
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic metabolism for in vitro acetone biosynthesis driven by ATP regeneration†
Ekaterina
Kozaeva‡
,
Manuel
Nieto-Domínguez‡
 ,
Abril D.
Hernández
and
Pablo I.
Nikel
*
,
Abril D.
Hernández
and
Pablo I.
Nikel
*
The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800 Kongens Lyngby, Denmark. E-mail: pabnik@biosustain.dtu.dk; Tel: +93 51 19 18
First published on 16th September 2022
Abstract
In vitro ketone production continues to be a challenge due to the biochemical features of the enzymes involved—even when some of them have been extensively characterized (e.g. thiolase from Clostridium acetobutylicum), the assembly of synthetic enzyme cascades still face significant limitations (including issues with protein aggregation and multimerization). Here, we designed and assembled a self-sustaining enzyme cascade with acetone yields close to the theoretical maximum using acetate as the only carbon input. The efficiency of this system was further boosted by coupling the enzymatic sequence to a two-step ATP-regeneration system that enables continuous, cost-effective acetone biosynthesis. Furthermore, simple methods were implemented for purifying the enzymes necessary for this synthetic metabolism, including a first-case example on the isolation of a heterotetrameric acetate:coenzyme A transferase by affinity chromatography.
Introduction
Acetone [(CH3)2CO, propanone or dimethyl ketone] is one of the most commonly-used industrial solvents as well as a platform chemical widely used in multiple industrial sectors that include the manufacturing of plastics, pharmaceutics and textiles, together with applications in agriculture.1–5 Mostly produced as a by-product of phenol chemical synthesis in the cumene process, acetone production continues to rely on energy-intensive, oil-derived processes that result in the generation of hazardous waste and greenhouse gas emissions.6–9 This scenario prompted the development of alternative processes for acetone production, including bacterial fermentation. Indeed, the ‘acetone-butanol-ethanol’ (ABE) fermentation by Clostridium strains isolated by Chaim Weizmann in the first half of the 20th century has been recognized as one of the most efficient processes for acetone (and structurally-related ketones) bioproduction.10–13 Due to issues in connection with climate change and fossil sources availability, biotechnological chemical production received renewed interest14–18—and sustainable, carbon-neutral acetone biomanufacturing has been recently demonstrated at the industrial scale.7Multiple research efforts have been deployed towards improving ketone production by microbes, with promising acetone titers (up to 122 mM in cultures of engineered Escherichia coli strains1,19). Supported by dedicated strain engineering approaches, previous studies shed light on the molecular mechanisms underlying the regulation of the acetone biosynthetic pathway.6,19–23 Enhanced ketones and alcohols production has been demonstrated by using engineered bacteria, adapted to consume different feedstocks under optimized fermentation setups.24 However, limited knowledge on the biocatalysts involved often resulted in relatively low yields, which remain to be a major hurdle for the development of economically-feasible ABE fermentation and related bioprocesses.25,26 A detailed understanding of the reaction properties and the biochemical features of the individual enzymes is necessary to address this issue. A simple, one-pot system composed by purified enzymes could accelerate the implementation and optimization of alternative bioproduction processes.
In this vein, in vitro systems, based either on purified enzymes or cell-free extracts, constitute a powerful approach to quantitatively evaluate pathway performance.27–29 Such approaches enable efficient bioproduction without the interference brought about by the presence of competing pathways in vivo, while avoiding potential toxicity issues exerted on the production host by product accumulation. Moreover, the precise control of reaction conditions allows for gaining useful insight on specifics about the pathway and its component enzymes—ultimately supporting the optimization of in vivo biosynthesis. Nevertheless, a limitation of in vitro approaches is the relatively high cost of the biocatalysts, together with the need of supplementing (equally expensive) cofactors to sustain enzyme-based catalysis.30,31 Along this line of reasoning, could the implementation of novel in vitro strategies help solve some of the current limitations for optimizing acetone biosynthesis? Synthetic metabolism in vitro, based on purified components, offers the possibility of exploring a number of practical aspects relevant for high-titer production of value-added compounds, including pathway bottlenecks,32 which cannot be assessed directly in vivo.
In this study, we describe the development of a synthetic metabolism for efficient acetone biosynthesis that overcomes the most relevant bottlenecks previously reported for the cognate pathway. On the one hand, we developed a platform to study parameters affecting both the reaction and key enzymes involved in the acetone biosynthetic pathway. On the other hand, the issue of cost-effectiveness in cell-free bioproduction was addressed by coupling the enzymatic cascade to a highly-efficient ATP-regeneration system. By rationally combining these approaches, we demonstrate maximal theoretical yields for acetate-dependent acetone biosynthesis without ATP supplementation in a self-sustaining enzymatic cascade.
Materials and methods
Bacterial strains, construction of plasmids and culture conditions
Bacterial strains and plasmids used in this study are presented in Table 1. Bacterial cultures [i.e. E. coli DH5α λpir or BL21(DE3)] were incubated at 37 °C and agitated at 200 rpm (MaxQ™ 8000 incubator; Thermo Fisher Scientific, Waltham, MA, USA). For general cloning procedures, lysogeny broth (LB medium, containing 10 g L−1 tryptone, 5 g L−1 yeast extract and 10 g L−1 NaCl) was used for cell growth; solid culture media additionally contained 15 g L−1 agar. For protein production experiments, cells were grown in 2 × YT medium,33 containing 16 g L−1 tryptone, 10 g L−1 yeast extract and 10 g L−1 NaCl; solid culture media additionally contained 15 g L−1 agar. Kanamycin (Km) was added whenever needed at 50 μg mL−1. The optical density of cell cultures was measured at 600 nm (OD600) to estimate biomass concentrations, and recorded in a Genesys 20 spectrophotometer (Thermo Fisher Scientific).| Bacterial strain | Relevant characteristicsa | Reference or source |
|---|---|---|
| a Antibiotic markers: Km, kanamycin and Nal, nalidixic acid. The source of relevant genes is indicated with a superscript as follows: Ca, Clostridium acetobutylicum; Ec, Escherichia coli; Aj, Acinetobacter johnsonii and Sm, Sinorhizobium meliloti. | ||
| Escherichia coli | ||
| DH5α λpir | Cloning host; F− λ−endA1 glnX44(AS) thiE1 recA1 relA1 spoT1 gyrA96(NalR) rfbC1 deoR nupG Φ80(lacZΔM15) Δ(argF-lac)U169 hsdR17(rK−mK+), λpir lysogen | Hanahan and Meselson69 |
| BL21(DE3) | Protein production host; F−ompT gal [dcm and lon] hsdS(rB−mB+) with DE3, a λ prophage carrying the T7 RNA polymerase gene and lacIQ | Jeong et al.70 |
| Plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| pET-28a(+)-TEV | Expression vector; oriV(pBR322), T7 promoter; KmR | GeneScript |
| pET28a(+)-NtHis6×-TEV-Thl | Derivative of vector pET-28a(+)-TEV harboring the thlCa gene in phase with a N-terminal 6×His-tag and the TEV cleavage site; PT7 →His6× → TEV-site → thlCa; KmR | This work |
| pET28a(+)-NtHis6×-TEV-AtoDA | Derivative of vector pET-28a(+)-TEV harboring the atoDAEc genes in phase with a N-terminal 6×His-tag and the TEV cleavage site; PT7 → His6× → TEV-site→atoAEc; KmR | This work |
| pET28a(+)-NtHis6×-TEV-Adc | Derivative of vector pET-28a(+)-TEV harboring the adcCa gene in phase with a N-terminal 6×His-tag and the TEV cleavage site; PT7 → His6× → TEV-site → adcCa; KmR | This work |
| pET28a(+)-CtHis6×-Adc | Derivative of vector pET-28a(+)-TEV harboring the adcCa gene in phase with a C-terminal 6×His-tag; PT7 → adcCa → His6×; KmR | This work |
| pET28a(+)-Adc | Derivative of vector pET-28a(+)-TEV harboring the adcCa gene; PT7 → adcCa; KmR | This work |
| pET28a(+)-PpkA | Derivative of vector pET-28a(+)-TEV harboring the ppk2Aj gene in phase with a N-terminal 6×His-tag and the TEV cleavage site; PT7 → His6× → TEV-site → ppk2Aj; KmR | This work |
| pET28a(+)-PpkS | Derivative of vector pET-28a(+)-TEV harboring the ppk2Sm gene in phase with a N-terminal 6×His-tag and the TEV cleavage site; PT7 → His6× → TEV-site → ppk2Sm; KmR | This work |
General cloning procedures and construction of plasmids
Oligonucleotides and gene fragments used in this work are listed in Table S1 and S2 in the ESI†. The gene fragments encoding the two polyphosphate (polyP) kinases34 used in this study [i.e. from Acinetobacter johnsonii (PPK2Aj) and Sinorhizobium meliloti (PPK2Sm)] were synthesized by Twist Bioscience (San Francisco, CA, USA). Genes encoding thiolase (Thl) and acetoacetate decarboxylase (Adc) from Clostridium acetobutylicum, as well as the fragment encoding acetate: coenzyme A (CoA) transferase (AtoDA) from E. coli, were amplified from plasmid pS4318·MKs.1 These fragments were inserted into a modified pET-28a(+) vector as N-terminal His6×-tag fusions by uracil-excision (USER) cloning using well-established protocols.35–37 The AMUSER tool was employed for designing oligonucleotides.36,38 In case of Adc, the variants (i) without a tag, (ii) with N-terminal or (iii) with C-terminal His6×-tag fusions were constructed. Phusion™ U high-fidelity DNA polymerase (ThermoFisher Scientific, Waltham, MA, USA) was used according to the manufacturer's specifications in amplifications intended for USER cloning. The vectors constructed herein encode a TEV cleavage site between the His6×-tag and the gene sequence encoding the protein of interest, and the native START codon was removed in all cases towards creating gene fusions. For colony PCR amplifications, the commercial OneTaq™ master mix (New England BioLabs, Ipswich, MA, USA) was used according to the supplier's instructions. E. coli DH5α λpir (Table 1) was employed as the bacterial host for general cloning purposes. Chemically-competent E. coli cells were prepared and transformed with plasmids using the Mix and Go™ commercial kit (Zymo Research, Irvin, CA, USA) according to the manufacturer's indications. The DNA sequence of all used plasmids was verified by Mix2Seq sequencing (Eurofins Genomics, Ebersberg, Germany). Following sequence verification, the plasmids were transformed into E. coli BL21(DE3) for protein production experiments (Table 1).Protein production and purification procedures
A single E. coli BL21(DE3) colony carrying individual pET-28a(+) constructs of interest (Table 1) was used to inoculate 5 mL of LB medium supplemented with Km and grown overnight at 37 °C with agitation at 200 rpm. Next, 500 mL (final volume) of 2 × YT medium supplemented with Km was inoculated with the overnight culture at a 1% (v/v) ratio and incubated at 37 °C and 200 rpm until OD600 reached 0.5–0.7. At this point, protein production was induced by addition of 0.5 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG; Sigma-Aldrich Co., St. Louis, MO, USA). Cultures were then allowed to grow overnight at 20 °C and 200 rpm, after which the cells were harvested by centrifugation (4000 × g, 20 min, 4 °C). Cell pellets were stored at −20 °C prior to protein extraction and purification.All samples were kept on ice throughout all purification processes. To this end, bacterial pellets were resuspended in 20 mL of buffer A (20 mM sodium phosphate buffer, pH = 7.5, 300 mM NaCl and 20 mM imidazole). The resulting cell suspensions were lysed by three passes through an Emulsiflex C5 high-pressure homogenizer39 at 1.0–1.5 kbar (Avestin Europe GmbH, Mannheim, Germany). Subsequently, 25 U mL−1 of Pierce™ universal nuclease for cell lysis was added to each of the samples, incubated for 30 min at room temperature and centrifuged (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 20 min, 4 °C) to remove cell debris. Following centrifugation and filtration of cell extracts through 0.2 μm membranes, protein purification was carried out using 1 mL of HisPur Ni-NTA Resin (Thermo Scientific) in 10 mL PierceTM disposable columns. Non-bound proteins were washed with 20 mL of buffer A before elution with 4 mL of buffer B (20 mM sodium phosphate buffer, pH = 7.5, 300 mM NaCl and 500 mM imidazole). The buffer of the eluted fraction was exchanged to 20 mM sodium phosphate, pH = 7.5, 300 mM NaCl and 1 mM EDTA, using PD-10 desalting columns (Cytiva, Marlborough, MA, USA) according to the instructions of the manufacturer. Protein concentrations were determined by measuring the absorbance at 280 nm in a NanoDropTM 2000 spectrophotometer (ThermoFisher Scientific) and using the theoretical extinction coefficient (ε) of the proteins as calculated by protparam (available at https://web.expasy.org/protparam; Table S3 in the ESI†). The degree of purification of each enzyme was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 4–20% precast polyacrylamide gels (Bio-Rad, Hercules, CA, USA) and loading 1 μg protein sample per lane (unless indicated otherwise). Purified enzyme fractions were aliquoted and stored at 4 °C until further use or, in some cases, flash-frozen in liquid N2 with 20% (v/v) glycerol for prolonged storage at −80 °C.
000 × g, 20 min, 4 °C) to remove cell debris. Following centrifugation and filtration of cell extracts through 0.2 μm membranes, protein purification was carried out using 1 mL of HisPur Ni-NTA Resin (Thermo Scientific) in 10 mL PierceTM disposable columns. Non-bound proteins were washed with 20 mL of buffer A before elution with 4 mL of buffer B (20 mM sodium phosphate buffer, pH = 7.5, 300 mM NaCl and 500 mM imidazole). The buffer of the eluted fraction was exchanged to 20 mM sodium phosphate, pH = 7.5, 300 mM NaCl and 1 mM EDTA, using PD-10 desalting columns (Cytiva, Marlborough, MA, USA) according to the instructions of the manufacturer. Protein concentrations were determined by measuring the absorbance at 280 nm in a NanoDropTM 2000 spectrophotometer (ThermoFisher Scientific) and using the theoretical extinction coefficient (ε) of the proteins as calculated by protparam (available at https://web.expasy.org/protparam; Table S3 in the ESI†). The degree of purification of each enzyme was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 4–20% precast polyacrylamide gels (Bio-Rad, Hercules, CA, USA) and loading 1 μg protein sample per lane (unless indicated otherwise). Purified enzyme fractions were aliquoted and stored at 4 °C until further use or, in some cases, flash-frozen in liquid N2 with 20% (v/v) glycerol for prolonged storage at −80 °C.
The untagged, native Adc protein was produced and purified as previously described.40 In this case, recombinant cells were grown in 200 mL of auto-induction media41 with Km (200 mg L−1) and incubated at 37 °C and 200 rpm for 24 h. An additional bacterial culture in 2 × YT was also prepared following the method referenced above in order to compare production yields. After cell lysis and nuclease treatment, the proteins of the bacterial host were precipitated by incubation at 65 °C for 2 h. The heat-treated cell extract was centrifuged (12000 × g, 20 min, 4 °C). The buffer was exchanged and the samples were stored as detailed above. In this case, protein quantification was carried out with the commercial QubitTM protein assay according to the manufacturer's instructions to avoid interference with any remaining nucleic acids.
Colorimetric analysis for acetone quantification
A vanillin-based spectrophotometric method was adopted for acetone determination.1 Reliant on the specific reaction of acetone with vanillin in a basic milieu, the samples (100 μL) were mixed with 60 μL of 130 mM vanillin, followed by addition of 40 μL of 5 M NaOH. After a 10 min incubation at 60 °C for 10 min and cooling the samples at room temperature for 10 min, the change in absorbance was measured at 430 nm in a plate reader. The reagent blanks (prepared with water or the corresponding buffer, depending on the type of experiment) and proper calibration curves were done in parallel in each set of experiments.In vitro assays for acetone production
All enzymatic reactions were carried out in 200 μL working volume. Reactions contained 5 μM of each enzyme (i.e. Acs, Thl, AtoDA and Adc), 50 mM phosphate buffer (pH = 7.0), acetate as a substrate, ATP or AMP as energy cofactors, MgCl2 (all of these components were added at the concentration specified in the text, depending on the experiment), 1 mM CoA and MilliQ water as necessary. Acs was added as the final component in the mixture to launch the reaction. When present, polyP kinases were included at 5 μM, together with polyP at the concentrations indicated in the text depending on the assay (calculated as single phosphate residues). The resulting samples were incubated at 30 °C for 16 h (unless indicated otherwise) with shaking at 600 rpm in an Eppendorf SmartBlockTM. In some cases, reactions were stopped by prompt addition of 10 μL of 1 g mL−1 trichloroacetic acid (TCA)7 [at a final concentration of 5% (w/v)], followed by centrifugation (15000 × g, 5 min) to separate the supernatant from precipitated proteins. These supernatant samples were subjected to HPLC analysis as detailed in the next section.
HPLC determination of acetone production and acetate consumption
The concentrations of acetone and acetate in the assays were measured by HPLC as described by Liew et al.7 with minor modifications. Samples were analyzed in a Dionex UltiMate 3000 HPLC system equipped with an AminexTM HPX-87X ion exclusion (300 × 7.8 mm) column (BioRad) coupled to RI-150 refractive index and UV (260, 277, 304 and 210 nm) detectors.42 The column was maintained at 30 °C with a run length of 30 min; the mobile phase comprised 5 mM H2SO4 in Milli-Q water at a flow rate of 0.6 mL min−1. The eluted compounds were detected by a HPLC Waters 481 UV-visible detector at 214 nm. This detector was connected in series to an RI detector (model 410). HPLC data were processed using the ChromeleonTM chromatography data system software 7.1.3 (Thermo Fisher Scientific). The detection of acetone and acetate was monitored at RI and compound concentrations were calculated from peak areas using a calibration curve (Fig. S1 and S2 in the ESI†) prepared with authentic acetone and acetate standards (99% HPLC standards, Sigma-Aldrich Co.). A control experiment was performed to rule out any potential interference in the detection method caused by the shift of pH induced by the addition of TCA to terminate the reactions. Standards of acetone (10, 35 and 70 mM) and acetate (25, 50 and 150 mM) were prepared in water and the areas under the peaks were compared to equivalent samples containing TCA at 5% (v/v) (Table S4 in the ESI†).Data and statistical analysis
All the experiments reported in this article were independently repeated at least twice (as indicated in the corresponding figure or table legend), and the mean value of the corresponding parameter ± standard deviation is presented.Results and discussion
Enzyme selection for assembly of an enzymatic cascade in vitro for acetone production
Acetate-dependent production of acetone relies on the enzymes of the canonical pathway from Clostridium acetobutylicum ATCC 824 (Fig. 1A). We adopted this enzymatic sequence in our study, where acetate is firstly converted into acetyl-coenzyme A (CoA) through the reaction of acetyl-CoA synthetase (Acs). Then, 2 mol of acetyl-CoA are condensed by thiolase (Thl) to generate 1 mol of acetoacetyl-CoA. Acetoacetyl-CoA transferase relocates the CoA moiety from acetoacetyl-CoA to acetate and forms acetoacetate, a reaction catalyzed by acetoacetate carboxylase (Adc) that finally yields acetone. Previous studies on pathway optimization in vivo indicated that AtoDA, an acetoacetyl-CoA transferase from E. coli, can mediate a 10-fold improvement in acetone production using acetate as the main carbon substrate.19 Based on this observation, we also chose included AtoDA in our designs towards establishing an optimal enzyme combination. Thus, the complete biosynthetic pathway from acetate to acetone includes a commercially available Acs from Bacillus subtilis (purchased from MegazymeTM), the Thl thiolase and the Adc acetoacetate decarboxylase from C. acetobutylicum ATCC 824 and the AtoDA acetoacetyl-CoA transferase from E. coli MG1655. These recombinant enzymes were produced in E. coli and purified as indicated in the Materials and methods section, and used to assemble enzymatic cascades as explained below.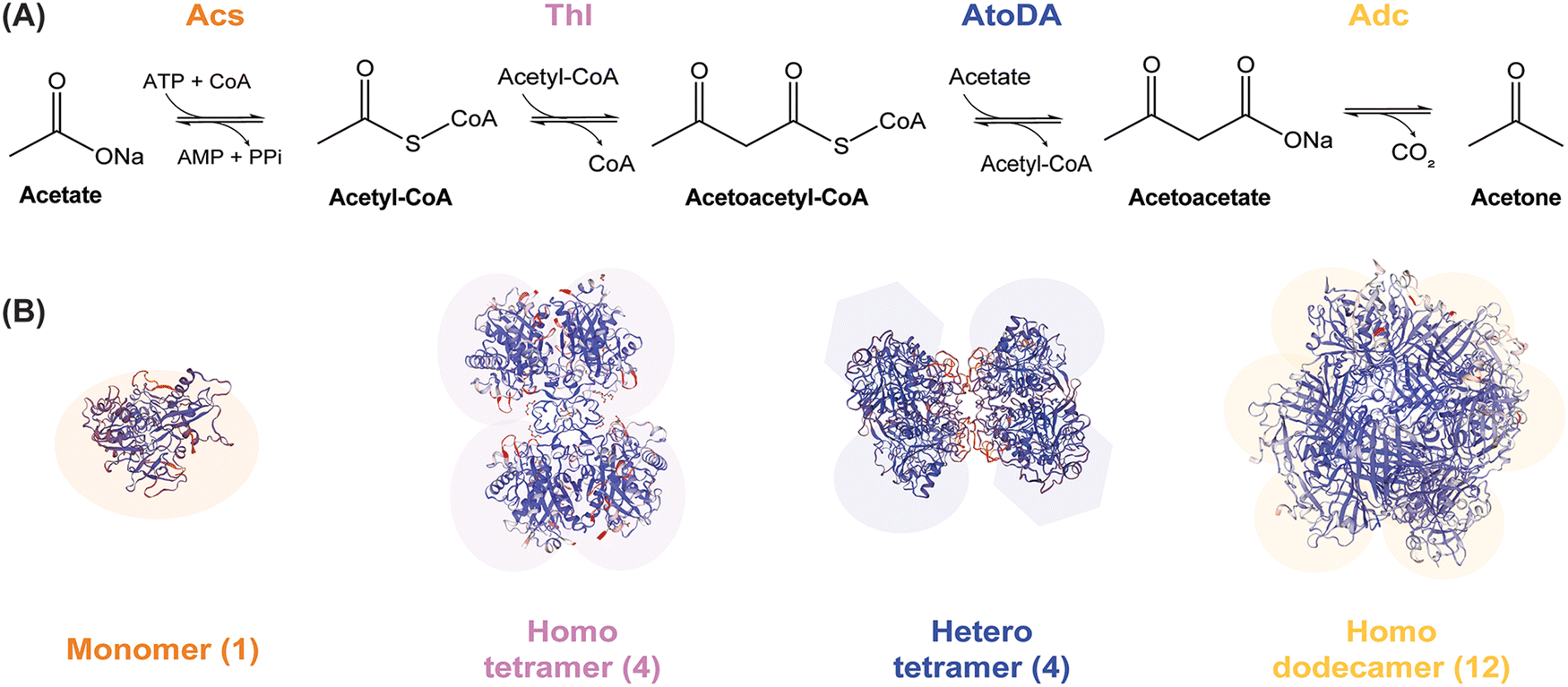 | ||
| Fig. 1 Scheme of the pathway adopted as a template to optimize acetone biosynthesis and oligomerization status of the individual enzymes therein. (A) Acetate and ATP-dependent enzymatic cascade proposed for in vitro acetone biosynthesis. Abbreviations: CoA, coenzyme A; PPi, inorganic pyrophosphate. (B) Structures of the enzymes catalyzing different steps of the ketone pathway generated by using the SWISS-MODEL71 platform. Monomeric acetyl-CoA synthase (Acs); homotetrameric thiolase (Thl);11 heterotetrameric acetoacetyl-CoA transferase (AtoDA; PDB DOI: 10.2210/pdb5DBN/pdb); and homododecameric acetoacetate decarboxylase (Adc).49 | ||
Optimizing an enzyme purification protocol to assemble an enzymatic cascade for acetone production
Two approaches were adopted for the purification of the proteins relevant to the enzymatic cascade. One of them was a classical strategy of a His6×-tag integration into the protein sequence (N-terminal), followed by production and purification according to the standard procedure described in Materials and methods. By applying this protocol, we could obtain Thl (a single subunit, forming a homotetramer) and AtoDA (two subunits, forming a heterotetramer; Fig. 1B). The feasibility of purifying the heterotetrameric AtoDA by affinity chromatography was of special relevance as the acetate:CoA transferase activity is essential for acetone production, yet the only approach reported for the purification of this enzyme required three chromatographic steps and fractional (NH4)2SO4 precipitation.43,44 After testing a few combinations, the optimal design of the pET-28a(+)–based protein production system was the one encoding the native atoD and atoA sequences from E. coli MG1655, with only one subunit (AtoD) tagged in the N-terminal position; the formation of the complex during protein production allowed for the successful recovery of an active enzyme complex (AtoDA). The possibility of fusing a His-tag without causing a significant effect on the enzyme activity not only eases the purification process but may also be a promising starting point towards developing an enzyme immobilization strategy in the future.45 Together with cofactor requirements (e.g. ATP), the cost of the biocatalysts (i.e. enzymes) is one of the major barriers for the commercial application of cell-free biosynthesis—and enzyme immobilization could help reducing the impact of such costs.Although the methodology applied for Thl, AtoDA and the PPK enzymes facilitated the recovery of these biocatalysts, a similar method could not be applied for Adc purification (a single subunit, forming a homododecamer, Fig. 1B). For this biocatalyst, a negative impact on the activity had been reported when the enzyme is fused to an N-terminal 6xHis-tag.40 The same approach was followed in other studies46 without indicating decreased activity, and no data is available for an enzyme variant with the 6xHis-tag at the C-terminal position. To clarify this potential interference, both His6x-tagged versions of the enzyme were assayed herein, and we verified a significant loss of activity in each case. Additionally, the complexity of the predicted dodecameric structure (Fig. S3A in the ESI†), and a potential intermolecular interference for the modelled His-tagged protein variant (Fig. S3B in the ESI†), could be identified using PyMOL and AlphaFold.47 Interestingly, removing the tag from the purified protein by treatment with a commercially-available TEV protease (Bionordika, Herlev, Denmark) did not improve enzyme recovery or performance. This observation indicates a role for the subunit terminals to foster protein folding, exacerbated by secondary protein structure disturbance and multicomponent complexity.48 Thus, we adopted and optimized an alternative protein production strategy for thermostable enzymes—a category where Adc belongs.40
Thermostable enzymes allow for His6×-tag free protein purification by heat-aided degradation and precipitation of the most abundant host [E. coli BL21(DE3)] proteins. In its native state, Adc is a 365-kDa homodecamer,49 and it is possible that the tag interferes with the protein assembly or substrate access to the active site.40 Different incubation times were tested to find the best conditions for purification of Adc from the cell extract through at 65 °C treatment, as monitored by SDS-PAGE gel analysis. The optimal incubation time was found to be 2 h, as no significant improvement in the protein purity was observed by prolonging this incubation time—in fact, longer incubation times seemed to negatively affect protein stability. Even though C. acetobutylicum is a mesophilic organism, this enzyme is stable above 70 °C, which does not come as a surprise considering that C. acetobutylicum spores can tolerate temperatures up to 80 °C without losing viability.50 In line with these observations, a previous study51 determined that the activity of the enzyme improved after an hour-long incubation at 70 °C. Hence, this purification protocol was retained for isolating Adc from C. acetobutylicum.
We further compared protein production profiles in 2 × YT medium and an auto-induction strategy for growth of recombinant E. coli BL21(DE3) towards improving Adc yields. Protein suspensions from different cell cultures grown under these conditions were purified with the previously established heat-treatment conditions and compared by means of SDS-PAGE analysis. The best condition, resulting in the highest yields of the Adc enzyme, were achieved by incubating the E. coli BL21(DE3) cells at 37 °C in auto-induction medium (Table S3 in the ESI†). The main components in this medium are glucose, glycerol and lactose. Glucose is consumed by the cells first, allowing to reach the desired cell density for sufficient protein production, which is activated by lactose while glycerol is supporting cell growth during the protein production phase.41 Indeed, when compared to the biomass obtained on 2 × YT medium and induced by IPTG addition (as indicated in Materials and methods), E. coli BL21(DE3) grown under the auto-induction conditions reached a 5-fold increase in Adc yields. Thus, the conditions described herein allowed for the efficient synthesis and purification of all recombinant proteins needed to assemble a synthetic metabolism for in vitro acetone formation (Fig. S4 in the ESI†).
Setting the enzymatic cascade in action: optimizing physicochemical conditions
With the purified enzymes at hand, we next explored the pathway performance for acetone production in vitro, and we first aimed at screening optimal pH and temperature conditions. To this end, the in vitro cascade was incubated with 10 mM acetate as the only carbon substrate and 10 mM ATP under different combinations of pH and temperature. In this first set of assays, we tested pH values between 6 and 8 and incubation temperatures between 25 and 37 °C. The short incubation period adopted in these experiments (i.e. 1 h) allowed for a quick comparison across all conditions by evaluating both acetone biosynthesis and substrate consumption by HPLC. We observed that the pathway was active under all tested conditions, with decreased activity at acidic pH and low temperatures (Fig. 2). The higher performance at alkaline pH might seem surprising, since Adc from C. acetobutylicum was reported to have an optimal pH about 6 and neutrality,52 and a substantial decrease of activity at high pH.46 Additionally, the measurements of the internal pH of C. acetobutylicum indicated a range of values from 4 to 6 depending on the culture conditions.24,53 Thl was determined to display a wide range of optimal activity (pH 5.5–7.4),54 whereas no information is available on the pH preference of AtoDA. However, the optimal pH of the Acs from B. subtilis was reported to be 8.4 by the manufacturer, suggesting that the optimal value for the pathway was a compromise between the performances of Adc and Acs. One way or the other, the results of our optimization efforts showed a slightly higher activity at pH 8 and 37 °C compared to pH 7 and 30 °C. The latter conditions were kept for further experiments, due to the general advantages of running the reaction at a low temperature. In this sense, we reasoned that lower temperatures would lead to a more energy-efficient process while minimizing potential losses of acetone due to evaporation in long incubations. Thus, the enzymatic cascade was set at 30 °C and pH 7 with all the enzymes of the pathway (i.e. Acs, Thl, AtoDA and Adc) added in equimolar amounts (5 μM). Under these conditions, acetone biosynthesis reached was close to the maximal theoretical yield (50% mol/mol of the initial substrate) at a titer of ca. 5 mM (Fig. 2).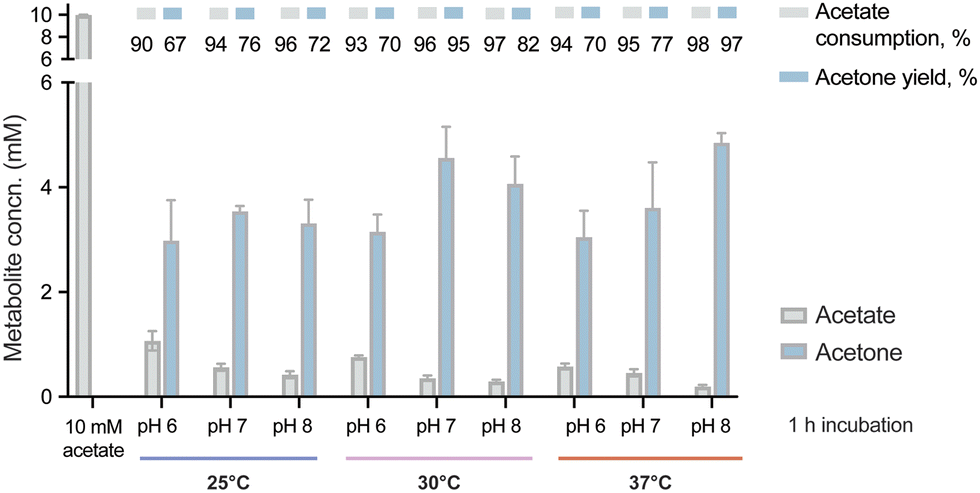 | ||
| Fig. 2 Optimizing physicochemical conditions for in vitro acetone biosynthesis. The reactions were incubated for 1 h at different pH values (6, 7 and 8) and temperatures (25, 30 and 37 °C) as indicated in the figure. The performance of the synthetic metabolism was evaluated under these conditions by HPLC analysis of acetone formation and acetate consumption as indicated in Materials and methods. Acetone yields on substrate are indicated as a % of the theoretical maximum. The bars represent average values from independent triplicates, with standard deviations indicated by error bars. Concn., concentration. | ||
Notably, the amount of ATP required by the pathway is equimolar to the acetone produced. Specifically, 2 molecules of acetyl-CoA are needed to generate 1 molecule of acetone. Only one acetyl-CoA unit is obtained by the ATP-dependent Acs enzyme, while another one is continuously regenerated from acetoacetyl-CoA by the cofactor-independent AtoDA enzyme. In fact, ATP supplementation is a critical (and expensive) feature to ensure a proper level of pathway activity. To find an alternative solution to this high-cost bottleneck, we investigated the possibility of providing ATP indirectly towards establishing energy-efficient acetone biosynthesis.
Design and implementation of an ATP-regeneration system
Cofactor regeneration is a crucial step for the development of cost-effective synthetic metabolism in vitro. In this regard, several approaches to replenish ATP for biocatalysis have been described over the last years55 and, among these, the implementation of polyP kinases is receiving increasing attention due to its versatility.56–58 Based on a previously reported biomimetic, polyP-based, cyclic enzymatic cascades involving methyltransferases,58 we designed an ATP-regeneration (ATPREG) system for our synthetic metabolism (Fig. 3). The ATPREG system relies on the activity of 2 PPK enzymes. PolyP kinase 2 from A. johnsonii (PPK2Aj) catalyzes the phosphoconversion of adenosine monophosphate (AMP) to adenosine diphosphate (ADP). Next, polyP kinase 2 from S. meliloti (PPK2Sm) forms adenosine triphosphate (ATP) from ADP. Both reactions use polyP as a phosphate donor (Fig. 3), a class of polymeric phosphate salt (widespread in nature59–61) that has been flagged as a nearly ideal substrate for ATP regeneration due to its low cost and stability in aqueous solutions.62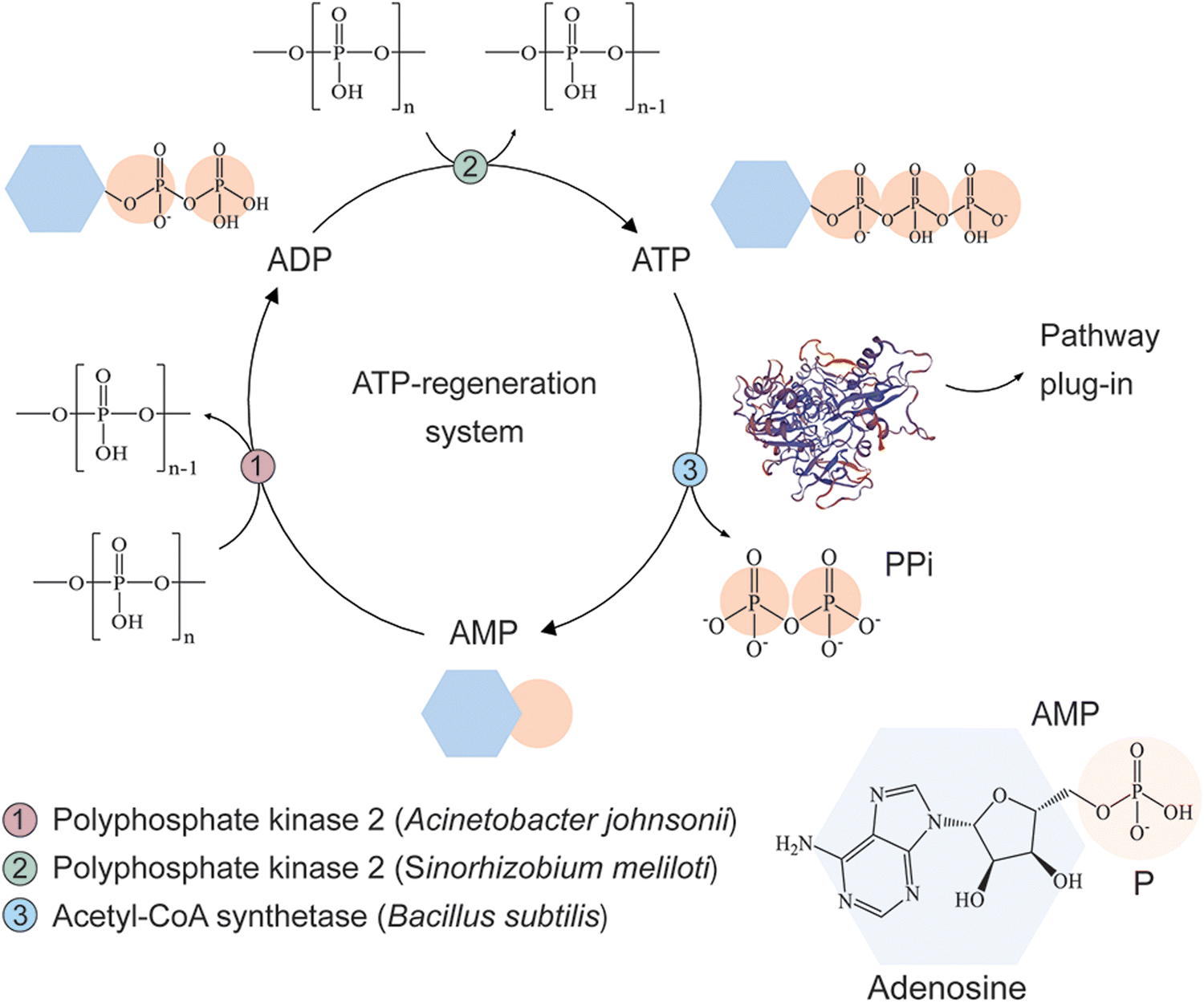 | ||
| Fig. 3 Establishing an ATP regeneration (ATPREG) system to support an energy-dependent synthetic metabolism in vitro. Polyphosphate kinase 2 from A. johnsonii (PPK2Aj) catalyzes the phosphoconversion of adenosine monophosphate (AMP) to adenosine diphosphate (ADP). Polyphosphate kinase 2 from S. meliloti (PPK2Sm) subsequently mediates the formation of adenosine triphosphate (ATP) from ADP. Both enzymatic reactions use polyphosphate as a phosphate donor; inorganic pyrophosphate (PPi) is generated by ATP hydrolysis catalyzed by Acs. | ||
In a first step, an assay was carried out to assess the potential of the designed ATPREG system to increase the production of acetone with a limited starting amount of ATP. The experiment was meant to compare acetone biosynthesis from 25 mM acetate using either (i) 2 and 10 mM ATP without ATP regeneration or (ii) 2 mM ATP and 10 mM AMP (i.e. no initial ATP supply) in the presence of the ATPREG system. In condition (ii), 60 mM polyP and 10 mM MgCl2 were supplemented to the synthetic metabolism to promote ATP recycling. Quantification of acetate and acetone concentrations at the end of these experiments (Fig. 4A) indicated that the pathway produced acetone at 1.4 mM and 7.8 mM when ATP replenishment was not possible—flagging the availability of an energy source as a major bottleneck to high-titer ketone biosynthesis by the synthetic metabolism. Based on the stoichiometry of the route, these results represent 70% and 78% of the maximal production yield (i.e. 2 and 10 mM acetone, respectively). Adding PPK2Aj and PPK2Sm, in contrast, had a dramatic effect of ATP regeneration. An initial amount of 2 mM ATP mediated the formation of 7.6 mM acetone (i.e. 5-fold enhancement as compared to the control conditions), whereas the assay where 10 mM AMP and the ATPREG system were present yielded 11.5 mM acetone—corresponding to nearly full substrate conversion (i.e. 12.5 mM acetone). Analyzing the results from the perspective of acetate consumption mirrored this trend, although the yields were slightly higher, probably due to minor loss of acetone due to evaporation.
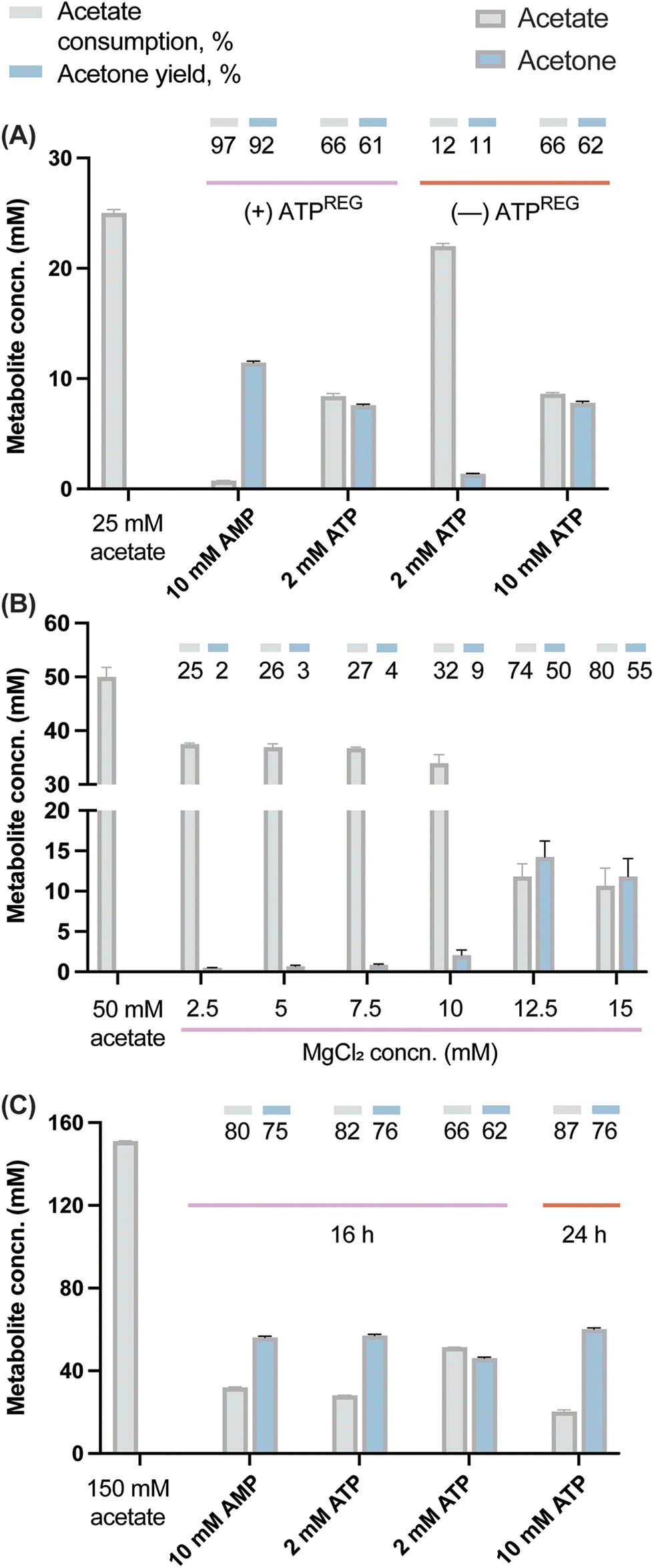 | ||
| Fig. 4 Fine-tuning of the synthetic metabolism in vitro. (A) Coupling acetone biosynthesis with an ATP-regeneration (ATPREG) system. (B) and (C) Enzymatic assays were incubated for 16 h (or, in some cases, 24 h) in the presence of different MgCl2 concentrations and the ATP-regeneration (ATPREG) system added with AMP or ATP at the concentrations indicated in the figure. In all cases, the performance of the synthetic metabolism was evaluated under these conditions by HPLC analysis of acetone formation and acetate consumption as indicated in Materials and methods. Acetone yields on substrate are indicated as a % of the theoretical maximum. The bars represent average values from independent triplicates, with standard deviations indicated by error bars. Concn., concentration. | ||
Once the feasibility of producing acetone in vitro with no initial supply of ATP was tested, substrate conversion was debottlenecked by adjusting the amount of polyP needed to support efficient ketone biosynthesis. In this regard, a similar system based on PPK enzymes suggested that 40 mM was the minimum amount of polyP necessary to regenerate 10 mM ATP.63 However, increasing the amount of polyP may be detrimental for the reaction yield64 due to its ability of sequestering Mg2+ (which could lead to both precipitation of insoluble mineral complexes and limited enzyme activity due to a dearth of metallic cofactors). The mild chelating properties of orthophosphate and phosphate oligomers are in fact a well-known phenomenon.65,66 Therefore, an assay was performed to determine the optimal Mg2+:polyP ratio in our operating conditions. Acetate was added in excess (at 50 mM), using a fixed initial polyP concentration of 60 mM and a range of MgCl2 concentrations from 2.5 to 15 mM. We observed an improvement of acetone production from 2.5 to 14.5 mM (Fig. 4B), indicating more significant effect with 12.5 and 15 mM of MgCl2. This dynamic suggests a threshold, where a minimal amount of MgCl2 is necessary to reach optimal pathway activity. Once the divalent cation is sufficiently available, the reaction rate rapidly reaches its maximum. The fact that high conversion yields were reached in similar conditions with 10 mM MgCl2 (Fig. 4A) suggests that in addition to phosphate and oligophosphates, acetate might also contribute to withdraw Mg2+.
Based on the former assays, new reactions were designed to explore the conversion of 150 mM acetate. The selected concentration required a stoichiometric amount of 75 mM ATP for which polyP was added at 360 mM and the concentration of MgCl2 was increased up to 75 mM. The production of acetone was assayed with 10 mM AMP, 10 mM ATP or 2 mM ATP. High conversion was verified in the presence of both 10 mM ATP and AMP, attaining 81% and 79% acetate consumption, respectively (Fig. 4C). The acetone concentration with 10 mM AMP reached 56 mM. In the case of the 10 mM ATP condition, the value increased up to 84% (130 mM acetate consumed) when the incubation time was prolonged to 24 h, with a final acetone concentration of 62 mM (i.e. 3 g L−1). To the best of our knowledge, this is the highest acetone production from acetate reported for a purified system (84% of the theoretical yield). In a recent work, 87 mM acetone was produced in a crude cell extract supplemented with glucose as the main carbon substrate.7 The conversion yields reported in our study highlight the robustness of the developed enzymatic platform, which is able to operate efficiently even under high-ionic-strength conditions. The slight increase in acetone biosynthesis from 16 to 24 h further suggests that the enzyme cascade is still active—although working at a suboptimal rate. This situation could be a consequence of biocatalyst denaturation but also an inhibitory effect of the acetone or pyrophosphate as the end-metabolites of the route. Specifically, pyrophosphate is known to exert product inhibition on Acs.67,68
The pipeline developed herein for the pathway assembly in vitro provided a highly efficient self-sustaining enzymatic cascade that, under optimal conditions, produced 62 mM of acetone from acetate, allowing cost-improved acetone biosynthesis in vitro. The system is compatible with a recently reported high-throughput assays for detection of ketones by cell factories.1 Thus, we expect this platform to be an asset not only for gaining insight on the mechanics of the pathway (including various substrate combinations), but also for a broad biosynthesis application of other related ketones and alcohols.
Conclusion
This work provides a first case-example of assembling one-pot synthetic metabolism in vitro for acetone biosynthesis from acetate with close-to-theoretical yield. Our approach includes straightforward production and purification strategies for the components of the enzymatic cascade, navigating through protein specifics, from hetero-mers to complex homo-oligomers. This in vitro platform for ketone production may also serve as a simple and efficient blueprint for studying acetone biosynthesis under different conditions (e.g. pH, temperature and ionic strength). Moreover, combined with an efficient ATP regeneration system, this strategy allowed to reach 84–94% of the maximum theoretical yield of acetone without external ATP supplementation. Using 150 mM of acetate as the feedstock, the fine-tuning of the reaction conditions led to 56 mM of acetone using AMP as the sole energy cofactor. In summary, these results will enable continuous cost-effective biosynthesis of acetone and potentially other products, bringing more opportunities for sustainable manufacturing powered by biochemistry.Author contributions
E. K. and M. N. D.: conceptualization, investigation, methodology, data curation, visualization, validation, writing – original draft. A. D. H.: methodology, validation, data curation. P. I. N.: supervision, resources, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank Dr Mikkel Madsen (Department of Biotechnology and Biomedicine, Technical University of Denmark) for helpful discussions on protein complex formation and subunit modeling. The financial support from The Novo Nordisk Foundation (NNF10CC1016517) and from the European Union's Horizon2020 Research and Innovation Programme under grant agreement No. 814418 (SinFonia) to P. I. N. and Villum Experiment (40979) to M. N. D. is gratefully acknowledged. E. K. is the recipient of a fellowship from the Novo Nordisk Foundation as part of the Copenhagen Bioscience PhD Programme, supported through grant NNF18CC0033664. The authors declare that there are no competing interests associated with the contents of this article.References
- E. Kozaeva, V. Mol, P. I. Nikel and A. T. Nielsen, High-throughput colorimetric assays optimized for detection of ketones and aldehydes produced by microbial cell factories, Microb. Biotechnol., 2022, 15, 2426–2438 CrossRef CAS PubMed.
- E. I. Lan, Y. Dekishima, D. S. Chuang and J. C. Liao, Metabolic engineering of 2-pentanone synthesis in Escherichia coli, AIChE J., 2013, 59, 3167–3175 CrossRef CAS.
- Y. Liu and J. Nielsen, Recent trends in metabolic engineering of microbial chemical factories, Curr. Opin. Biotechnol, 2019, 60, 188–197 CrossRef CAS PubMed.
- D. Yang, S. Y. Park, Y. S. Park, H. Eun and S. Y. Lee, Metabolic engineering of Escherichia coli for natural product biosynthesis, Trends Biotechnol., 2020, 38, 745–765 CrossRef CAS PubMed.
- R. Young, M. Haines, M. Storch and P. S. Freemont, Combinatorial metabolic pathway assembly approaches and toolkits for modular assembly, Metab. Eng., 2021, 63, 81–101 CrossRef CAS PubMed.
- D. Liu, Z. Yang, P. Wang, H. Niu, W. Zhuang, Y. Chen, J. Wu, C. Zhu, H. Ying and P. Ouyang, Towards acetone-uncoupled biofuels production in solventogenic Clostridium through reducing power conservation, Metab. Eng., 2018, 47, 102–112 CrossRef CAS PubMed.
- F. E. Liew, R. Nogle, T. Abdalla, B. J. Rasor, C. Canter, R. O. Jensen, L. Wang, J. Strutz, P. Chirania, S. De Tissera, A. P. Mueller, Z. Ruan, A. Gao, L. Tran, N. L. Engle, J. C. Bromley, J. Daniell, R. Conrado, T. J. Tschaplinski, R. J. Giannone, R. L. Hettich, A. S. Karim, S. D. Simpson, S. D. Brown, C. Leang, M. C. Jewett and M. Köpke, Carbon-negative production of acetone and isopropanol by gas fermentation at industrial pilot scale, Nat. Biotechnol., 2022, 40, 335–344 CrossRef CAS PubMed.
- P. Dürre and B. J. Eikmanns, C1-carbon sources for chemical and fuel production by microbial gas fermentation, Curr. Opin. Biotechnol, 2015, 35, 63–72 CrossRef PubMed.
- S. Hoffmeister, M. Gerdom, F. R. Bengelsdorf, S. Linder, S. Flüchter, H. Öztürk, W. Blümke, A. May, R. J. Fischer, H. Bahl and P. Dürre, Acetone production with metabolically engineered strains of Acetobacterium woodii, Metab. Eng., 2016, 36, 37–47 CrossRef CAS PubMed.
- M. Sauer, Industrial production of acetone and butanol by fermentation—100 years later, FEMS Microbiol. Lett., 2016, 363, fnw134 CrossRef.
- L. L. Bermejo, N. E. Welker and E. T. Papoutsakis, Expression of Clostridium acetobutylicum ATCC 824 genes in Escherichia coli for acetone production and acetate detoxification, Appl. Environ. Microbiol., 1998, 64, 1079–1085 CrossRef CAS.
- E. T. Papoutsakis, Engineering solventogenic clostridia, Curr. Opin. Biotechnol, 2008, 19, 420–429 CrossRef CAS.
- C. Weizmann and B. Rosenfeld, The activation of the butanol-acetone fermentation of carbohydrates by Clostridium acetobutylicum (Weizmann), Biochem. J., 1937, 31, 619–639 CrossRef CAS PubMed.
- P. Calero and P. I. Nikel, Chasing bacterial chassis for metabolic engineering: A perspective review from classical to non-traditional microorganisms, Microb. Biotechnol., 2019, 12, 98–124 CrossRef CAS PubMed.
- C. Zhao, Y. Zhang and Y. Li, Production of fuels and chemicals from renewable resources using engineered Escherichia coli, Biotechnol. Adv., 2019, 37, 107402 CrossRef CAS PubMed.
- A. Sánchez-Pascuala, V. de Lorenzo and P. I. Nikel, Refactoring the Embden-Meyerhof-Parnas pathway as a whole of portable GlucoBricks for implantation of glycolytic modules in Gram-negative bacteria, ACS Synth. Biol., 2017, 6, 793–805 CrossRef.
- A. Sánchez-Pascuala, L. Fernández-Cabezón, V. de Lorenzo and P. I. Nikel, Functional implementation of a linear glycolysis for sugar catabolism in Pseudomonas putida, Metab. Eng., 2019, 54, 200–211 CrossRef PubMed.
- D. C. Volke and P. I. Nikel, Getting bacteria in shape: Synthetic morphology approaches for the design of efficient microbial cell factories, Adv. Biosyst., 2018, 2, 1800111 CrossRef.
- H. Yang, B. Huang, N. Lai, Y. Gu, Z. Li, Q. Ye and H. Wu, Metabolic engineering of Escherichia coli carrying the hybrid acetone-biosynthesis pathway for efficient acetone biosynthesis from acetate, Microb. Cell Fact., 2019, 18, 6 CrossRef PubMed.
- J. Park, M. Rodríguez-Moyá, M. Li, E. Pichersky, K. Y. San and R. Gonzalez, Synthesis of methyl ketones by metabolically engineered Escherichia coli, J. Ind. Microbiol. Biotechnol., 2012, 39, 1703–1712 CrossRef CAS PubMed.
- A. J. Shaw, F. H. Lam, M. Hamilton, A. Consiglio, K. MacEwen, E. E. Brevnova, E. Greenhagen, W. G. LaTouf, C. R. South, H. van Dijken and G. Stephanopoulos, Metabolic engineering of microbial competitive advantage for industrial fermentation processes, Science, 2016, 353, 583–586 CrossRef CAS PubMed.
- S. C. Nies, T. B. Alter, S. Nölting, S. Thiery, A. N. T. Phan, N. Drummen, J. D. Keasling, L. M. Blank and B. E. Ebert, High titer methyl ketone production with tailored Pseudomonas taiwanensis VLB120, Metab. Eng., 2020, 62, 84–94 CrossRef CAS PubMed.
- K. Srirangan, X. Liu, L. Akawi, M. Bruder, M. Moo-Young and C. P. Chou, Engineering Escherichia coli for microbial production of butanone, Appl. Environ. Microbiol., 2016, 82, 2574–2584 CrossRef PubMed.
- C. A. Vees, C. S. Neuendorf and S. Pflügl, Towards continuous industrial bioprocessing with solventogenic and acetogenic clostridia: Challenges, progress and perspectives, J. Ind. Microbiol. Biotechnol., 2020, 47, 753–787 CrossRef CAS PubMed.
- S. Li, L. Huang, C. Ke, Z. Pang and L. Liu, Pathway dissection, regulation, engineering and application: Lessons learned from biobutanol production by solventogenic clostridia, Biotechnol. Biofuels, 2020, 13, 39 CrossRef CAS PubMed.
- C. Cheng, T. Bao and S. T. Yang, Engineering Clostridium for improved solvent production: Recent progress and perspective, Appl. Microbiol. Biotechnol., 2019, 103, 5549–5566 CrossRef CAS PubMed.
- T. J. Erb, Structural organization of biocatalytic systems: The next dimension of synthetic metabolism, Emerging Top. Life Sci., 2019, 3, 579–586 CrossRef CAS.
- A. Danchin, In vivo, in vitro and in silico: An open space for the development of microbe-based applications of synthetic biology, Microb. Biotechnol., 2021, 15, 42–64 CrossRef PubMed.
- B. J. Rasor, B. Vögeli, G. M. Landwehr, J. W. Bogart, A. S. Karim and M. C. Jewett, Toward sustainable, cell-free biomanufacturing, Curr. Opin. Biotechnol, 2021, 69, 136–144 CrossRef CAS PubMed.
- Q. M. Dudley, A. S. Karim and M. C. Jewett, Cell-free metabolic engineering: Biomanufacturing beyond the cell, Biotechnol. J., 2015, 10, 69–82 CrossRef CAS PubMed.
- N. J. Claassens, S. Burgener, B. Vögeli, T. J. Erb and A. Bar-Even, A critical comparison of cellular and cell-free bioproduction systems, Curr. Opin. Biotechnol, 2019, 60, 221–229 CrossRef CAS PubMed.
- E. Orsi, N. J. Claassens, P. I. Nikel and S. N. Lindner, Growth-coupled selection of synthetic modules to accelerate cell factory development, Nat. Commun., 2021, 12, 5295 CrossRef CAS PubMed.
- J. Sambrook and D. W. Russell, Molecular cloning: A laboratory manual, Cold Spring Harbor Laboratory, 2001 Search PubMed.
- A. E. Parnell, S. Mordhorst, F. Kemper, M. Giurrandino, J. P. Prince, N. J. Schwarzer, A. Hofer, D. Wohlwend, H. J. Jessen, S. Gerhardt, O. Einsle, P. C. F. Oyston, J. N. Andexer and P. L. Roach, Substrate recognition and mechanism revealed by ligand-bound polyphosphate kinase 2 structures, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3350–3355 CrossRef CAS.
- A. M. Cavaleiro, S. H. Kim, S. Seppälä, M. T. Nielsen and M. H. Nørholm, Accurate DNA assembly and genome engineering with optimized uracil excision cloning, ACS Synth. Biol., 2015, 4, 1042–1046 CrossRef CAS PubMed.
- D. C. Volke, R. A. Martino, E. Kozaeva, A. M. Smania and P. I. Nikel, Modular (de)construction of complex bacterial phenotypes by CRISPR/nCas9-assisted, multiplex cytidine base-editing, Nat. Commun., 2022, 13, 3026 CrossRef CAS.
- D. C. Volke, J. Turlin, V. Mol and P. I. Nikel, Physical decoupling of XylS/Pm regulatory elements and conditional proteolysis enable precise control of gene expression in Pseudomonas putida, Microb. Biotechnol., 2020, 13, 222–232 CrossRef CAS PubMed.
- H. J. Genee, M. T. Bonde, F. O. Bagger, J. B. Jespersen, M. O. A. Sommer, R. Wernersson and L. R. Olsen, Software-supported USER cloning strategies for site-directed mutagenesis and DNA assembly, ACS Synth. Biol., 2015, 4, 342–349 CrossRef CAS PubMed.
- P. I. Nikel, D. Pérez-Pantoja and V. de Lorenzo, Pyridine nucleotide transhydrogenases enable redox balance of Pseudomonas putida during biodegradation of aromatic compounds, Environ. Microbiol., 2016, 18, 3565–3582 CrossRef CAS PubMed.
- B. M. Zeldes, C. T. Straub, J. K. Otten, M. W. W. Adams and R. M. Kelly, A synthetic enzymatic pathway for extremely thermophilic acetone production based on the unexpectedly thermostable acetoacetate decarboxylase from Clostridium acetobutylicum, Biotechnol. Bioeng., 2018, 115, 2951–2961 CrossRef CAS.
- F. W. Studier, Protein production by auto-induction in high-density shaking cultures, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS PubMed.
- E. Kozaeva, S. Volkova, M. R. A. Matos, M. P. Mezzina, T. Wulff, D. C. Volke, L. K. Nielsen and P. I. Nikel, Model-guided dynamic control of essential metabolic nodes boosts acetyl-coenzyme A–dependent bioproduction in rewired Pseudomonas putida, Metab. Eng., 2021, 67, 373–386 CrossRef CAS PubMed.
- S. J. Sramek and F. E. Frerman, Purification and properties of Escherichia coli coenzyme A-transferase, Arch. Biochem. Biophys., 1975, 171, 14–26 CrossRef CAS.
- S. Korolev, O. Koroleva, K. Petterson, M. Gu, F. Collart, I. Dementieva and A. Joachimiak, Autotracing of Escherichia coli acetate CoA-transferase α-subunit structure using 3.4 Å MAD and 1.9 Å native data, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 2116–2121 CrossRef CAS.
- M. Romero-Fernández and F. Paradisi, Protein immobilization technology for flow biocatalysis, Curr. Opin. Chem. Biol., 2020, 55, 1–8 CrossRef.
- K. Min, S. Kim, T. Yum, Y. Kim, B. I. Sang and Y. Um, Conversion of levulinic acid to 2-butanone by acetoacetate decarboxylase from Clostridium acetobutylicum, Appl. Microbiol. Biotechnol., 2013, 97, 5627–5634 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS.
- T. Kittilä, P. Calero, F. Fredslund, P. T. Lowe, D. Tezé, M. Nieto-Domínguez, D. O'Hagan, P. I. Nikel and D. H. Welner, Oligomerization engineering of the fluorinase enzyme leads to an active trimer that supports synthesis of fluorometabolites in vitro, Microb. Biotechnol., 2022, 15, 1622–1632 CrossRef.
- M. C. Ho, J. F. Ménétret, H. Tsuruta and K. N. Allen, The origin of the electrostatic perturbation in acetoacetate decarboxylase, Nature, 2009, 459, 393–397 CrossRef CAS PubMed.
- M. A. Al-Hinai, S. W. Jones and E. T. Papoutsakis, σK of Clostridium acetobutylicum is the first known sporulation-specific sigma factor with two developmentally separated roles, one early and one late in sporulation, J. Bacteriol., 2014, 196, 287–299 CrossRef PubMed.
- A. P. Autor and I. Fridovich, The thermal inactivation of acetoacetate decarboxylase, J. Biol. Chem., 1970, 245, 5223–5227 CrossRef CAS.
- L. A. Highbarger, J. A. Gerlt and G. L. Kenyon, Mechanism of the reaction catalyzed by acetoacetate decarboxylase. Importance of lysine 116 in determining the pKa of active-site lysine 115, Biochemistry, 1996, 35, 41–46 CrossRef CAS PubMed.
- L. Huang, L. N. Gibbins and C. W. Forsberg, Transmembrane pH gradient and membrane potential in Clostridium acetobutylicum during growth under acetogenic and solventogenic conditions, Appl. Environ. Microbiol., 1985, 50, 1043–1047 CrossRef CAS PubMed.
- D. P. Wiesenborn, F. B. Rudolph and E. T. Papoutsakis, Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents, Appl. Environ. Microbiol., 1988, 54, 2717–2722 CrossRef CAS.
- H. Chen and Y. P. J. Zhang, Enzymatic regeneration and conservation of ATP: Challenges and opportunities, Crit. Rev. Biotechnol., 2021, 41, 16–33 CrossRef CAS PubMed.
- S. Mordhorst and J. N. Andexer, Round, round we go—Strategies for enzymatic cofactor regeneration, Nat. Prod. Rep., 2020, 37, 1316–1333 RSC.
- J. N. Andexer and M. Richter, Emerging enzymes for ATP regeneration in biocatalytic processes, ChemBioChem, 2015, 16, 380–386 CrossRef CAS.
- S. Mordhorst, J. Siegrist, M. Müller, M. Richter and J. N. Andexer, Catalytic alkylation using a cyclic S-adenosylmethionine regeneration system, Angew. Chem., Int. Ed. Engl., 2017, 56, 4037–4041 CrossRef CAS.
- M. Q. Bowlin and M. J. Gray, Inorganic polyphosphate in host and microbe biology, Trends Microbiol., 2021, 29, 1013–1023 CrossRef CAS PubMed.
- P. I. Nikel, M. Chavarría, E. Martínez-García, A. C. Taylor and V. de Lorenzo, Accumulation of inorganic polyphosphate enables stress endurance and catalytic vigour in Pseudomonas putida KT2440, Microb. Cell Fact., 2013, 12, 50 CrossRef CAS PubMed.
- H. Rosigkeit, L. Kneißle, S. Obruča and D. Jendrossek, The multiple roles of polyphosphate in Ralstonia eutropha and other bacteria, Microb. Physiol., 2021, 31, 163–177 CrossRef PubMed.
- L. Butler, A suggested approach to ATP regeneration for enzyme technology applications, Biotechnol. Bioeng., 1977, 19, 591–593 CrossRef CAS PubMed.
- G. A. Strohmeier, I. C. Eiteljörg, A. Schwarz and M. Winkler, Enzymatic one-step reduction of carboxylates to aldehydes with cell-free regeneration of ATP and NADPH, Chemistry, 2019, 25, 6119–6123 CrossRef CAS PubMed.
- F. L. Oetting and R. A. McDonald, The thermodynamic properties of magnesium orthophosphate and magnesium pyrophosphate, J. Phys. Chem., 1963, 67, 2737–2743 CrossRef CAS.
- S. J. Knabel, H. W. Walker and P. A. Hartman, Inhibition of Aspergillus flavus and selected Gram-positive bacteria by chelation of essential metal cations by polyphosphates, J. Food Prot., 1991, 54, 360–365 CrossRef CAS.
- P. Gopinath, V. Ramalingam and R. Breslow, Magnesium pyrophosphates in enzyme mimics of nucleotide synthases and kinases and in their prebiotic chemistry, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12011–12014 CrossRef CAS PubMed.
- G. G. Preston, J. D. Wall and D. W. Emerich, Purification and properties of acetyl-CoA synthetase from Bradyrhizobium japonicum bacteroids, Biochem. J., 1990, 267, 179–183 CrossRef CAS PubMed.
- J. O'Sullivan and L. Ettlinger, Characterization of the acetyl-CoA synthetase of Acetobacter aceti, Biochim. Biophys. Acta, 1976, 450, 410–417 CrossRef.
- D. Hanahan and M. Meselson, Plasmid screening at high colony density, Methods Enzymol., 1983, 100, 333–342 CAS.
- H. Jeong, H. J. Kim and S. J. Lee, Complete genome sequence of Escherichia coli strain BL21, Genome Announc., 2015, 3, e00134–15 Search PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, SWISS-MODEL: Homology modelling of protein structures and complexes, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00170e |
| ‡ These authors contributed equally and should be considered joint first authors. |
| This journal is © The Royal Society of Chemistry 2022 |