| IVT | Nuclease-free water | N/A | 18.2 |
| MgCl2 | 1 M | 1 | |
| Tris–HCl, pH 8.0 | 1 M | 4 | |
| rNTP mix | 10 mM | 3 | |
| Spermidine | 0.1 M | 1 | |
| DTT | 1 M | 0.1 | |
| Pyrophosphatase (NEB) | 100 U μL−1 | 0.2 | |
| DNA from PCR | 28–35 ng μL−1 | 10 | |
| T7 polymerase (Tolbert lab) | 50000 U mL−1 |
2.5 | |
| DMSO | N/A | 10 | |
The DNA template was then amplified for 30 cycles in an Eppendorf Echo thermocycler. A Zymo DNA-clean-up kit was then utilized to clean up the desired DNA sequence. A solution of amplified DNA in water was made to reach 20 ng mL−1. The sequence was then in vitro transcribed (IVT) using the protocol detailed in Table 2 for each 50 μL reaction.
The reaction is then incubated at 25 °C for 12 hours. Following incubation, the reaction is then incubated at 37 °C and treated with 3 μL of DNase I (1000 units, NEB) and 6 μL DNase I buffer (NEB) twice in intervals of 30 minutes, followed by addition of 10% of the reaction volume of EDTA. The desired RNA is then extracted using phenol chloroform extraction and the aqueous layer was then subjected to ethanol precipitation. The concentrated product is then re-constituted in 1 mL of water and vortexed to obtain a fully dissolved heterogeneous solution, which is further purified by FPLC (BioRad). The solution is loaded on a 70 SEC column (BioRad) and run via isocratic protocol in 50 mM HEPES-KOH, 50 mM KCl, 0.1 mM EDTA at pH 7.5. Purity and size of the RNA construct is confirmed by Small RNA chip on Agilent Bioanalyzer and 10% TBE (Fig. S2, ESI†).
MALAT1 two step RT-qPCR
RNA was synthesized and purified as previously described. For a given reverse transcription reaction RNA was incubated in water with DMSO or small molecules at room temperature for 20 min. After incubation the PCR strip was placed on an ice block. A mastermix of reverse transcription reaction reagents was prepared and aliquoted in each reaction to reach a total of 40 μL Components were added to the reverse transcription reaction to reach the final concentration listed in Table 3. PCR strip was then incubated at 37 °C in a thermocycler (Eppendorf, Nexus Gradient) for 15 minutes before inactivating the RT enzyme for 5 minutes at 98 °C. The PCR strip was then placed on an ice block. In the meantime, a qPCR mix was prepared according to the concentrations listed in Table 3. The qPCR mastermix was aliquotted in each well of a 96-well lightcycler plate (Roche 96) and 1 μL of RT reaction was added to each. Amplification protocol was run by incubating at 95 °C for 3 minutes followed by a 3-step amplification for 25–45 cycles and a final cooling to 40 °C over the course of 10 minutes. Results were analyzed using the Roche 96 light cycler software v1.1.| Component | Volume added to reaction (μL) | Final concentration | |
|---|---|---|---|
| Reverse transcription reaction | DMSO/small molecule | 0.8 | 10 μM |
| RNA | 4 | 10 nM | |
| Reverse primer | 3 | 150 nM | |
| SSIV | 3 | 15 units | |
| dNTPs | 2.4 | 600 nM | |
| MgCl2 | 0.6 | 300 nM | |
| Nuclease free water | 26.2 | ||
| qPCR reaction | RT reaction | 1 | |
| Reverse primer | 0.9 | 300 nM | |
| Forward primer | 0.9 | 300 nM | |
| SYBR mix | 10 | ||
| Nuclease free water | 17.2 | ||
SARS-CoV-2 pseudoknot two step RT-qPCR
RNA was synthesized and purified as previously described. The two-step procedure was optimized starting with the conditions used for the MALAT1 triple helix, further showcasing the efficient adaptability of this assay to different RNA structures. For a given reverse transcription reaction RNA was incubated in water with DMSO or small molecules at room temperature for 20 min. After incubation the PCR strip was placed on an ice block. A mastermix of reverse transcription reaction reagents was prepared and aliquoted in each reaction to reach a total of 40 μL Components were added to the reverse transcription reaction to reach the final concentration listed in Table 4. PCR strip was then incubated at 37 °C in a thermocycler (Eppendorf, Nexus Gradient) for 15 minutes before inactivating the RT enzyme for 5 minutes at 98 °C. The PCR strip was then placed on an ice block. In the meantime, a qPCR mix was prepared according to the concentrations listed in Table 4. The qPCR mastermix was aliquoted in each well of a 96-well lightcycler plate (Roche 96) and 1 μL of RT reaction was added to each. Amplification protocol was run by incubating at 95 °C for 3 minutes followed by a 3-step amplification for 25–45 cycles and a final cooling to 40 °C over the course of 10 minutes. Results were analyzed using the Roche 96 light cycler software v1.1.| Component | Volume added to reaction (μL) | Final concentration | |
|---|---|---|---|
| Reverse transcription reaction | DMSO/small molecule | 2 | 25 μM |
| RNA | 2 | 3.75 nM | |
| Reverse primer | 3 | 150 nM | |
| SSIV | 0.5 | 2.5 units | |
| dNTPs | 2.4 | 600 nM | |
| MgCl2 | 0.6 | 300 nM | |
| Nuclease free water | 29 | ||
| qPCR reaction | RT reaction | 1 | |
| Reverse primer | 0.9 | 300 nM | |
| Forward primer | 0.9 | 300 nM | |
| SYBR mix | 10 | ||
| Nuclease free water | 17.2 | ||
One pot RT-qPCR
RNA was synthesized and purified as previously described. For a given RT-qPCR reaction 10 nM of RNA were incubated in water with DMSO or 10 μM of small molecules at room temperature for 20 min in a 96 well lightcycler plate (Roche). During incubation an RT-qPCR mastermix was 1 step SYBR mastermix (QuantaBio), qScript RT enzyme (QuantaBio 1-step RTqPCR kit), and forward and reverse primer solution according to Table 5 for a total of 50 μL for each reaction. The mastermix of reverse transcription reaction reagents was prepared and aliquoted in each well by adding 31 μL of the master mix to each RNA-DMSO or RNA-Small molecule well. The plate was then sealed with optically clear foils and inserted in a Roche 96 light cycler. The RT step was performed by incubating at 37 °C for 10 minutes, followed by an inactivation at 95 °C for 5 minutes. The qPCR step immediately followed with a 3-step amplification carried for 30–35 cycles, which was followed by cooling to 40 °C over the course of 10 minutes. Results were analyzed using the Roche 96 light cycler software v1.1.| Component | Volume added to reaction (μL) | Final concentration |
|---|---|---|
| Nuclease free water | 13 | |
| RNA | 5 | 10 nM |
| DMSO/small molecule | 1 | 10 μM |
| Forward primer | 2.5 | 500 nM |
| Reverse primer | 2.5 | 500 nM |
| qScript TaqMan | 1 | 1× |
| 1-step SYBR mix | 25 | 1× |
Funding
U.S. National Institutes of Health R35 GM124785 (A. E. H.; M. Z.); Research Corporation for Science Advancement COVID Initiative, Award 27340 (A. E. H.; M. Z); Duke University Chemistry Department Burroughs Wellcome Fellowship (M. Z.): Duke University Dean's Summer Research Fellowship (D. M.) Duke University's Biological Sciences Undergraduate Research Fellowship (N. M.).Conflicts of interest
Nothing to declare.Acknowledgements
We are grateful to past Hargrove lab members, especially to Dr Emily McFadden and Dr Sarah Wicks, for their feedback and input. We would like to give a special thanks to current members of the Hargrove lab, Marek Zorawski, Justin Martyr, and Emily Swanson, who helped finalized the structure of this manuscript. We are grateful to the Tolbert lab for their generous donation of the T7 polymerase used for in vitro transcription. We would also like to thank Dr Jessica Brown for kindly sharing illustrator files to make 2D-renderings of the MALAT1 triple helix and the Duke Biology Department for sharing the use of their Roche light cycler instrument. Figures in this manuscript were made in BioRender.References
- A. M. Mustoe, S. Busan, G. M. Rice, C. E. Hajdin, B. K. Peterson, V. M. Ruda, N. Kubica, R. Nutiu, J. L. Baryza and K. M. Weeks, Pervasive regulatory functions of mRNA structure revealed by high-resolution shape probing., Cell, 2018, 173(1), 181–195.e18 CrossRef CAS PubMed.
- J. Sztuba-Solinska, J. W. Rausch, R. Smith, J. T. Miller, D. Whitby and S. F.-J. Le Grice, Kaposi's sarcoma-associated herpesvirus polyadenylated nuclear RNA: A structural scaffold for nuclear, cytoplasmic and viral proteins., Nucleic Acids Res., 2017, 45(11), 6805–6821 CrossRef CAS PubMed.
- J. A. Brown, Unraveling the structure and biological functions of RNA triple helices, WIREs RNA, 2020, 11(6), e1598 CAS.
- X. Zhang, M. H. Hamblin and K.-J. Yin, The long noncoding RNA Malat1: Its physiological and pathophysiological functions., RNA Biol., 2017, 14(12), 1705–1714 CrossRef PubMed.
- J. A. West, C. P. Davis, H. Sunwoo, M. D. Simon, R. I. Sadreyev, P. I. Wang, M. Y. Tolstorukov and R. E. Kingston, The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites., Mol. Cell, 2014, 55(5), 791–802 CrossRef CAS PubMed.
- B. L. Gudenas and L. Wang, Prediction of LncRNA subcellular localization with deep learning from sequence features., Sci. Rep., 2018, 8(1), 16385 CrossRef PubMed.
- M. C. Bridges, A. C. Daulagala and A. Kourtidis, LNCcation: lncRNA localization and function., J. Cell Biol., 2021, 220(2), e202009045 CrossRef CAS PubMed.
- N. K. Conrad, The emerging role of triple helices in RNA biology, WIREs RNA, 2014, 5(1), 15–29 CrossRef CAS PubMed.
- A. Donlic, M. Zafferani, G. Padroni, M. Puri and A. E. Hargrove, Regulation of MALAT1 triple helix stability and in vitro degradation by diphenylfurans., Nucleic Acids Res., 2020, 48(14), 7653–7664 CrossRef CAS PubMed.
- F. A. Abulwerdi, W. Xu, A. A. Ageeli, M. J. Yonkunas, G. Arun, H. Nam, J. S. Schneekloth Jr., T. K. Dayie, D. Spector, N. Baird and S. F.-J. Le Grice, Selective small-molecule targeting of a triple helix encoded by the long noncoding RNA, MALAT1., ACS Chem. Biol., 2019, 14(2), 223–235 CrossRef CAS PubMed.
- A. Donlic, B. S. Morgan, J. L. Xu, A. Liu, C. Roble Jr. and A. E. Hargrove, Discovery of small molecule ligands for MALAT1 by tuning an RNA-binding scaffold., Angew. Chem., Int. Ed., 2018, 57(40), 13242–13247 CrossRef CAS PubMed.
- M. Zafferani and A. E. Hargrove, Small molecule targeting of biologically relevant RNA tertiary and quaternary structures., Cell Chem. Biol., 2021, 28(5), 594–609 CrossRef CAS PubMed.
- A. Das, K. Bhadra and G. Suresh Kumar, Targeting RNA by small molecules: Comparative structural and thermodynamic aspects of aristololactam-β-D-glucoside and daunomycin binding to tRNAphe., PLoS One, 2011, 6(8), e23186 CrossRef CAS PubMed.
- H. Yu, W. Yang, O. Alkhamis, J. Canoura, K.-A. Yang and Y. Xiao, In vitro isolation of small-molecule-binding aptamers with intrinsic dye-displacement functionality., Nucleic Acids Res., 2018, 46(8), e43 CrossRef PubMed.
- J. E. Sokoloski and P. C. Bevilacqua, Analysis of RNA folding and ligand binding by conventional and high-throughput calorimetry., Methods Mol. Biol., 2012, 905, 145–174 CAS.
- R. Silvers, H. Keller, H. Schwalbe and M. Hengesbach, Differential scanning fluorimetry for monitoring RNA stability., ChemBioChem, 2015, 16(7), 1109–1114 CrossRef CAS PubMed.
- M. M. Fay, S. M. Lyons and P. Ivanov, RNA G-quadruplexes in biology: Principles and molecular mechanisms., J. Mol. Biol., 2017, 429(14), 2127–2147 CrossRef CAS PubMed.
- C. K. Kwok, G. Marsico and S. Balasubramanian, Detecting RNA G-Quadruplexes (rG4s) in the Transcriptome., Cold Spring Harbor Perspect. Biol., 2018, 10(7), a032284 CrossRef PubMed.
- C. K. Kwok and S. Balasubramanian, Targeted Detection of G-Quadruplexes in Cellular RNAs., Angew. Chem., Int. Ed., 2015, 54(23), 6751–6754 CrossRef CAS PubMed.
- J. Jamroskovic, I. Obi, A. Movahedi, K. Chand, E. Chorell and N. Sabouri, Identification of putative G-quadruplex DNA structures in S. pombe genome by quantitative PCR stop assay., DNA Repair, 2019, 82, 102678 CrossRef CAS PubMed.
- X. Yang, J. Cheema, Y. Zhang, H. Deng, S. Duncan, M. I. Umar, J. Zhao, Q. Liu, X. Cao, C. K. Kwok and Y. Ding, RNA G-quadruplex structures exist and function in vivo in plants., Genome Biol., 2020, 21(1), 226 CrossRef CAS PubMed.
- Y. Katsuda, S.-i Sato, L. Asano, Y. Morimura, T. Furuta, H. Sugiyama, M. Hagihara, M. Uesugi and A. Small, Molecule that represses translation of G-quadruplex-containing mRNA., J. Am. Chem. Soc., 2016, 138(29), 9037–9040 CrossRef CAS PubMed.
- J. A. Brown, M. L. Valenstein, T. A. Yario, K. T. Tycowski and J. A. Steitz, Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENβ noncoding RNAs, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(47), 19202–19207 CrossRef CAS PubMed.
- J. A. Brown, D. Bulkley, J. Wang, M. L. Valenstein, T. A. Yario, T. A. Steitz and J. A. Steitz, Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix., Nat. Struct. Mol. Biol., 2014, 21(7), 633–640 CrossRef CAS PubMed.
- T. Gutschner, M. Hämmerle, M. Eissmann, J. Hsu, Y. Kim, G. Hung, A. Revenko, G. Arun, M. Stentrup, M. Gross, M. Zörnig, A. R. MacLeod, D. L. Spector and S. Diederichs, The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells., Cancer Res., 2013, 73(3), 1180–1189 CrossRef CAS PubMed.
- T. Gutschner, M. Hämmerle, M. Eißmann, J. Hsu, Y. Kim, G. Hung, A. Revenko, G. Arun, M. Stentrup, M. Groß, M. Zörnig, A. R. MacLeod, D. L. Spector and S. Diederichs, The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells, Cancer Res., 2013, 73(3), 1180–1189 CrossRef CAS PubMed.
- I. Manfredonia and D. Incarnato, Structure and regulation of coronavirus genomes: State-of-the-art and novel insights from SARS-CoV-2 studies, Biochem. Soc. Trans., 2021, 49(1), 341–352 CrossRef CAS PubMed.
- C. P. Jones and A. R. Ferré-D’Amaré, Crystal structure of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) frameshifting pseudoknot., RNA, 2022, 28(2), 239–249 CrossRef CAS PubMed.
- P. R. Bhatt, A. Scaiola, G. Loughran, M. Leibundgut, A. Kratzel, R. Meurs, R. Dreos, K. M. O’Connor, A. McMillan and J. W. Bode, Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome., Science, 2021, 372(6548), 1306–1313 CrossRef CAS PubMed.
- A. E. Firth and I. Brierley, Non-canonical translation in RNA viruses., J. Gen. Virol., 2012, 93(7), 1385–1409 CrossRef CAS PubMed.
- J. E. Wilusz, C. K. JnBaptiste, L. Y. Lu, C. D. Kuhn, L. Joshua-Tor and P. A. Sharp, A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails., Genes Dev., 2012, 26(21), 2392–2407 CrossRef CAS PubMed.
- C. E. Hajdin, S. Bellaousov, W. Huggins, C. W. Leonard, D. H. Mathews and K. M. Weeks, Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots., Proc. Natl. Acad. Sci. U. S. A., 2013, 110(14), 5498–5503 CrossRef CAS PubMed.
- K. A. Wilkinson, E. J. Merino and K. M. Weeks, Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution, Nat. Protoc., 2006, 1(3), 1610–1616 CrossRef CAS PubMed.
- B. Zhang, Y. S. Mao, S. D. Diermeier, I. V. Novikova, E. P. Nawrocki, T. A. Jones, Z. Lazar, C.-S. Tung, W. Luo, S. R. Eddy, K. Y. Sanbonmatsu and D. L. Spector, Identification and characterization of a class of MALAT1-like genomic loci., Cell Rep., 2017, 19(8), 1723–1738 CrossRef CAS PubMed.
- M. Troutman, The Significance of Early Ct's – Ask TaqMan. https://www.thermofisher.com/blog/behindthebench/the-significance-of-early-cts-ask-taqman-35/.
- C. G.-B. Caraguel, H. Stryhn, N. Gagné, I. R. Dohoo and K. L. Hammell, Selection of a cutoff value for real-time polymerase chain reaction results to fit a diagnostic Purpose: Analytical and epidemiologic approaches., J. Vet. Diagn. Invest., 2011, 23(1), 2–15 CrossRef PubMed.
- S. I. Omar, M. Zhao, R. V. Sekar, S. A. Moghadam, J. A. Tuszynski and M. T. Woodside, Modeling the structure of the frameshift-stimulatory pseudoknot in SARS-CoV-2 reveals multiple possible conformers., PLoS Comput. Biol., 2021, 17(1), e1008603 CrossRef CAS PubMed.
- W. D. Penn, H. R. Harrington, J. P. Schlebach and S. Mukhopadhyay, Regulators of viral frameshifting: More than RNA influences translation events., Annu. Rev. Virol., 2020, 7(1), 219–238 CrossRef CAS PubMed.
- E. P. Plant and J. D. Dinman, The role of programmed-1 ribosomal frameshifting in coronavirus propagation., Front. Biosci., 2008, 13, 4873–4881 CrossRef CAS PubMed.
- A. E. Firth, B. Y.-W. Chung, M. N. Fleeton and J. F. Atkins, Discovery of frameshifting in Alphavirus 6 K resolves a 20 year enigma., Virol. J., 2008, 5(1), 108 CrossRef PubMed.
- T. Jacks and H. E. Varmus, Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting., Science, 1985, 230(4731), 1237–1242 CrossRef CAS PubMed.
- R. J. Riegger and N. Caliskan, Thinking outside the frame: Impacting genomes capacity by programmed ribosomal frameshifting., Front. Mol. Biosci., 2022, 9 Search PubMed.
- I. Brierley, S. Pennell and R. J.-C. Gilbert, Viral RNA pseudoknots: Versatile motifs in gene expression and replication., Nat. Rev. Microbiol., 2007, 5(8), 598–610 CrossRef CAS PubMed.
- E. P. Plant, R. Rakauskaitė, D. R. Taylor and J. D. Dinman, Achieving a golden mean: Mechanisms by which coronaviruses ensure synthesis of the correct stoichiometric ratios of viral proteins., J. Virol., 2010, 84(9), 4330–4340 CrossRef CAS PubMed.
- D. B. Ritchie, J. Soong, W. K.-A. Sikkema and M. T. Woodside, Anti-frameshifting ligand reduces the conformational plasticity of the SARS virus pseudoknot., J. Am. Chem. Soc., 2014, 136(6), 2196–2199 CrossRef CAS PubMed.
- K. Zhang, I. N. Zheludev, R. J. Hagey, R. Haslecker, Y. J. Hou, R. Kretsch, G. D. Pintilie, R. Rangan, W. Kladwang, S. Li, M. T.-P. Wu, E. A. Pham, C. Bernardin-Souibgui, R. S. Baric, T. P. Sheahan, V. D’Souza, J. S. Glenn, W. Chiu and R. Das, Cryo-EM and antisense targeting of the 28 kDa frameshift stimulation element from the SARS-CoV-2 RNA genome., Nat. Struct. Mol. Biol., 2021, 28(9), 747–754 CrossRef CAS PubMed.
- Y. Sun, L. Abriola, R. O. Niederer, S. F. Pedersen, M. M. Alfajaro, V. S. Monteiro, C. B. Wilen, Y.-C. Ho, W. V. Gilbert, Y. V. Surovtseva, B. D. Lindenbach and J. U. Guo, Restriction of SARS-CoV-2 replication by targeting programmed −1 ribosomal frameshifting., Proc. Natl. Acad. Sci. U. S. A., 2021, 118(26), e2023051118 CrossRef CAS PubMed.
- S. Munshi, K. Neupane, S. M. Ileperuma, M. T.-J. Halma, J. A. Kelly, C. F. Halpern, J. D. Dinman, S. Loerch and M. T. Woodside, Identifying inhibitors of −1 programmed ribosomal frameshifting in a broad spectrum of coronaviruses, Viruses, 2022, 14(2), 177 CrossRef CAS PubMed.
- D.-G. Ahn, G. Y. Yoon, S. Lee, K. B. Ku, C. Kim, K.-D. Kim, Y.-C. Kwon, G.-W. Kim, B.-T. Kim, S.-J. Kim and A. Novel, Frameshifting inhibitor having antiviral activity against zoonotic coronaviruses, Viruses, 2021, 13(8), 1639 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00015f |
| This journal is © The Royal Society of Chemistry 2022 |