
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[4 + 2] and [2 + 4] cycloaddition reactions on single- and double-stranded DNA: a dual-reactive nucleoside†
Anna
Bujalska
a,
Kaleena
Basran
b and
Nathan W.
Luedtke
 *ab
*ab
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland
bDepartment of Chemistry, McGill University, 801 Sherbrooke St. West, Montréal, Québec H3A 0B8, Canada. E-mail: nathan.luedtke@mcgill.ca
First published on 5th May 2022
Abstract
Here we report dual reactivity of diene-modified duplex DNA containing 5-(1,3-butadienyl)-2′-deoxyuridine “BDdU”. Normal-electron demand [4 + 2] cycloaddition proceeded upon addition of a maleimide, whereas inverse-electron demand [2 + 4] cycloaddition occurred upon addition of a tetrazine to give a novel, photoswitchable product.
Bioorthogonal “Click” reactions1 are widely used for bioconjugation of modified nucleic acids in vitro2 and in vivo.3 Post-synthetic labeling of nucleic acids using nucleophilic addition,4 normal-electron-demand Diels–Alder reactions (DA),5 and strain-promoted inverse-electron-demand Diels–Alder reactions (invDA)6 enable numerous bioanalytical, material and diagnostic techniques. The introduction of bulky modifications onto nucleosides and nucleotides is common practice for such labeling strategies, however, the resulting steric hindrance can interfere with duplex stability and/or nucleoside metabolism.7 The development of new strategies utilizing smaller, non-interfering bioorthogonal functional groups therefore remains a high priority.
Terminal alkenes have recently emerged as minimalistic modifications that can serve as reactive groups in catalyst-free bioconjugation reactions in vitro,8 on cell surfaces,9 in whole cells10 and animals.11 In 2014, our group reported the metabolic incorporation of 5-vinyl-2′-deoxyuridine (VdU) into cellular DNA,10b and later synthetic oligonucleotides,4g as well as its modification by means of invDA reactions with tetrazines. Herein, we report the synthesis and reactivity of 5-(1,3-butadienyl)-2′-deoxyuridine (BDdU) bearing a 1,3-butadienyl moiety attached to C5 position of 2′-deoxyuridine. We postulated that the expansion of the conjugated π system should increase the HOMO energy level of the molecule, allowing for improved HOMO–LUMO overlap5c and increased tetrazine reactivity as compared to VdU. In addition, the inclusion of a butadienyl group was envisioned to allow dual modality of DNA modification in both inverse- and normal-electron-demand Diels–Alder cycloaddition reactions with electron-poor dienes or dienophiles, respectively (Scheme 1).
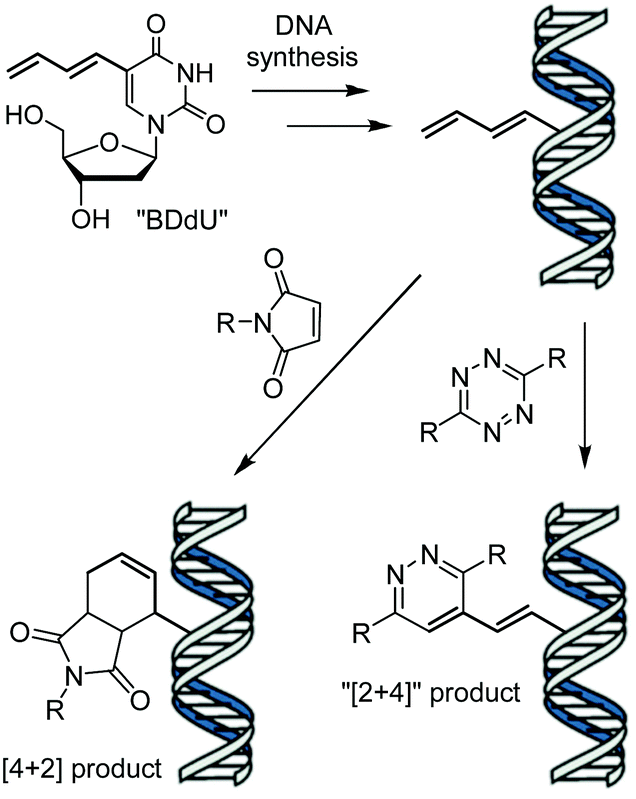 | ||
| Scheme 1 DNA containing 5-(1,3-butadienyl)-2′-deoxyuridine (BDdU) can react as a diene or dienophile upon addition of a maleimide or tetrazine, respectively. | ||
To synthesize 5-(1,3-butadienyl)-2′-deoxyuridine (BDdU, 2), we utilized a Stille cross coupling reaction between bromovinyl deoxyuridine (BVDU, 1) and tri-n-butylvinylstanne (Scheme 2), the resulting BDdU nucleoside was obtained in a 54% yield and fully characterized by 1H NMR, 13C NMR, and high resolution ESI MS (see ESI†). To prepare materials for DNA synthesis, BDdU (2) was treated with DMTrCl to give the corresponding DMTr-protected nucleoside (3) which was subsequently reacted with chlorophosphoramidite under basic conditions to give BDdU phosphoramidite (4) in a 28% overall isolated yield.
 | ||
| Scheme 2 Synthesis of BDdU nucleoside and phosphoramidite where THF = tetrahydrofuran, Pyr. = pyridine, DMTrCl = 4,4′-dimethoxytriphenylmethyl chloride, and DIPEA = N,N-diisopropylethylamine. See the ESI† for the synthesis and characterization of these compounds. | ||
To investigate the chemical reactivity of BDdU in an inverse-electron demand Diels–Alder reaction, BDdU (2) and 3,6-di-2-pyridyl-1,2,4,5-tetrazine were reacted in an aqueous solvent mixture, reaching completion after 3 hours with a 60% isolated yield of the oxidized product (6) containing a trans alkene (Scheme 3). In contrast, when the same reaction was conducted using VdU (7) under ambient conditions, the main product was the dihydrodiazine product (8).10b To evaluate their relative reaction rates, a series of BDdU-tetrazine reactions were conducted under pseudo-first-order conditions by monitoring the consumption of tetrazine (5) upon addition of a large excess of VdU or BDdU. The progress of each reaction was tracked by the disappearance of tetrazine's absorbance maximum at 530 nm (see Fig. S1, ESI†). The invDA cycloaddition reaction between BDdU (2) and tetrazine (5) exhibited an apparent, second-order reaction rate constant (k) = 7.4 × 10−2 M−1 s−1. This value was over 3-fold higher than that of VdU-tetrazine (5) with a rate constant (k) = 2.1 × 10−2 M−1 s−1 (Fig. S1, ESI†).
 | ||
| Scheme 3 The reaction between BDdU (2) and 3,6-di-2-pyridyl-1,2,4,5-tetrazine in the presence of ambient oxygen gives diazine (6), whereas VdU (7) gives mostly dihydrodiazine (8).10b | ||
The framework of oxidized product 6 resembles photoswitchable trans-stilbenes which have been reported to be fluorescent molecular rotors12 that can undergo UV-light mediated isomerization to give their corresponding cis isomers.13 Indeed, irradiation of a solution of trans-6 with 365 nm UV light at room temperature for 15 minutes resulted in the formation of two sets of peaks by 1H NMR that were assigned according to the coupling constants at 16 Hz for trans and 12 Hz for the cis isomer (see Fig. S4, ESI†). This trans-cis isomerization resulted in the steady-state formation of 71% of the cis isomer after 30 min of irradiation. Trans-cis photoswitching was accompanied by changes in the molecule's absorbance and fluorescence spectra (Fig. S2 and S3, ESI†).
To evaluate product formation in DNA, we synthesized oligonucleotides modified with BDdU (2) or VdU (7) using standard, solid-phase supported synthesis. The VdU phosphoramidite was synthesized according to published procedures.4g,14 We introduced BDdU (2) or VdU (7) into oligonucleotides using standard phosphoramidite chemistry and purified the products using HPLC (Fig. S6, ESI†). The modified single-stranded (ss) or double stranded (ds) DNA was reacted with a fluorescent, BODIPY-tetrazine conjugate “Bodipy-Tz” (Jena Bioscience) and analyzed using denaturing gel electrophoresis. Both ss and ds DNA containing BDdU exhibited a higher reactivity towards Bodipy-Tz than VdU in both ssDNA and dsDNA (Scheme 4). The products for each reaction were confirmed using MALDI-TOF-MS (Table S1, ESI†).
 | ||
| Scheme 4 Reactions of oligonucleotides modified with VdU (7) or BDdU (2) with Bodipy-Tz on single- (A) and double-stranded DNA (B). Oligonucleotides were incubated with a 10-, 100- or 250-fold excess of Bodipy-Tz for 2 h at 37 °C in 10 mM sodium cacodylate buffer (pH 7.4). The gels (22.5% PAGE) were stained with SYBR Gold in 1X TBE buffer and scanned using Typhoon FLA 9500. See ESI† for Bodipy-Tz structure. | ||
Oligonucleotides containing VdU exhibited moderate yields (41%) in single-stranded DNA (ssDNA) treated with a 250-fold excess of tetrazine for 2 hours at 37 oC. There was a drop to 24% conversion for the same reaction in double-stranded DNA (dsDNA) modified with VdU (7) after 2 hours of incubation with an excess of Bodipy-Tz. In contrast, the invDA reaction on oligonucleotides containing BDdU (2) was less impacted by being ssDNA or dsDNA. The conversion after 2 hours of incubation with 250-fold excess of tetrazine was 63% and 56% for ssDNA and dsDNA, respectively. These results suggest that BDdU (2) is more reactive towards tetrazines as compared to VdU (7) due to both electronic and steric effects in DNA.
To evaluate its reactivity towards maleimides, BDdU (2) and the fluorogenic maleimide (9) were incubated in a mixture of DMSO and water for 12 h at room temperature (Scheme 5). The reaction afforded a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture of products (10a + 10b) with a 37% overall isolated yield. The progress of the reaction could be monitored according to fluorescence increases over time (λex = 310 nm, λem = 366 nm, Fig. S5c, ESI†). Reactions conducted by adding 9 to BDdU-containing DNA gave a modest yield of approximately 45% for both the single-stranded and duplex DNA (Scheme 5 and Fig. S7, ESI†).
:1 diastereomeric mixture of products (10a + 10b) with a 37% overall isolated yield. The progress of the reaction could be monitored according to fluorescence increases over time (λex = 310 nm, λem = 366 nm, Fig. S5c, ESI†). Reactions conducted by adding 9 to BDdU-containing DNA gave a modest yield of approximately 45% for both the single-stranded and duplex DNA (Scheme 5 and Fig. S7, ESI†).
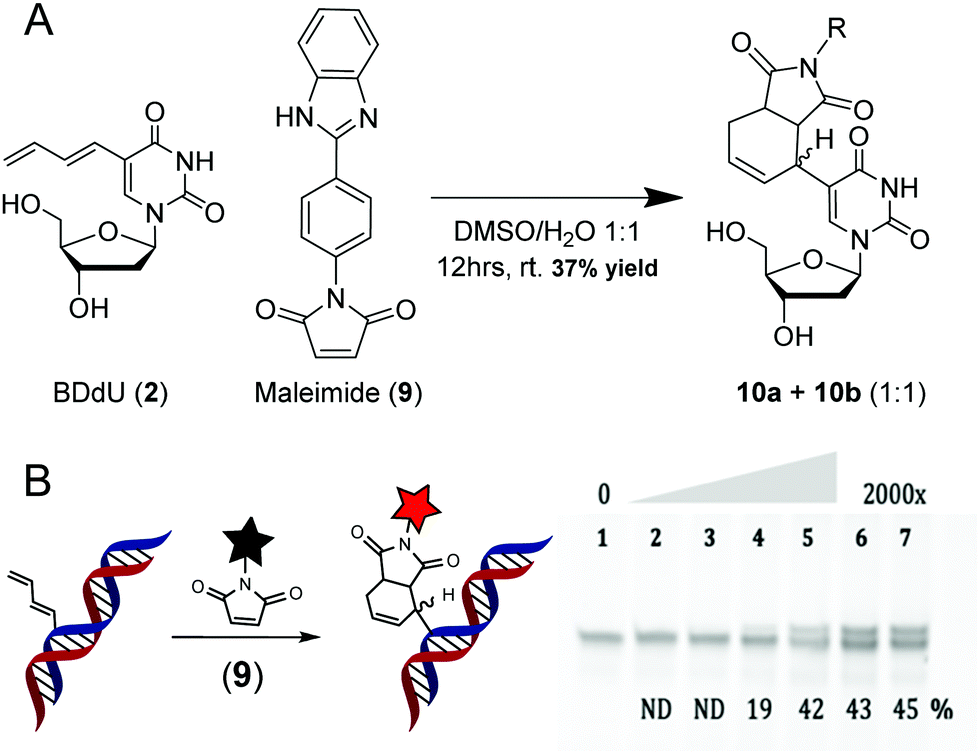 | ||
| Scheme 5 A. Reaction between BDdU nucleoside (2) and fluorogenic maleimide (9). B. Reactions between BDdU-containing duplex DNA with a 10-, 100-, 250-, 500-, 1000- or 2000-fold excess of 9 for 16 hours at 37 °C in 10 mM sodium cacodylate buffer (pH 7.4). The gels (22.5% denaturing PAGE) were stained with SYBR Gold solution in TBE buffer and scanned using Typhoon FLA 9500. Reaction yields are indicated at the bottom of the gel. The product identity for the DNA was confirmed using MALDI-TOF-MS (Table S1, ESI†). | ||
In summary, here we report the synthesis of a new, base-conjugated butadiene (BDdU) phosphoramidite and its incorporation into DNA where it exhibits dual reactivity, undergoing normal-electron demand [4 + 2] cycloaddition upon addition of a maleimide, or inverse-electron demand [2 + 4] cycloaddition upon addition of a tetrazine. In both cases, the reaction can be fluorogenic, where a fluorescence-quenching maleimide15 or a tetrazine is consumed in the reaction.6j Preliminary results suggest that BDdU will show good stability in the presence of cellular thiols (Fig. S8, ESI†), however, it may require installation of a membrane-permeable nucleotide triester “pro-phosphate” group to be metabolically incorporated into cellular DNA.7e
BDdU exhibited faster reaction rates with tetrazines as compared to VdU. These results demonstrate that a nucleobase-conjugated diene can exhibit a faster tetrazine reaction rate and give a product that is more prone to oxidation as compared to a simple terminal alkene. Following initial product formation and oxidation, trans-(6) undergoes a photo-isomerization reaction upon irradiation with 365 nm light to give cis-(6). The future development of light-mediated control of nucleobase structure and function may enable new strategies for manipulating biological processes.16
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the University of Zurich, McGill University, the Natural Sciences and Engineering Research Council of Canada (grant #05048), and the Canada Foundation for Innovation/John R. Evans Leaders Fund (#39168/253033) for funding.Notes and references
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and B. K. Sharpless, Angew. Chem., Int. Ed., 2002, 41(14), 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67(9), 3057–3064 CrossRef PubMed.
- (a) J. Gierlich, G. A. Burley, P. M.-E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8(17), 3639–3642 CrossRef CAS PubMed; (b) F. Seela, V. R. Sirivolu and P. Chittepu, Bioconjugate Chem., 2008, 19(1), 211–224 CrossRef CAS PubMed; (c) A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39(4), 1388 RSC; (d) E. Paredes and S. R. Das, ChemBioChem, 2011, 12(1), 125–131 CrossRef CAS PubMed; (e) I. Ivancová, D.-L. Leone and M. Hocek, Curr. Opin. Chem. Biol., 2019, 52, 136–144 CrossRef PubMed.
- (a) A. Salic and T. J. Mitchison, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(7), 2415–2420 CrossRef CAS PubMed; (b) A. B. Neef and N. W. Luedtke, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(51), 20404–20409 CrossRef CAS PubMed; (c) P. Shieh, V. T. Dien, B. J. Beahm, J. M. Castellano, T. Wyss-Coray and C. R. Bertozzi, J. Am. Chem. Soc., 2015, 137(22), 7145–7151 CrossRef CAS PubMed.
- (a) H. Eberhard, F. Diezmann and O. Seitz, Angew. Chem., Int. Ed., 2011, 50(18), 4146–4150 CrossRef CAS PubMed; (b) J. Dadová, P. Orság, R. Pohl, M. Brázdová, M. Fojta and M. Hocek, Angew. Chem., Int. Ed., 2013, 52(40), 10515–10518 CrossRef PubMed; (c) A. Sánchez, E. Pedroso and A. Grandas, Chem. Commun., 2013, 49(3), 309–311 RSC; (d) L. L.-G. Carrette, E. Gyssels, N. De Laet and A. Madder, Chem. Commun., 2016, 52(8), 1539–1554 RSC; (e) J. Matyašovský, P. Perlíková, V. Malnuit, R. Pohl and M. Hocek, Angew. Chem., Int. Ed., 2016, 55(51), 15856–15859 CrossRef PubMed; (f) A. Olszewska, R. Pohl, M. Brázdová, M. Fojta and M. Hocek, Bioconjugate Chem., 2016, 27(9), 2089–2094 CrossRef CAS PubMed; (g) A. Naik, J. Alzeer, T. Triemer, A. Bujalska and N. W. Luedtke, Angew. Chem., Int. Ed., 2017, 56(36), 10850–10853 CrossRef CAS PubMed.
- (a) V. Borsenberger and S. Howorka, Nucleic Acids Res., 2009, 37(5), 1477–1485 CrossRef CAS PubMed; (b) C. Paris, O. Brun, E. Pedroso and A. Grandas, Molecules, 2015, 20(4), 6389–6408 CrossRef CAS PubMed; (c) B. Milián-Medina and J. Gierschner, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 513–524 CrossRef.
- (a) J. Schoch, M. Wiessler and A. Jäschke, J. Am. Chem. Soc., 2010, 132(26), 8846–8847 CrossRef CAS PubMed; (b) J. Šečkutė, J. Yang and N. K. Devaraj, Nucleic Acids Res., 2013, 41(15), e148 CrossRef PubMed; (c) P. N. Asare-Okai, E. Agustin, D. Fabris and M. Royzen, Chem. Commun., 2014, 50(58), 7844–7847 RSC; (d) A. M. Pyka, C. Domnick, F. Braun and S. Kath-Schorr, Bioconjugate Chem., 2014, 25(8), 1438–1443 CrossRef CAS PubMed; (e) H. Wu, B. T. Cisneros, C. M. Cole and N. K. Devaraj, J. Am. Chem. Soc., 2014, 136(52), 17942–17945 CrossRef CAS PubMed; (f) C. Domnick, F. Eggert and S. Kath-Schorr, Chem. Commun., 2015, 51(39), 8253–8256 RSC; (g) X. Ren, A. H. El-Sagheer and T. Brown, Analyst, 2015, 140(8), 2671–2678 RSC; (h) K. Wang, D. Wang, K. Ji, W. Chen, Y. Zheng, C. Dai and B. Wang, Org. Biomol. Chem., 2015, 13(3), 909–915 RSC; (i) M. Merkel, S. Arndt, D. Ploschik, G. B. Cserép, U. Wenge, P. Kele and H.-A. Wagenknecht, J. Org. Chem., 2016, 81(17), 7527–7538 CrossRef CAS PubMed; (j) N. W. Luedtke and M. O. Loehr, Angew. Chem., Int. Ed., 2022, e202112931 Search PubMed.
- (a) F. Seela, E. Feiling, J. Gross, F. Hillenkamp, N. Ramzaeva, H. Rosemeyer and M. Zulauf, J. Biotechnol., 2001, 86(3), 269–279 CrossRef CAS PubMed; (b) A. B. Neef, L. Pernot, V. N. Schreier, L. Scapozza and N. W. Luedtke, Angew. Chem., Int. Ed., 2015, 54(27), 7911–7914 CrossRef CAS PubMed; (c) A. Hottin and A. Marx, Acc. Chem. Res., 2016, 49(3), 418–427 CrossRef CAS PubMed; (d) V. Raindlová, M. Janoušková, M. Slavíčková, P. Perlíková, S. Boháčová, N. Milisavljevič, H. Šanderová, M. Benda, I. Barvík, L. Krásný and M. Hocek, Nucleic Acids Res., 2016, 44(7), 3000–3012 CrossRef PubMed; (e) M. Tera, S. M.-K. Glasauer and N. W. Luedtke, ChemBioChem, 2018, 19(18), 1939–1943 CrossRef CAS PubMed.
- (a) H. Bußkamp, E. Batroff, A. Niederwieser, O. S. Abdel-Rahman, R. F. Winter, V. Wittmann and A. Marx, Chem. Commun., 2014, 50(74), 10827–10829 RSC; (b) A. R. Kore, B. Yang and B. Srinivasan, Tetrahedron Lett., 2015, 56(6), 808–811 CrossRef CAS.
- (a) A. Niederwieser, A.-K. Späte, L. D. Nguyen, C. Jüngst, W. Reutter and V. Wittmann, Angew. Chem., Int. Ed., 2013, 52(15), 4265–4268 CrossRef CAS PubMed; (b) Y.-J. Lee, Y. Kurra, Y. Yang, J. Torres-Kolbus, A. Deiters and W. R. Liu, Chem. Commun., 2014, 50(86), 13085–13088 RSC; (c) A.-K. Späte, V. F. Schart, S. Schöllkopf, A. Niederwieser and V. Wittmann, Chem. – Eur. J., 2014, 20(50), 16502–16508 CrossRef PubMed.
- (a) Q. Li, T. Dong, X. Liu and X. Lei, J. Am. Chem. Soc., 2013, 135(13), 4996–4999 CrossRef CAS PubMed; (b) U. Rieder and N. W. Luedtke, Angew. Chem., Int. Ed., 2014, 53(35), 9168–9172 CrossRef CAS PubMed; (c) J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6(2), 1362–1369 RSC; (d) H. Wu, S. C. Alexander, S. Jin and N. K. Devaraj, J. Am. Chem. Soc., 2016, 138(36), 11429–11432 CrossRef CAS PubMed.
- Y. Wu, G. Guo, J. Zheng, D. Xing and T. Zhang, ACS Sens., 2019, 4(1), 44–51 CrossRef CAS PubMed.
- A. Karimi, R. Börner, G. Mata and N. W. Luedtke, J. Am. Chem. Soc., 2020, 142(34), 14422–14426 CrossRef CAS PubMed.
- D. Schulte-Frohlinde, H. Blume and H. Güsten, J. Phys. Chem., 1962, 66(12), 2486–2491 CrossRef CAS.
- K. Fujimoto, S. Matsuda, N. Takahashi and I. Saito, J. Am. Chem. Soc., 2000, 122(23), 5646–5647 CrossRef CAS.
- J. Guy, K. Caron, S. Dufresne, S. W. Michnick, W. G. Skene and J. W. Keillor, J. Am. Chem. Soc., 2007, 129(39), 11969–11977 CrossRef CAS PubMed.
- (a) S. Ogasawara, ChemBioChem, 2014, 15, 2652–2655 CrossRef CAS PubMed; (b) K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118(21), 10710–10747 CrossRef PubMed; (c) M. L. Hammill, G. Islam and J. P. Desaulniers, ChemBioChem, 2020, 21(16), 2367–2372 CrossRef CAS PubMed; (d) Y. Wu, Z. Yang and Y. Lu, Curr. Opin. Chem. Biol., 2020, 57, 95–104 CrossRef CAS PubMed; (e) L. Yang, D. Trentini, H. Kim, J. Y. Sul, J. H. Eberwine and I. J. Dmochowski, ChemPhotoChem, 2021, 5(10), 940–946 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00062h |
| This journal is © The Royal Society of Chemistry 2022 |