
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImitate to illuminate: labeling of bacterial peptidoglycan with fluorescent and bio-orthogonal stem peptide-mimicking probes
Huibin
Lin
a,
Chaoyong
Yang
 *ab and
Wei
Wang
*a
*ab and
Wei
Wang
*a
aInstitute of Molecular Medicine, Shanghai Key Laboratory for Nucleic Acid Chemistry and Nanomedicine, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200127, China. E-mail: cyyang@xmu.edu.cn; wwang@shsmu.edu.cn
bThe MOE Key Laboratory of Spectrochemical Analysis and Instrumentation, Key Laboratory for Chemical Biology of Fujian Province State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
First published on 5th August 2022
Abstract
Because of its high involvement in antibiotic therapy and the emergence of drug-resistance, the chemical structure and biosynthesis of bacterial peptidoglycan (PGN) have been some of the key topics in bacteriology for several decades. Recent advances in the development of fluorescent or bio-orthogonal stem peptide-mimicking probes for PGN-labeling have rekindled the interest of chemical biologists and microbiologists in this area. The structural designs, bio-orthogonal features and flexible uses of these peptide-based probes allow directly assessing, not only the presence of PGN in different biological systems, but also specific steps in PGN biosynthesis. In this review, we summarize the design rationales, functioning mechanisms, and microbial processes/questions involved in these PGN-targeting probes. Our perspectives on the limitations and future development of these tools are also presented.
Introduction
A stiff envelope that counters the cytoplasmic turgor pressure and protects bacterial cells from a hostile environment, bacterial peptidoglycan (PGN) also acts as an interface interacting with the external environments. PGN's distinct biosynthesis pathway is nearly absent in eukaryotic cells, and the indispensability of these structures to bacteria make PGN an ideal target of antibiotics. As a result, understanding the structural and biosynthetic features of PGN has long been a fundamental topic in microbiology.1–4 Highly conserved among the great variety of bacterial species, PGN is an organized three-dimensional network, composed of a repeating disaccharide building unit which is attached with a stem peptide cross-linked to its neighboring peptides. The disaccharide is β-(1,4) linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc), and the stem peptide sequence is generally L-Ala-D-Glx-(L-Lys/m-DAP)-D-Ala-D-Ala, which is linked to MurNAc by its N-terminal (Glx stands for iso-glutamate or iso-glutamine and m-DAP stands for meso-2,6-diaminopimelic acid). The 3rd amino acid position can differ between different bacterial species, often L-Lys in most Gram-positive bacteria and m-DAP in Gram-negative bacteria. What is worth noting is that a small group of Gram-positive bacteria, including Bacilli, Clostridia, and Lactobacilli (all belonging to the phylum Firmicutes), are reported to have m-DAP on this position.3,5,6The de novo biosynthetic steps of PGN are briefly illustrated in Fig. 1a. The synthesis of the UDP-MurNAc-L-Ala-D-Glx-(L-Lys/m-DAP)-D-Ala-D-Ala (UDP-MurNAc-pentapeptide) subunit is catalyzed by a series of enzymes in the cytoplasm;7 then the UDP-MurNAc-pentapeptide is transferred to the cell membrane, followed by the formation of lipid II via a phosphotransferase MraY and a glycosyltransferase MurG. Lipid II is flipped into the periplasm by the flippase MurJ, where it is inserted into the growth point of cell wall by transglycosylases to extend the polysaccharide chain. The adjacent stem peptides are then cross-linked by penicillin binding proteins (PBPs) which catalyze 3–4 cross-links and are also known as D,D-transpeptidases (Ddts), and L,D-transpeptidases (Ldts, catalyze 3–3 cross-links) to form a dense network (Fig. 1b).8 Of note, the transglycosylase and transpeptidase activities are often carried out by different domains of the same PBP.9
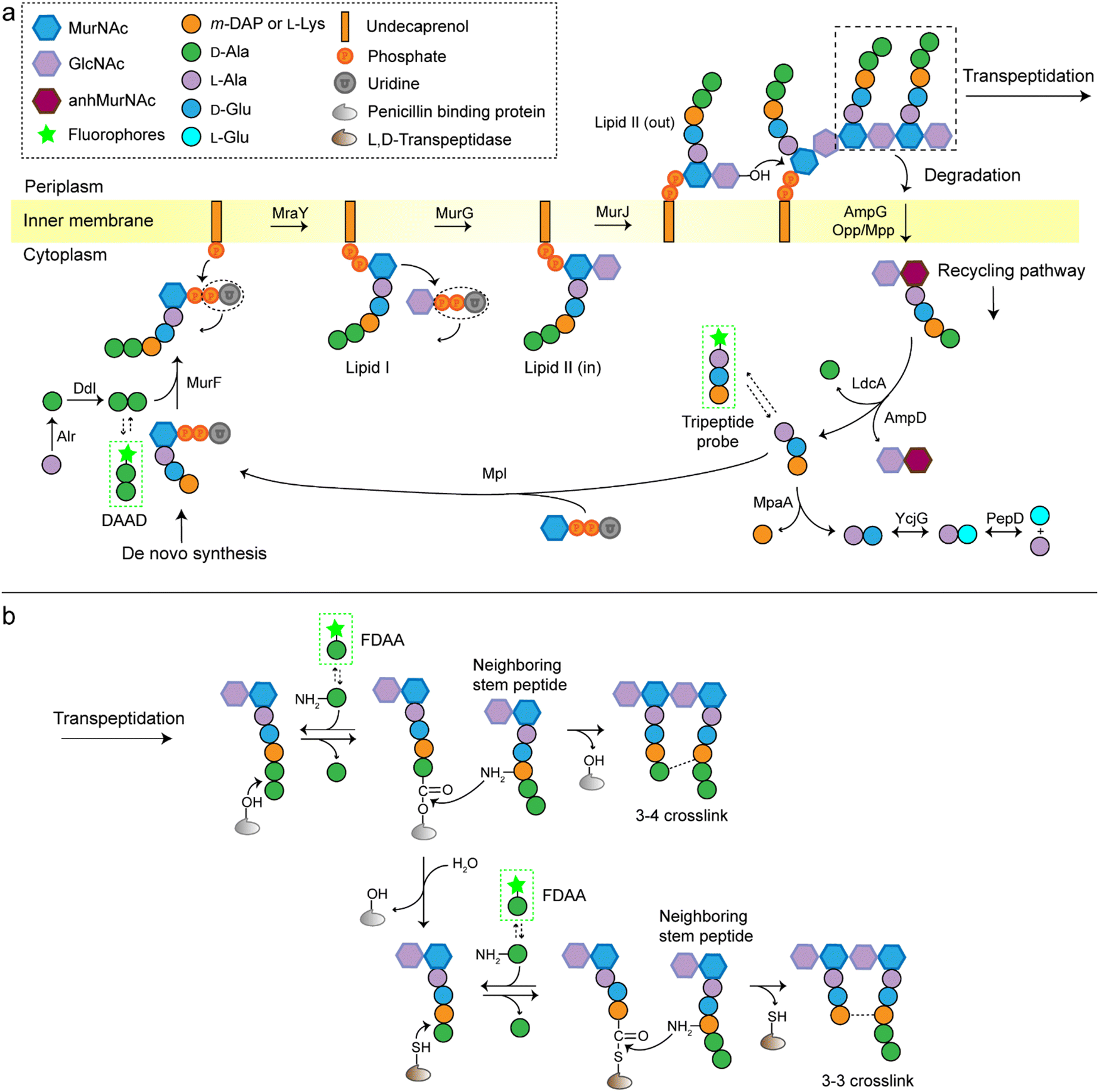 | ||
| Fig. 1 Schematic illustration of the biosynthesis and cross-linking of PGN. (a) The de novo pathway and recycling pathway of PGN synthesis. The D-Ala-D-Ala dipeptide is conjugated to UDP-MurNAc-L-Ala-D-Glx-(L-Lys/m-DAP) (UDP-MurNAc-tripeptide) to form the UDP-MurNAc-pentapeptide by MurF. Then catalyzed by phosphotransferase MraY, the UDP-MurNAc-pentapeptide is incorporated into a transport lipid (undecaprenyl phosphate) to form lipid I. Following catalysis of glycosyltransferase MurG, GlcNAc is linked to lipid I to form lipid II. Flippase MurJ then translocates lipid II to the outer face of the inner membrane, where its disaccharide core is linked into a growing glycan strand by PBPs (transglycosylase activity). The mature PGN can be degraded and the degradation product is transported into the cytoplasm by AmpG or Opp/Mpp permease system; then enzymatic hydrolysis by carboxypeptidase LdcA and amidase AmpD to form a tripeptide. The tripeptide can be further degraded by a series of enzymes into single amino acids or incorporated into a UDP-MurNAc by Mpl and reenter the de novo synthesis process of PGN. (b) Overview of PGN cross-linking. The PBPs can form 3–4 cross-links between adjacent peptide strands (D,D-transpeptidase activity), or cleave the terminal D-Ala residue from the peptide (D,D-carboxypeptidase activity). The product of the carboxypeptidase may then by catalyzed by L,D-transpeptidases (Ldts) to form 3–3 cross-links. GlcNAc, N-acetylglucosamine; MurNAc, N-acetylmuramic acid; anhMurNAc, 1,6-anhydro-N-acetylmuramic acid; m-DAP, meso-2,6-diaminopimelic acid. Alr, Ala racemase; Ddl, D-Ala–D-Ala ligase; MurF, UDP-MurNAc-tripeptide-D-alanyl-D-Ala ligase; MraY, phospho-UDP-MurNAc-pentapeptide transferase; MurG, undecaprenyldiphosphomuramoylpentapeptide N-acetylglucosaminyltransferase; MurJ, lipid II flippase; AmpG, GlcNAc-anhMurNAc permease; Opp, oligopeptide permease; Mpp, murein peptide permease; AmpD, anhMurNAc-L-Ala amidase; LdcA, L,D-carboxypeptidase; MpaA, γ-D-Glu-Dap amidase; YcjG, L-Ala-D/L-Glu epimerase; PepD, L-Ala-L-Glu dipeptidase; Mpl, UDP-MurNAc-L-Ala-D-Glu-m-Dap ligase. The green frames indicate the fluorescent probes for labelling of PGN. | ||
Traditional approaches for studying PGN structures and constructions include nuclear magnetic resonance and mass spectrometry-based analysis often aided by bacterial labeling with isotopic PGN precursors,10–12 and genetic engineering methods used to define the functions of specific enzymes.13,14 In the past decade, we have witnessed a rapid progress in PGN biology, partially because of the invention of new chemical probes for monitoring PGN constructions and modifications in living bacteria. Among these probes, fluorescent D-amino acids (FDAAs) have played a critical role,15–17 the applications of which have been thoroughly reviewed.18–20 FDAAs are anchored onto PGN by exchanging with one of the two terminal D-Ala, a process that is mediated by Ldts or Ddts (Fig. 1b). Because of the ease and versatility of probe synthesis and use, FDAA has been employed in labeling PGN in a variety of bacterial systems.20 Other categories of chemical probes used in PGN labeling, which were designed by mimicking a part of the stem peptide structures, have been recently proposed by several groups. The functioning of these probes relies on different endogenous enzymes involved in the PGN biosynthesis, thus providing opportunities to investigate specific steps in the process without the need for any genetic manipulations. In this review, we discuss the different types of stem peptide-mimicking probes, categorized based on their functioning mechanisms, for labeling and investigating the biosynthesis processes of PGN.
D-Amino acid dipeptide-based probes (DAADs)
During PGN synthesis in the cytoplasm, the D-Ala-D-Ala dipeptide formed by an alanine racemase (Alr) and a D-Ala-D-Ala ligase (Ddl) is incorporated into the UDP-MurNAc-tripeptide by a ligase MurF to form the UDP-MurNAc-pentapeptide (Fig. 1a). These steps are the targets of some experimental/commercial antibiotics (D-cycloserine, for example). In 2006, Bugg's group first showed the substrate promiscuity of MurF for unnatural dipeptides by incorporating a D-Cys-containing dipeptide into UDP-MurNAc-tripeptide,21 indicative of the possibility of using bio-orthogonal dipeptide probes in labeling PGN. In 2014, the Maurelli and VanNieuwenhze groups used this strategy and explored a long-standing question “chlamydial anomaly”,22 namely, Chlamydia contained genes for PGN biosynthesis and was susceptible to “anti-PGN” antibiotics23–26 but attempts to detect PGN in any Chlamydia species had been proved unsuccessful.27–29 Both D-Ala and D-Ala-D-Ala dipeptide could be incorporated by Chlamydia trachomatis based on the observation that sensitivity of C. trachomatis to D-cycloserine (D-Ala-D-Ala ligase inhibitor) could be reversed by addition of exogenous D-Ala and D-Ala–D-Ala but not by other D- or L-amino acids.24,30 However, ethynyl-D-Ala (EDA, a bio-orthogonal D-amino acid probe) could not label C. trachomatis, probably because the potentially existing PGN synthesis machinery in Chlamydia could not recognize side-chain modified D-amino acid.22 They then tested two clickable DAADs, ethynyl-D-Ala–D-Ala (EDA-DA) and DA-EDA (structures shown in Fig. 2a), in labeling Chlamydia. After the DAADs were anchored, reporter fluorophores were then installed in the ethynyl tag through a click reaction. Only the EDA-DA probe could successfully label C. trachomatis (Fig. 2b), probably due to the cleavage of the fifth amino acid from the pentapeptide during transpeptidation/carboxypeptidation.22 Therefore, via labeling with bio-orthogonal DAADs, the authors successfully proved the presence of functional ring-like PGN in C. trachomatis.22 Based on this finding, they further explored the characteristics of chlamydial PGN. By using three-dimensional super-resolution microscopy, they identified the PGN as a dynamic ring limited to the division plane of the cell, which occurred only in the replicative reticulate body phase of Chlamydia (Fig. 2c).31 They hypothesized that the limited ring-like PGN of pathogenic Chlamydia was the result of adaptation to its intracellular lifestyle, since PGN was a type of pathogen-associated molecular patterns (PAMPs) recognizable by the host immune system.31Via pulse-chase labeling with DAADs and immunostaining with MreB (an actin-like protein in bacteria), they proposed a synthesis model of chlamydial PGN, in which MreB moved along the ring plane to start the non-uniform synthesis of new PGN and meanwhile affected the functioning of a currently unknown degradation machinery which continuously removed older materials.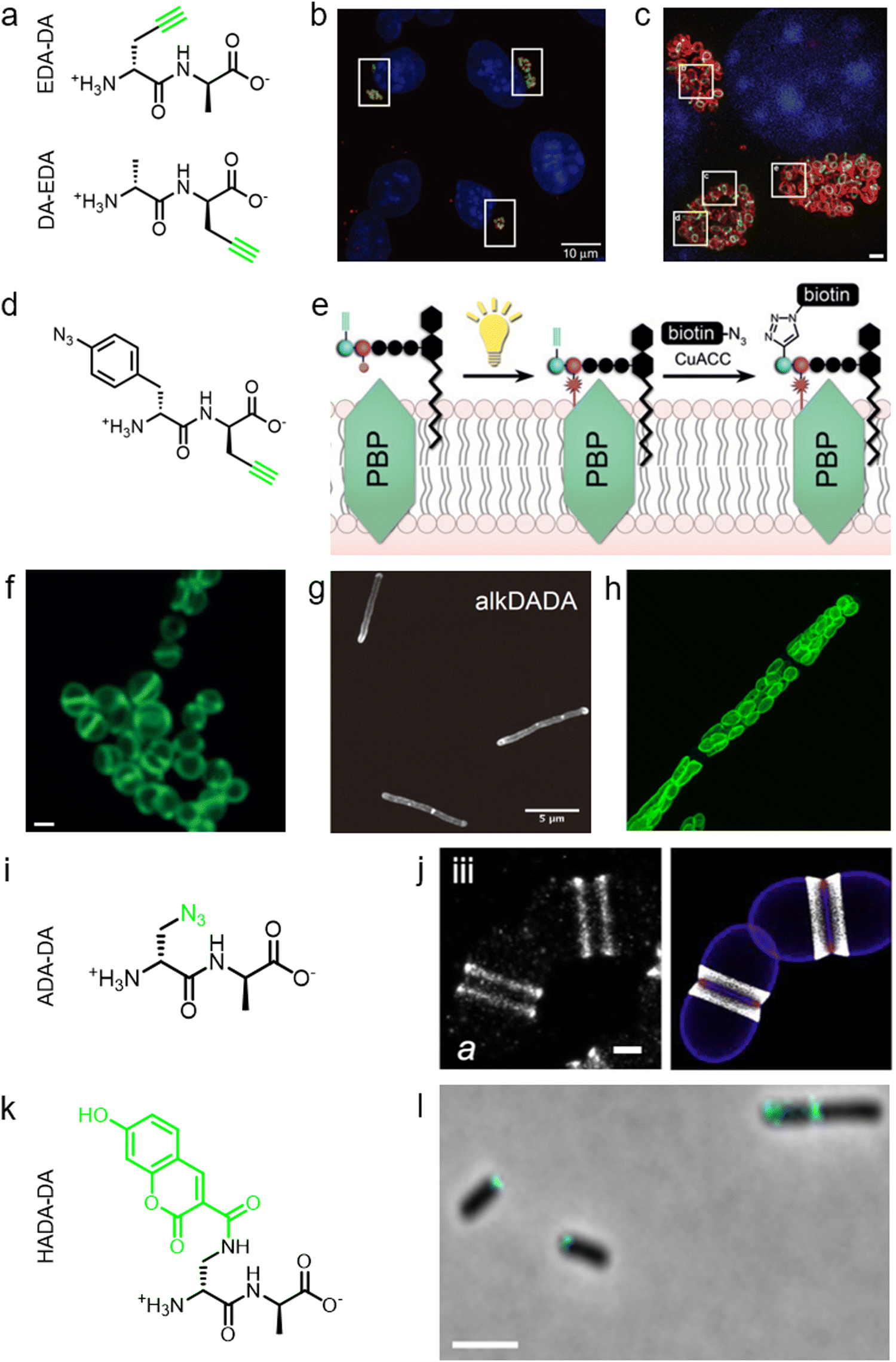 | ||
| Fig. 2 The use of DAADs in labeling various microbial cells. (a) Chemical structures of the dipeptide probe EDA-DA and DA-EDA. EDA, ethynyl-D-alanine, DA, D-alanine. (b) Fluorescence imaging of C. trachomatis (infecting L2 cells) labeled with EDA-DA (green). Reproduced with permission from ref. 22. Copyright 2013 Springer Nature. (c) Fluorescence imaging of the EDA-DA labeled ring-like PGN (green) on the middle plane of C. trachomatis. The figure was adapted from Liechti et al.31 (d) Chemical structure of the bifunctional dipeptide probe that contained a photo-crosslinking and a bio-orthogonal clickable handle. (e) Schematic illustration of the bifunctional dipeptide probe anchored onto the stem peptides and used to capture PBP-lipid II association. Reproduced with permission from ref. 41. Copyright 2016 John Wiley and Sons. (f) Fluorescence imaging of Enterococci labeled with EDA-DA (green). Reproduced with permission from ref. 44. Copyright 2017 John Wiley and Sons. (g) Fluorescence imaging of Mycobacterium smegmatis labeled with EDA-DA (abbreviated as alkDADA in ref. 45). The figure was adapted from García-Heredia et al.45 (h) Fluorescence imaging of moss chloroplasts labeled with EDA-DA (green). Reproduced with permission from ref. 46. Copyright 2016 Oxford University Press. (i) Chemical structure of the dipeptide probe ADA-DA. ADA, azido-D-alanine. (j) Reconstructed dSTORM image of S. pneumoniae labeled with ADA-DA (bright white). Reproduced with permission from ref. 47. Copyright 2021 Elsevier. (k) Chemical structure of the dipeptide probe HADA-DA. HADA, hydroxycoumarin-modified D-amino acid. (l) Fluorescence imaging of labeled A. tumefaciens with HADA-DA (cyan). The figure was adapted from Williams et al.48 | ||
Lipid II is a critical limiting precursor in PGN biosynthesis owing to its extremely low present levels.32,33 Many antibiotics function by interfering with PGN biosynthesis via binding and blocking lipid II processing, such as lantibiotics, vancomycin and teixobactin.34 Lipid II is incorporated into a growing glycan strand by PBPs’ transglycosylase activity.35 The in vitro interaction of lipid II and PBPs had been thoroughly studied,36–39 but their association within cells had not been directly characterized. Pires's group proposed a bifunctional DAAD that contained a photo-crosslinking40 and a clickable ethynyl group (structure shown Fig. 2d) and was still tolerable by MurF.41 Under irradiation, the photoaffinity handle of the probe could be activated into a highly reactive intermediate which quickly captured the surrounding associations by forming a covalent adduct (Fig. 2e). Then the adducts were subjected to click reaction with an azido-conjugated biotin handle, and the biotin-modified adducts (protein/lipid II-biotin) were subsequently analyzed by western blotting. Using this strategy, the authors captured the in situ interactions between lipid II and PBPs for the first time.
As a critical last-line antibiotic, vancomycin binds the D-Ala–D-Ala motif of lipid II in the periplasm to hinder lipid II from further transglycosylation and/or transpeptidation.42 To gain resistance to vancomycin, some pathogens can synthesize and utilize D-alanine–D-lactic (D-Ala–D-Lac) while hydrolyzing the original D-Ala–D-Ala,43 which resulted in the reprograming of lipid II termini from D-Ala to D-Lac, and greatly reduced the binding affinity of vancomycin. The hydrolysis of D-Ala–D-Ala dipeptide is catalyzed by VanX,43 the expression of which can be induced by treatment of vancomycin in resistant pathogens.44 Based on this principle, Pires's group used the EDA-DA probe to investigate the vancomycin-resistant kinetics of Enterococci.44 In sensitive stains, the probe was anchored into PGN in a normal PGN biosynthesis manner, and the labeled cells showed strong labeling after click reaction with fluorescent dyes (Fig. 2f). Conversely, in vancomycin-resistant Enterococci, the probe could be hydrolyzed by VanX, leading to a significant reduction of labeling signals.44 Therefore, by using this strategy, they could directly monitor the vancomycin-resistant phenotype switch of pathogens in real time.44
Furthermore, DAADs have also been used in investigating the PGN syntheses of many other cellular systems. In a report by Siegrist's lab, it was demonstrated again that the EDA-DA probe was incorporated into lipid II precursor through MurF.45 They combined the use of EDA and EDA-DA and showed that mycobacterial PGN construction was localized in the pole and along the sidewall during the synthetic and repair processes (Fig. 2g).45 In addition to bacteria, chloroplasts also have cell wall structures, and DAADs have been successfully used in probing PGN of moss chloroplasts (Fig. 2h).46 In another recent report by Morlot's group, to gain access to nanoscale details of PGN assembly in Streptococcus pneumoniae, the authors adopted a pulse-chase labeling strategy with a clickable probe ADA-DA (azido-D-Ala–D-Ala, Fig. 2i).47Via imaging with dSTORM (direct stochastic optical reconstruction microscopy), they proposed a model that both septal and peripheral PGN syntheses first occurred within a single ring-shaped zone which later divided into two concentric regions, and the elongation continued after the septation was finished (Fig. 2j).
In addition to bio-orthogonal DAADs, fluorescent-DAAD, which could eliminate the need for subsequent click reaction, was also recently developed by Brown's group (Fig. 2k).48 By combining the use of fluorescent-DAAD with FDAAs, they successfully visualized the unipolar growth of a plant pathogen Agrobacterium tumefaciens (Fig. 2l), a species that lacked cell elongation synthases, and also showed that a single class A PBP (PBP1a) was essential for polar PGN synthesis. These studies showcase the broad and intriguing applications of dipeptide-based probes, which provide more space for chemical modifications than single D-amino acid-based probes and enable studying specific PGN-related questions that could not be directly investigated by FDAAs.
Recycling pathway-based peptide probe
In addition to the de novo synthesis, PGN can also be constructed by cell-wall recycling pathway in many bacteria (Fig. 1). In E. coli, for example, a considerable amount of PGN is decomposed by lytic transglycosylases, amidases and endopeptidases, and then recycled.49 The enzymatic hydrolysate anhydromuropeptides are transported into the cytoplasm by the permease AmpG or the oligopeptide permease (Opp)/murein peptide permease (Mpp) pathway (Fig. 1a),49 where the peptides are cleaved by AmpD amidase.49 The terminal D-Ala is then released by LdcA to form the tripeptide L-Ala-D-Glu-m-Dap, which is ligated to UDP-MurNAc by murein peptide ligase (Mpl) as a building block and reenter the PGN synthesis process.49 By taking advantage of this pathway, Blaauwen's group developed a fluorescent tripeptide probe mimicking the recycling building block to label E. coli.50 The probe was based on the tripeptide L-Ala-γ-D-Glu-L-Lys modified with a NBD fluorophore on the lysine residue (AeK-NBD) (Fig. 3a). Lysine rather than m-DAP was chosen, which was the original amino acid on the 3rd position in E. coli, owing to the chemical availability of the lysine building blocks. By using fluorescence imaging, thin-layer chromatography analysis of lipid I-and lipid II-containing extracts, and LC-MS/MS analysis of PGN digests, they showed that AeK-NBD was successfully incorporated into the stem peptide of E. coli (Fig. 3b).50 The use of this method, however, is limited for several reasons. First, the fluorescent probe must enter the cytoplasm to get incorporated, which might lead to a high fluorescence background because of the free probe. Second, the tripeptide probe could still be degraded by MpaA, compromising its labeling efficiency. Of special note, because of the diverse recycling mechanisms in different bacteria,51 the development of new recycling pathway-dependent probes merits further exploration.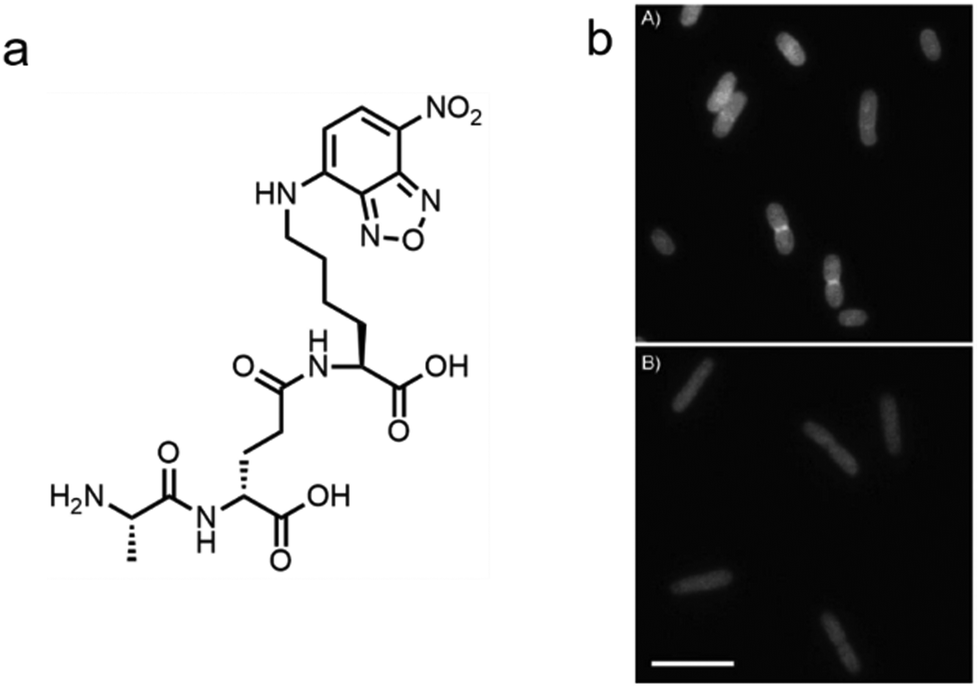 | ||
| Fig. 3 Tripeptide probe functioning via PGN-recycling labels E. coli in a Mpl dependent manner. (a) Chemical structure of the probe AeK-NBD. (b) Fluorescence imaging of AeK-NBD labeling of Mpl-deleted mutant E. coli strain transformed with plasmid expressing Mpl (above) and Mpl deleted mutant strain (below). Reproduced with permission from ref. 50. Copyright 2011 John Wiley and Sons. | ||
PGN cross-link targeting peptide probes
As a key step in the construction of the dense mesh-like PGN, cross-linking of neighboring stem peptides provides the bacterial cell walls with tensile strength and resistance to turgor pressure. Essential to bacterial growth, these cross-links represent a major target for antibiotic development (β-lactams, for example). However, what we currently know about the spatiotemporal features of cross-linking in bacteria is still very limited, which is partially due to the lack of suitable tools for investigating transpeptidase activity in living bacteria. In 2015, Spiegel's group first developed a fluorescent peptide imitating the endogenous substrates of PBPs. Based on the structure of the Staphylococcus aureus stem peptide, they designed a fluorescent hexapeptide probe (HexaFI) containing a PBP recognition motif L-Lys-D-Ala-D-Ala conjugated to a fluorophore by a tripeptide linker L-Lys-Gly-Gly, which could be incorporated into PGN via a 3-4 cross-link (Fig. 4a).52S. aureus can express PBP1-4 in a methicillin-sensitive strain and an additional PBP2a in a resistant strain.53 By comparing the labeling of the PBP4-mutant and wild type (WT) strains they found that HexaFl was selectively recognized by PBP4. Based on a previous report that wall teichoic acid (WTA) intermediates could recruit PBP4 to the septum,54 they pulse-labeled WTA-null (ΔTagO, deficient in WTA synthesis54) and WT S. aureus strain with the probe. Unlike the converged labeling at the septum in WT, the labelling of ΔTagO diffused throughout the cell wall (Fig. 4b, Van-A488 is a fluorescent derivative of vancomycin used as a marker for staining PGN). Taken these data together, they concluded that PBP4 activity was restricted to the septum in a WTA-dependent manner in S. aureus.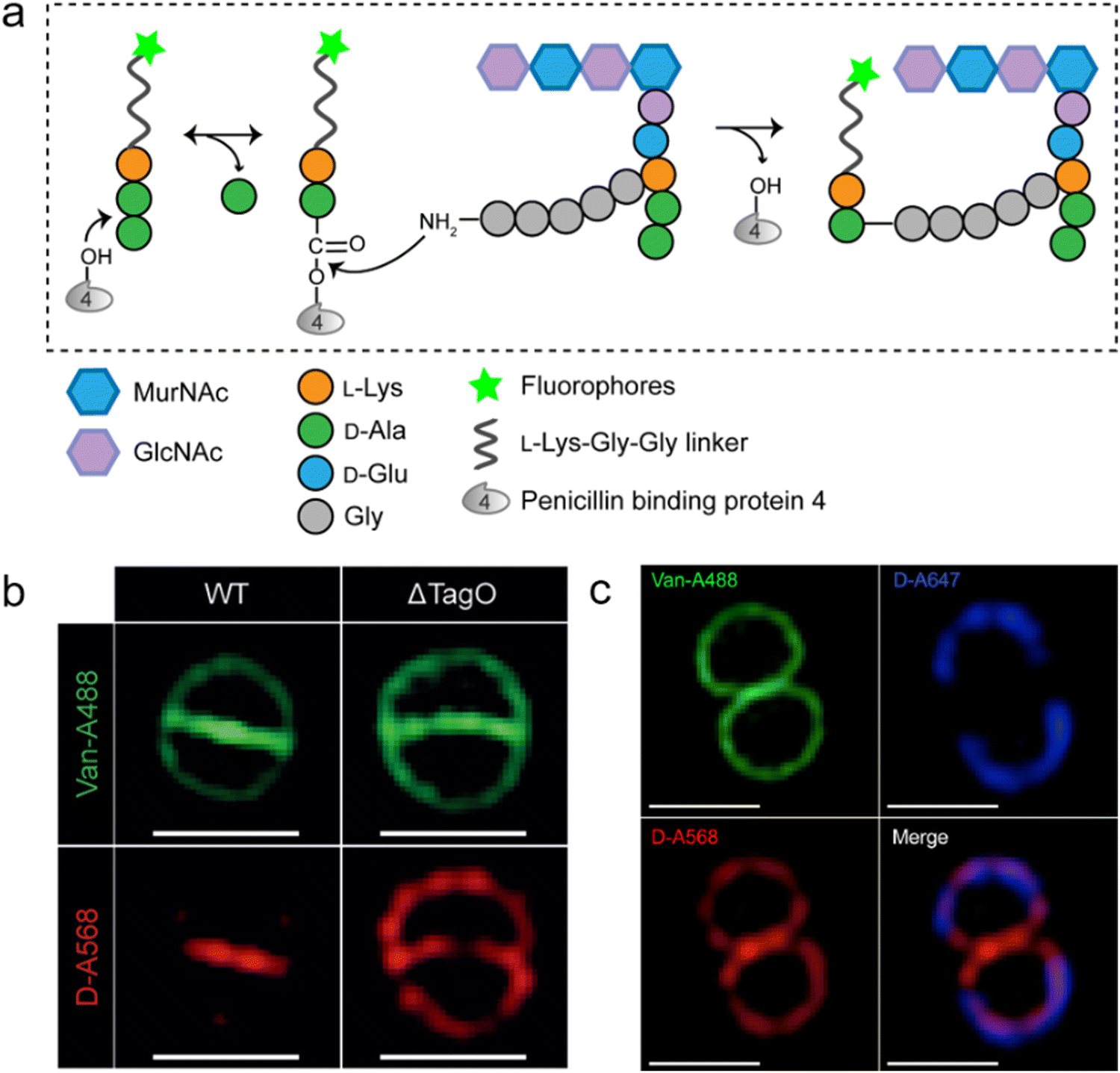 | ||
| Fig. 4 Fluorescent hexapeptide probe (HexaFI) interrogates cross-links mediated by PBP4 in S. aureus. (a) Schematic illustration of the functioning mechanism of HexaFI labeling. (b) WT and ΔTagO S. aureus strains labeled with Alexa Fluor 568 tagged HexaFI (abbreviated as D-A568, red) and stained with Van-A488 (Alexa Fluor 488 tagged vancomycin, green, indicating the whole cell wall). Reproduced with permission from ref. 52. Copyright 2015 John Wiley and Sons. (c) S. aureus treated with Alexa Fluor 647 tagged HexaFI (abbreviated as D-A647, blue) for 2 h. Free probe was then removed by washing and cells were cultured for 1 h before being labeled for 7.5 min with D-A568 (red) and stained with Van-A488 (green). Reproduced with permission from ref. 55. Copyright 2015 American Chemical Society. | ||
In a following report, the same group found that PBP4 was also functional outside the septum in non-separating S. aureus via a sequential labeling strategy.55S. aureus cells were first labeled uniformly with HexaFI-A647 (HexaFI tagged with Alexa Fluor 647) for 2 h to label the existing PGN (Fig. 4c, blue), and then cultured without probe for 1 h. Subsequently, the cells were pulse-labeled with HexaFI-A568 to mark the sites of newly synthesized PGN cross-links (red). As shown in Fig. 4c, HexaFI-A568 labeling overlapped with the HexaFI-A647 (blue) peripheral wall marker, indicating that PBP4 was also active outside the septum. They thus concluded that there were two spatiotemporally different cross-linking modes in S. aureus: one was located in the septum during cell division, and the other was in the sidewall between divisions.
In additional to the classical 3–4 cross-link formed by Ddts that exists in most bacteria, another 3–3 cross-link catalyzed by Ldts (Fig. 1b) can be found in a subgroup of bacterial species. It has been reported that 3–3 cross-links can function as supplement when the forming of 3–4 cross-links is inhibited by penicillin and cephalosporins; resistant to most β-lactams (except for carbapenems), Ldts were shown to help some bacteria develop drug resistance in vitro.56 Recently, chemical tools that could specifically probe the functioning of Ldts were proposed. In 2019, by mimicking the stem substrates of different transpeptidases, Pires's group constructed a series of fluorescent tetrapeptide (TetraFl) and pentapeptide (PentaFl) probes, which could label PGN via Ldts or Ddts, respectively (functioning mechanisms shown in Fig. 5a and b).57 The labeling of these probes against Enterococcus faecium (an Ldt-expressing bacterium) were thoroughly tested and compared. E. faecium treated with TetraFl led to a 5-fold fluorescence increase over PentaFl-treated cells (Fig. 5c and d), suggesting that the Ldt-mediated labeling was more efficient than that of Ddts. In addition to labeling under normal bacterial growth conditions, the author also evaluated how 3–3 cross-links were affected by different types of antibiotics. They found that treatment with penicillin-class of β-lactams led to a concentration-dependent increase in TetraFl labeling and decrease in PentaFl labeling. In contrast, carbapenems (meropenem and imipenem) led to a reduction of both TetraFl and PentaFl labeling, and antibiotics that were not β-lactams (vancomycin and erythromycin) caused no significant changes in fluorescence across all sublethal concentrations. The development of this tetrapeptide tool enabled direct probing of Ldts, a less-studied transpeptidase family potentially involved in the bacterial resistance to β-lactam antibiotics.
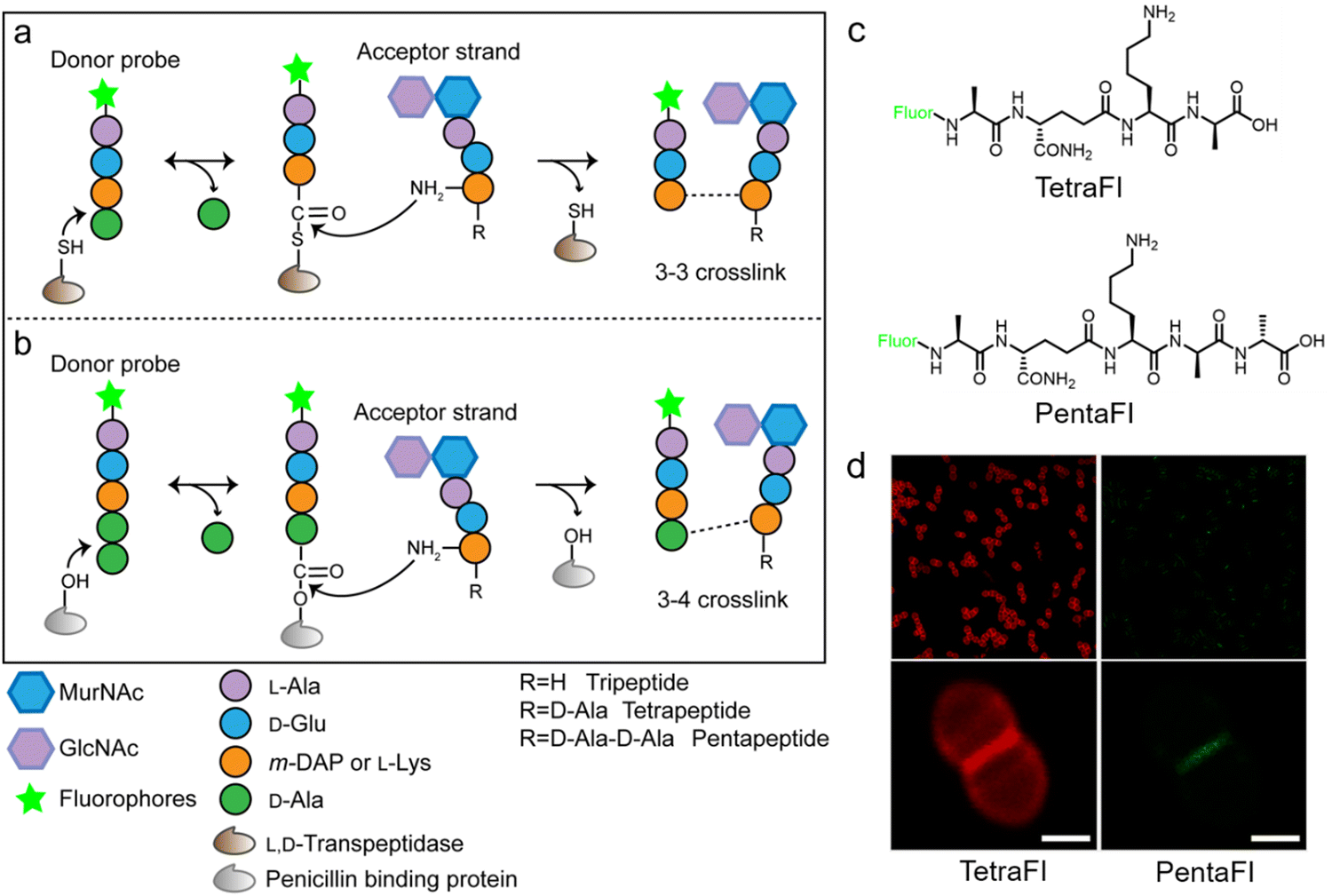 | ||
| Fig. 5 Acyl-donor peptide probes used in inquiring PGN cross-linking. (a and b) Schematic illustration of the functioning mechanisms of the acyl-donor peptide probes. (c) Chemical structures of TetraFl and PentaFl. (d) Fluorescence imaging of E. faecium labeled with TetraFl (red) and PentaFl (green). Reproduced with permission from ref. 57. Copyright 2019 American Chemical Society. | ||
As is shown in Fig. 1b, the transpeptidation occurs when the terminal amide bond of stem peptide is attacked by a hydroxyl group of Ddt or a sulfhydryl group of Ldt, forming an ester or a thioester bond. The acyl group of the PGN-enzyme intermediate was then attacked by the amino group on the side chain of m-DAP or L-Lys of a neighboring peptide. In this process, peptide chains which provide acyl groups are referred as the acyl-donor strand, and peptides providing amino groups as the acyl-acceptor strand.
The tetrapeptide-based probes described above are mostly processed as acyl-donor strands (FDAAs can thus also be considered as acyl acceptor probes). To evaluate how the acyl-acceptor strands were involved in cross-linking, Pires's group developed a new type of probe which could be processed exclusively as an acyl-acceptor (functioning mechanisms shown in Fig. 6a and b).58 In the probe, they installed an acyl-acceptor fragment (the cross-bridge D-iAsn or D-iAsn was included) and removed the terminal fragment recognizable by Ldt/Ddt as the acyl-donor site (terminal D-Ala or D-Ala–D-Ala) (structures shown in Fig. 6c).58 Therefore, the fluorescent tripeptide could only serve as an acyl-acceptor strand. Among different probe designs, the one containing iso-glutamine at the 2nd position and cross-bridge D-iAsn on the 3rd position showed the strongest labeling against E. faecium (Fig. 6c and d). Of note, this tripeptide probe could also attack the enzyme intermediate of Ddts and form a 3–4 cross-link (Fig. 6b). Taking these examples together, acyl-donor and -acceptor probes provide a versatile platform for dissecting PGN cross-linking processes in situ.
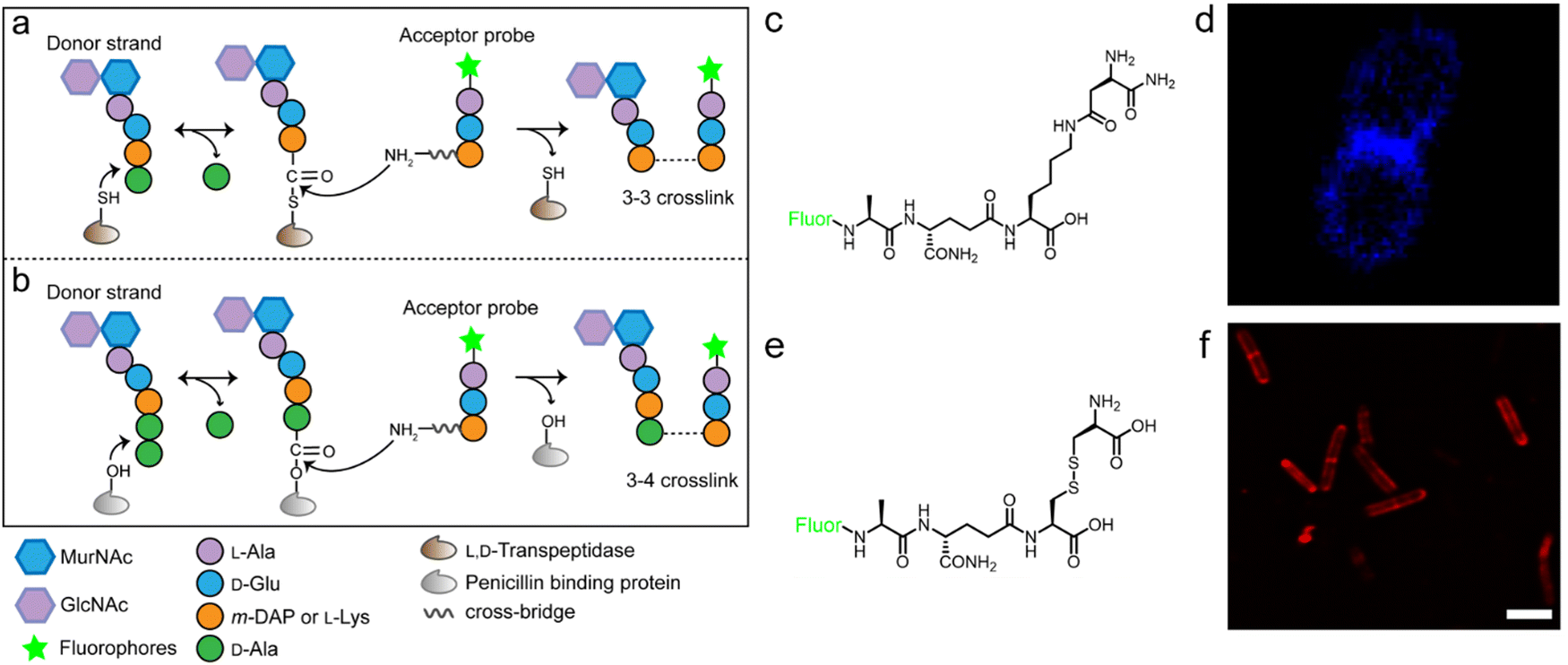 | ||
| Fig. 6 Acyl-acceptor peptide probes used in inquiring PGN cross-linking. (a and b) Schematic illustration of the functioning mechanisms of the acyl-acceptor peptide probe. (c) Chemical structures of acyl-acceptor probe. (d) Fluorescence image of E. faecium labeled with the acyl-acceptor probe (blue). Reproduced with permission from ref. 58. Copyright 2020 American Chemical Society. (e) Chemical structure of tripeptide probe containing m-CYT. (f) Fluorescence image of Mycobacterium smegmatis labeled with m-CYT tripeptide probe (red). Reproduced with permission from ref. 60. Copyright 2020 American Chemical Society. | ||
As described above, in most Gram-negative and a small portion of Gram-positive bacteria, the amino acid on the 3rd position of stem peptide is m-DAP. The difference of a carboxylic acid group on m-DAP relative to L-Lys has an important implication in the activation of the human innate immune system mediated by Nod1 and Nod2. For these two PAMP sensors, Nod2 recognizes the muramyl dipeptide, which exists in the PGN of nearly all bacteria, and yet Nod1 senses m-DAP-containing PGN fragments.59 Therefore, there is an urgent need to label bacteria that possess m-DAP-containing PGN and understand the interactions between their PGN fragments and Nod1. However, compared with the preparation of L-Lys-containing probes, the chemical synthesis of m-DAP-containing peptide is much more challenging. To circumvent this obstacle, Pires's group used meso-cystine (m-CYT) to mimic the structural features of m-DAP and constructed an acyl-acceptor probe to establish the recognition of the m-CYT side chain (structures shown in Fig. 6e).60 Incubated with Mycobacterium smegmatis (having m-DAP in their PGN), the m-CYT probes could be readily incorporated (Fig. 6f). Although this strategy provides a convenient protocol to study m-DAP-containing bacteria, the disulfide bond in m-CYT makes it vulnerable to various chemical or enzymatic environments, compromising its use in different settings.
Ldt-Catalyzed 3–3 cross-links have been shown in vitro to be associated with β-lactam resistance in some pathogens; however, the prevalence of Ldts in the mammalian gut microbiota and whether they could mediate resistance to β-lactams in vivo remained unknown. Considering the alternating composition of D- and L-amino acids in the stem peptides (potentially more resistant to peptidase hydrolysis in the intestines), our group recently exploited an in vivo labeling strategy, where the tetrapeptide acyl-donor probe (TetraAA-AcLys) was directly administered to mice, to investigate Ldts in their gut microbiota (scheme shown in Fig. 7a).61 After gavage, the probe efficiently labeled ∼18% of total gut bacteria, indicative of the high prevalence of Ldts and 3–3 cross-links in the mouse gut microbiota. With the aid of fluorescence in situ hybridization (FISH) staining, we further identified the cellular localizations of 3–3 cross-links in different bacterial species, including those that were still unculturable in the laboratory (Fig. 7b).61 Because only carbapenems in β-lactams could inhibit Ldts, the effects of meropenem (a carbapenem antibiotic) and ampicillin (a cephalosporin antibiotic) on Ldts-mediated cross-links in gut microbiota were compared. After oral administration of ampicillin or meropenem, not surprisingly, only the latter successfully inhibited the tetrapeptide labeling in the gut (Fig. 7c). These data suggested that the oral use of some β-lactam antibiotics might lead to microbiota dysbiosis caused by the biased growth of some Ldt-expressing bacteria. This strategy allows us to efficiently investigate the PGN structures of a large number of bacterial species and gain more knowledge of mammalian gut microbiota, endorsing the value of in vivo probing with chemical tools.
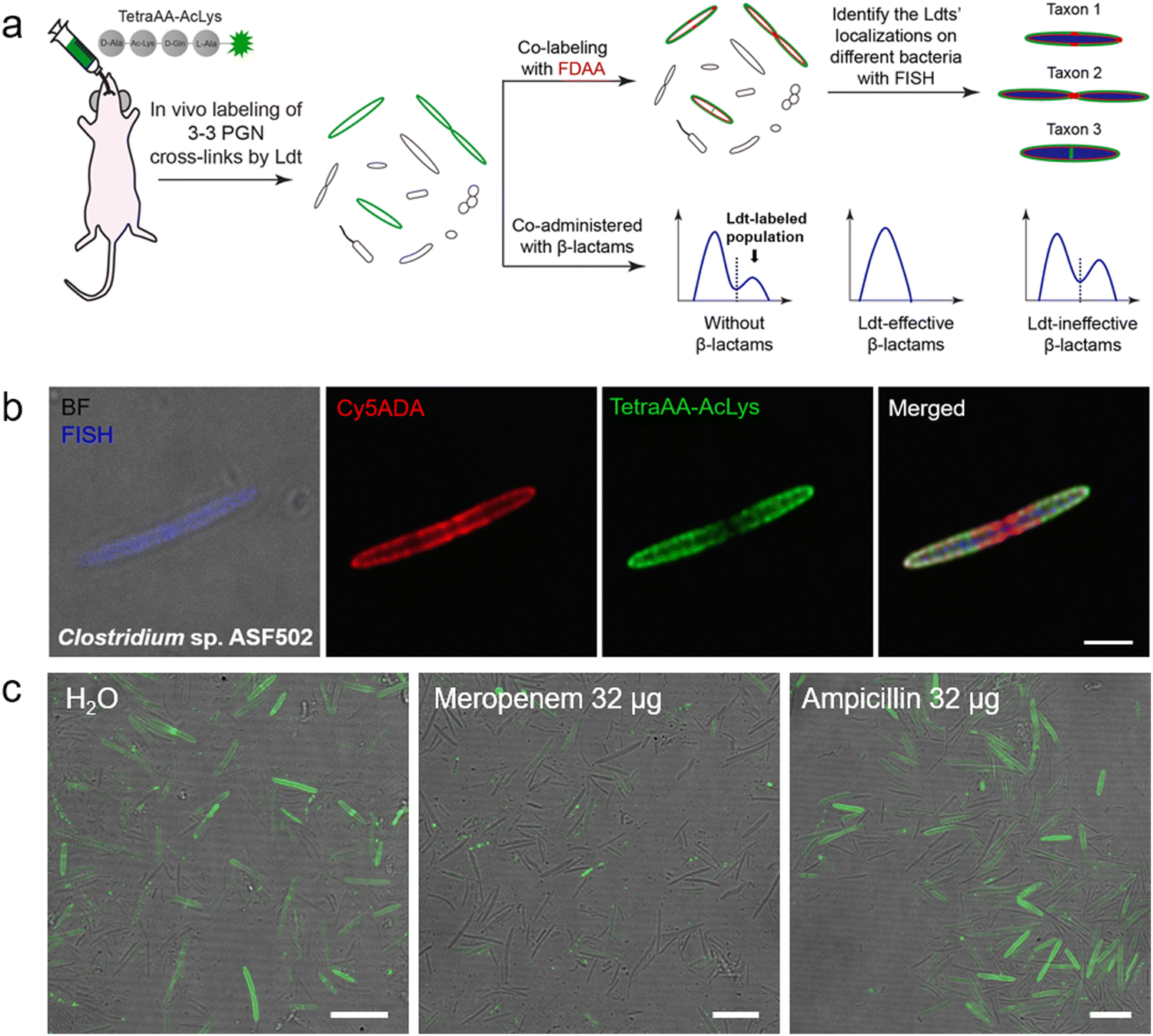 | ||
| Fig. 7 Tetrapeptide-based acyl-donor probes for investigating 3–3 cross-link in the mouse gut microbiota. (a) Schematic illustration of the in vivo Ldt-mediated labeling and probing workflow. (b) Fluorescence images revealing the relative localizations of 3–3 cross-links (green) on the cell surfaces of Clostridium sp. ASF502 (identified by FISH, blue), where the total PGN was labeled with Cy5ADA (a FDAA probe, red). (c) Fluorescence images of mouse gut microbiota treated with different antibiotics (meropenem or ampicillin) and labeled with the tetrapeptide probe (green) in vivo. Reproduced with permission from ref. 61. Copyright 2021 American Chemical Society. | ||
Conclusions and perspectives
Highly conserved among different bacteria, PGN's construction and degradation processes have been attracting wide attention from microbiologists for more than eight decades.2 However, the spatiotemporal features of PGN biosynthesis were still not well-understood until a few years ago, when Maurelli's group and collaborators discovered that MreB (bacterial homolog of actin) mediated a directional septum synthesis in Chlamydia through short pulse labeling with DAADs and immunostaining with MreB in 2016,31 and two other groups independently reported the FtsZ (bacterial homolog of tubulin)-guided treadmilling construction of a cell wall in model bacterial species via the use of sequential FDAA-labeling in 2017.62,63 There's no doubt that the development of PGN-targeting chemical probes has dramatically advanced our knowledge in this field by enabling facile and direct visualization of PGN localizations and dynamics. The multi-steps involved in the synthesis, modification and recycling of PGN provide space for chemical biologists to design new chemical tools targeting different stages of the construction. Here, we reviewed the different stem peptide-mimicking fluorescent probes, a major category of these labeling tools, including dipeptide probes designed for the de novo synthesis pathway, tripeptide probes for the recycling pathway, and tetra/penta-peptide probes for the cross-linking steps (summarized in Table 1). Structural variations of PGN among different bacteria, especially at the 3rd amino acid of the stem and the cross-bridges connecting neighboring peptides, may potentially allow selective labeling of certain groups of bacteria via a rational design of the peptide probes. The complementary use of these peptide tools with FDAAs (all function via endogenous enzymes of bacteria), and another MurNAc-based unnatural sugar probe (genetically engineered bacteria required) developed by the Grimes group64 will offer more versatility and facets for PGN studies. It needs to be mentioned that the high stability and the bio-orthogonal feature of these peptide probes make them powerful tools for in vivo probing of bacteria, including both pathogens at the infection sites and the commensal microbiotas. We envision that the development and use of these versatile tools will bring new insights into the broad microbiology society in the near future.| Probes | Figures | Reporter groups | Labeling mechanisms | Key enzymes | Bacteria typically used in labeling | Ref. |
|---|---|---|---|---|---|---|
| a NA, not available. | ||||||
| Single D-amino acid | NAa | Fluorescent or bio-orthogonal tag | Acyl-acceptor for 3–3 or 3–4 cross-link | Ldt; Ddt | Highly diverse | 15–20 |
| D-Amino acid-composed dipeptide | Fig. 2a | Alkyne | Lipid II synthesis | MurF | Chlamydia and others | 22, 31 and 44–46 |
| Fig. 2d | Alkyne | Bacillus subtilis | 41 | |||
| Fig. 2i | Azide | Streptococcus pneumoniae | 47 | |||
| Fig. 2k | Fluorophore | Agrobacterium tumefaciens | 48 | |||
| Stem tripeptide | Fig. 3a | Fluorophore | PGN recycling | Mpl | Escherichia coli | 50 |
| Stem tripeptide with a linker | Fig. 4a | Fluorophore | Acyl-donor for 3-4 cross-link | PBP4 | Staphylococcus aureus | 52 and 55 |
| Stem tetrapeptide | Fig. 5c | Fluorophore | Acyl-donor for 3–3 cross-link | Ldt | Enterococcus faecium; Gut microbiota | 57 and 61 |
| Stem pentapeptide | Fig. 5c | Fluorophore | Acyl-donor for 3–4 cross-link | Ddt | Enterococcus faecium | 57 |
| Stem tripeptide with a cross-bridge amino acid | Fig. 6c | Fluorophore | Acyl-acceptor for 3–3 or 3–4 cross-link | Ldt; Ddt | Enterococcus faecium | 58 |
| Stem tripeptide | Fig. 6e | Fluorophore | Acyl-acceptor for 3–3 or 3–4 cross-link | Ldt; Ddt | Mycobacterium smegmatis | 60 |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grants 22122702, 21735004, and 21775128) and the Innovative Research Team of High-Level Local Universities in Shanghai for financial support (SHSMU-ZLCX20212601).References
- M. J. Osborn, Annu. Rev. Biochem., 1969, 38, 501–538 CrossRef CAS PubMed.
- M. R. J. Salton, New Compr. Biochem., 1994, 27, 1–22 CAS.
- W. Vollmer, D. Blanot and M. A. de Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS.
- A. J. F. Egan, J. Errington and W. Vollmer, Nat. Rev. Microbiol., 2020, 18, 446–460 CrossRef CAS.
- K. H. Schleifer and O. Kandler, Bacteriol. Rev., 1972, 36, 407–477 CrossRef CAS.
- E. Work, Nature, 1957, 179, 841–847 CrossRef CAS.
- A. Typas, M. Banzhaf, C. A. Gross and W. Vollmer, Nat. Rev. Microbiol., 2011, 10, 123–136 CrossRef PubMed.
- A. Aliashkevich and F. Cava, FEBS J., 2021, 289, 4718–4730 CrossRef PubMed.
- X. Chen, C. H. Wong and C. Ma, ACS Infect. Dis., 2019, 5, 1493–1504 CrossRef CAS PubMed.
- P. J. Calabretta, H. L. Hodges, M. B. Kraft, V. M. Marando and L. L. Kiessling, J. Am. Chem. Soc., 2019, 141, 9262–9272 CrossRef PubMed.
- J. D. Chang, A. G. Wallace, E. E. Foster and S. J. Kim, Biochemistry, 2018, 57, 1274–1283 CrossRef CAS PubMed.
- J. A. H. Romaniuk and L. Cegelski, Biochemistry, 2018, 57, 3966–3975 CrossRef CAS.
- R. W. Bourdeau, A. Lee-Gosselin, A. Lakshmanan, A. Farhadi, S. R. Kumar, S. P. Nety and M. G. Shapiro, Nature, 2018, 553, 86–90 CrossRef CAS PubMed.
- A. Lakshmanan, Z. Jin, S. P. Nety, D. P. Sawyer, A. Lee-Gosselin, D. Malounda, M. B. Swift, D. Maresca and M. G. Shapiro, Nat. Chem. Biol., 2020, 16, 988–996 CrossRef CAS PubMed.
- E. Kuru, H. V. Hughes, P. J. Brown, E. Hall, S. Tekkam, F. Cava, M. A. de Pedro, Y. V. Brun and M. S. VanNieuwenhze, Angew. Chem., Int. Ed., 2012, 51, 12519–12523 CrossRef CAS PubMed.
- E. Kuru, A. Radkov, X. Meng, A. Egan, L. Alvarez, A. Dowson, G. Booher, E. Breukink, D. I. Roper, F. Cava, W. Vollmer, Y. Brun and M. S. VanNieuwenhze, ACS Chem. Biol., 2019, 14, 2745–2756 CrossRef CAS PubMed.
- M. S. Siegrist, S. Whiteside, J. C. Jewett, A. Aditham, F. Cava and C. R. Bertozzi, ACS Chem. Biol., 2013, 8, 500–505 CrossRef CAS PubMed.
- A. D. Radkov, Y. P. Hsu, G. Booher and M. S. VanNieuwenhze, Annu. Rev. Biochem., 2018, 87, 991–1014 CrossRef CAS PubMed.
- L. Lin, Y. Du, J. Song, W. Wang and C. Yang, Acc. Chem. Res., 2021, 54, 2076–2087 CrossRef CAS PubMed.
- Y. P. Hsu, G. Booher, A. Egan, W. Vollmer and M. S. VanNieuwenhze, Acc. Chem. Res., 2019, 52, 2713–2722 CrossRef CAS PubMed.
- J. A. Schouten, S. Bagga, A. J. Lloyd, G. de Pascale, C. G. Dowson, D. I. Roper and T. D. Bugg, Mol. BioSyst., 2006, 2, 484–491 RSC.
- G. W. Liechti, E. Kuru, E. Hall, A. Kalinda, Y. V. Brun, M. VanNieuwenhze and A. T. Maurelli, Nature, 2014, 506, 507–510 CrossRef CAS PubMed.
- J. W. Moulder, Infect. Agents Dis., 1993, 2, 87–99 CAS.
- J. W. Moulder, D. L. Novosel and J. E. Officer, J. Bacteriol., 1963, 85, 707–711 CrossRef CAS PubMed.
- A. Tamura and G. P. Manire, J. Bacteriol., 1968, 96, 875–880 CrossRef CAS PubMed.
- R. S. Stephens, S. Kalman, C. Lammel, J. Fan, R. Marathe, L. Aravind, W. Mitchell, L. Olinger, R. L. Tatusov, Q. Zhao, E. V. Koonin and R. W. Davis, Science, 1998, 282, 754–759 CrossRef CAS PubMed.
- A. Fox, J. C. Rogers, J. Gilbart, S. Morgan, C. H. Davis, S. Knight and P. B. Wyrick, Infect. Immun., 1990, 58, 835–837 CrossRef CAS PubMed.
- T. P. Hatch, J. Bacteriol., 1996, 178, 1–5 CrossRef CAS PubMed.
- A. G. Barbour, K. Amano, T. Hackstadt, L. Perry and H. D. Caldwell, J. Bacteriol., 1982, 151, 420–428 CrossRef CAS PubMed.
- A. J. McCoy and A. T. Maurelli, Mol. Microbiol., 2005, 57, 41–52 CrossRef CAS PubMed.
- G. Liechti, E. Kuru, M. Packiam, Y. P. Hsu, S. Tekkam, E. Hall, J. T. Rittichier, M. VanNieuwenhze, Y. V. Brun and A. T. Maurelli, PLoS Pathog., 2016, 12, e1005590 CrossRef PubMed.
- J. van Heijenoort, Microbiol. Mol. Biol. Rev., 2007, 71, 620–635 CrossRef CAS PubMed.
- D. Mengin-Lecreulx and J. van Heijenoort, J. Bacteriol., 1985, 163, 208–212 CrossRef CAS PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- S. A. Cochrane and C. T. Lohans, Eur. J. Med. Chem., 2020, 194, 112262 CrossRef CAS PubMed.
- A. Bouhss, A. E. Trunkfield, T. D. Bugg and D. Mengin-Lecreulx, FEMS Microbiol. Rev., 2008, 32, 208–233 CrossRef CAS PubMed.
- G. E. Jackson and J. L. Strominger, J. Biol. Chem., 1984, 259, 1483–1490 CrossRef CAS PubMed.
- Y. Qiao, M. D. Lebar, K. Schirner, K. Schaefer, H. Tsukamoto, D. Kahne and S. Walker, J. Am. Chem. Soc., 2014, 136, 14678–14681 CrossRef CAS PubMed.
- A. Zapun, J. Philippe, K. A. Abrahams, L. Signor, D. I. Roper, E. Breukink and T. Vernet, ACS Chem. Biol., 2013, 8, 2688–2696 CrossRef CAS PubMed.
- P. K. Mishra, C. M. Yoo, E. Hong and H. W. Rhee, ChemBioChem, 2020, 21, 924–932 CrossRef CAS PubMed.
- S. Sarkar, E. A. Libby, S. E. Pidgeon, J. Dworkin and M. M. Pires, Angew. Chem., Int. Ed., 2016, 55, 8401–8404 CrossRef CAS PubMed.
- P. E. Reynolds, Eur. J. Clin. Microbiol. Infect. Dis., 1989, 8, 943–950 CrossRef CAS PubMed.
- D. Kahne, C. Leimkuhler, W. Lu and C. Walsh, Chem. Rev., 2005, 105, 425–448 CrossRef CAS PubMed.
- S. E. Pidgeon and M. M. Pires, Angew. Chem., Int. Ed., 2017, 56, 8839–8843 CrossRef CAS PubMed.
- A. García-Heredia, A. A. Pohane, E. S. Melzer, C. R. Carr, T. J. Fiolek, S. R. Rundell, H. C. Lim, J. C. Wagner, Y. S. Morita, B. M. Swarts and M. S. Siegrist, eLife, 2018, 7, e37234 CrossRef PubMed.
- T. Hirano, K. Tanidokoro, Y. Shimizu, Y. Kawarabayasi, T. Ohshima, M. Sato, S. Tadano, H. Ishikawa, S. Takio, K. Takechi and H. Takano, Plant Cell, 2016, 28, 1521–1532 CrossRef CAS PubMed.
- J. Trouve, A. Zapun, C. Arthaud, C. Durmort, A. M. Di Guilmi, B. Soderstrom, A. Pelletier, C. Grangeasse, D. Bourgeois, Y. S. Wong and C. Morlot, Curr. Biol., 2021, 31, 2844–2856 CrossRef CAS PubMed.
- M. A. Williams, A. Aliashkevich, E. Krol, E. Kuru, J. M. Bouchier, J. Rittichier, Y. V. Brun, M. S. VanNieuwenhze, A. Becker, F. Cava and P. J. B. Brown, mBio, 2021, 12, e02346–21 CrossRef PubMed.
- J. T. Park and T. Uehara, Microbiol. Mol. Biol. Rev., 2008, 72, 211–227 CrossRef CAS PubMed.
- N. K. Olrichs, M. E. Aarsman, J. Verheul, C. J. Arnusch, N. I. Martin, M. Herve, W. Vollmer, B. de Kruijff, E. Breukink and T. den Blaauwen, ChemBioChem, 2011, 12, 1124–1133 CrossRef CAS PubMed.
- J. W. Johnson, J. F. Fisher and S. Mobashery, Ann. N. Y. Acad. Sci., 2013, 1277, 54–75 CrossRef CAS PubMed.
- S. Gautam, T. Kim, T. Shoda, S. Sen, D. Deep, R. Luthra, M. T. Ferreira, M. G. Pinho and D. A. Spiegel, Angew. Chem., Int. Ed., 2015, 54, 10492–10496 CrossRef CAS PubMed.
- M. G. Pinho, M. Kjos and J. W. Veening, Nat. Rev. Microbiol., 2013, 11, 601–614 CrossRef CAS PubMed.
- M. L. Atilano, P. M. Pereira, J. Yates, P. Reed, H. Veiga, M. G. Pinho and S. R. Filipe, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18991–18996 CrossRef CAS PubMed.
- S. Gautam, T. Kim and D. A. Spiegel, J. Am. Chem. Soc., 2015, 137, 7441–7447 CrossRef CAS PubMed.
- J. L. Mainardi, R. Legrand, M. Arthur, B. Schoot, J. van Heijenoort and L. Gutmann, J. Biol. Chem., 2000, 275, 16490–16496 CrossRef CAS PubMed.
- S. E. Pidgeon, A. J. Apostolos, J. M. Nelson, M. Shaku, B. Rimal, M. N. Islam, D. C. Crick, S. J. Kim, M. S. Pavelka, B. D. Kana and M. M. Pires, ACS Chem. Biol., 2019, 14, 2185–2196 CAS.
- A. J. Apostolos, S. E. Pidgeon and M. M. Pires, ACS Chem. Biol., 2020, 15, 1261–1267 CrossRef CAS PubMed.
- L. Franchi, N. Warner, K. Viani and G. Nunez, Immunol. Rev., 2009, 227, 106–128 CrossRef CAS PubMed.
- A. J. Apostolos, J. M. Nelson, J. R. A. Silva, J. Lameira, A. M. Achimovich, A. Gahlmann, C. N. Alves and M. M. Pires, ACS Chem. Biol., 2020, 15, 2966–2975 CrossRef CAS PubMed.
- H. Lin, L. Lin, Y. Du, J. Gao, C. Yang and W. Wang, ACS Chem. Biol., 2021, 16, 1164–1171 CrossRef CAS PubMed.
- A. W. Bisson-Filho, Y. P. Hsu, G. R. Squyres, E. Kuru, F. Wu, C. Jukes, Y. Sun, C. Dekker, S. Holden, M. S. VanNieuwenhze, Y. V. Brun and E. C. Garner, Science, 2017, 355, 739–743 CrossRef CAS PubMed.
- X. Yang, Z. Lyu, A. Miguel, R. McQuillen, K. C. Huang and J. Xiao, Science, 2017, 355, 744–747 CrossRef CAS PubMed.
- K. E. DeMeester, H. Liang, J. Zhou, K. A. Wodzanowski, B. L. Prather, C. C. Santiago and C. L. Grimes, Curr. Protoc. Chem. Biol., 2019, 11, e74 CAS.
| This journal is © The Royal Society of Chemistry 2022 |