
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-induced telomeric DNA damage in human cancer cells†
Justin
Weynand
 ab,
Harikleia
Episkopou
c,
Gabriel
Le Berre
c,
Martin
Gillard
a,
Jérôme
Dejeu
b,
Anabelle
Decottignies
*c,
Eric
Defrancq
*b and
Benjamin
Elias
*a
ab,
Harikleia
Episkopou
c,
Gabriel
Le Berre
c,
Martin
Gillard
a,
Jérôme
Dejeu
b,
Anabelle
Decottignies
*c,
Eric
Defrancq
*b and
Benjamin
Elias
*a
aUniversité catholique de Louvain (UCLouvain), Institut de la Matière Condensée et des Nanosciences (IMCN), Molecular Chemistry, Materials and Catalysis (MOST), Place Louis Pasteur 1, bte L4.01.02, B-1348 Louvain-la-Neuve, Belgium. E-mail: Benjamin.Elias@uclouvain.be
bUniversité Grenoble-Alpes (UGA), Département de Chimie Moléculaire, UMR CNRS 5250, CS 40700, 38058 Grenoble, France. E-mail: eric.defrancq@univ-grenoble-alpes.fr
cUniversité catholique de Louvain (UCLouvain), Genetic and Epigenetic Alterations of Genomes, de Duve Institute, Avenue Hippocrate 75, 1200 Brussels, Belgium. E-mail: anabelle.decottignies@uclouvain.be
First published on 31st October 2022
Abstract
Herein we report on the study of novel dinuclear ruthenium(II) complexes designed to target and to photo-react with G-quadruplex telomeric DNA. Upon irradiation, complexes efficiently generate guanine radical cation sites as photo-oxidation products. The compounds also display efficient cell penetration with localization to the nucleus and show strong photocytotoxicity toward osteosarcoma cells. Thanks to a microscopic-based telomere dysfunction assay, which allows the direct visualization of DNA damage in cells, we brought the first evidence of forming photo-oxidative damage at telomeres in cellulo. This emphasizes interesting prospects for the development of future cancer phototherapies.
In recent years, non-canonical DNA secondary structures have attracted much attention. Among them, G-quadruplex (G4) DNA is particularly appealing due to its multiple biological involvements.1–4 The most documented importance of these quadruple helical structures is their prevalence in human telomeric DNA. The telomeric sequences are composed of hundreds of 3′TTAGGG5′ repeats and play a crucial role in the development of cancer. The single-stranded overhang of telomeric DNA can potentially fold up to ten consecutive G4s linked by TTA spacers. After each cell division, they are shortened and once a limit is reached (i.e. the Hayflick limit), the cell enters senescence.5,6 However, this situation is almost never reached in cancer cells due to telomere maintenance processes.7,8 This telomeric length retention leads to cancer cell immortality. As G4 structures are believed to regulate telomere maintenance mechanisms, a large number of ligands have been designed to target these G-quadruplex architectures with a view to developing new cancer treatments.9,10 Most of the G4 ligands have been built from a rigid aromatic core able to interact with the G-quartet through π-stacking and decorated with substituents (often positively charged) that can interact with G-quadruplex grooves and/or loops, leading to improved affinity for G4s as well as selectivity over duplex DNA.11–14
Recently, smart G4 ligands were reported to trigger a subsequent reaction event in addition to interacting selectively with G-quadruplex versus duplex DNA. This approach is of great interest to better control the activity and to limit off-target side effects of the ligands. Thereby, covalent modification of the G-quadruplex architecture has been achieved through different strategies including G4-metalation,15,16 G4-alkylation,17 G4-scission18,19 and G4-oxidation.20 This latter process is particularly relevant as guanine is the nucleobase more prone to oxidation. In this context, a number of compounds including porphyrins, naphthalene, perylene diimide derivatives and metal complexes that can photo-oxidize G-quadruplex structures have been reported.19,21–26 However, none of them was shown to induce G-driven telomeric DNA lesions in cellulo. Herein, we report on the design of new smart G4 ligands based on Ru(II) complexes and combining (i) high affinity towards multimeric G4 structures of the telomeric sequence and (ii) photoreactivity to induce oxidative DNA damage. Thanks to telomere dysfunction-induced foci (TIF) experiments, we demonstrate that some complexes, with high affinity for G-quadruplex telomeric DNA, suitable cell penetration and photocytotoxic activity, are able to photo-induce DNA damage in the telomeric region of cancer cells. To the best of our knowledge, the present study demonstrates for the first time the ability of metal complexes to photo-induce telomeric damage in cancer cells. The synthesis of six dinuclear complexes 1a–c and 2a–c (Fig. 1(A); Scheme S1 and Fig. S1–S15, ESI†), bearing either 1,10-phenanthroline (phen) or 1,4,5,8-tetraazaphenanthrene (TAP) ancillary ligands, and a previously reported27 bridging phenanthroimidazole ligand with flexible polyether linkers, was achieved through the coordination of [Ru(phen)2Cl2] or [Ru(TAP)2Cl2] to the desired bridging ligand. The use of dinuclear compounds compared to mononuclear compounds was driven towards designing systems with higher affinities for multimeric G quadruplexes. Ru(II) complexes bearing at least two TAP ligands are well known to trigger PET (Photo-induced Electron Transfer – type I photoreaction) with guanine upon light irradiation, inducing subsequent strand cleavage and photo-adduct formation.28,29 Recently, a TAP containing dinuclear Ru(II) compound also proved to induce phototoxicity in hypoxic regions of melanoma cancer spheroids.30
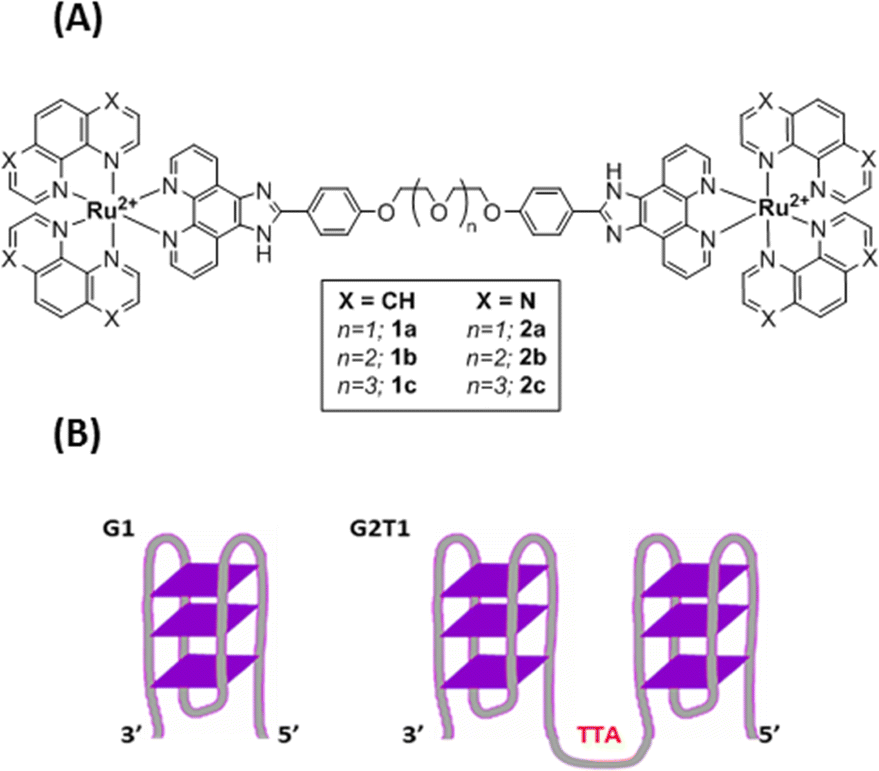 | ||
| Fig. 1 (A) Structures of Ru(II) complexes 1a–c and 2a–c (B) schematic representation of the G1 and G2T1 model G-quadruplex DNA structures. G1: 3′-A(GGGTTA)3GGG-5′; G2T1: 3′-A(GGGTTA)7GGG-5′. | ||
All complexes 1a–c and 2a–c exhibited ground-state absorption spectra and steady-state photoluminescence typical of charge transfer states occurring in Ru(II) polypyridyl complexes (Table S1 and Fig. S16, S17, ESI†). As anticipated, almost no difference was observed in the photo-physical data of complexes bearing different polyether-length linkers. In cyclic voltammetry, complexes 1a–c and 2a–c display successive one-electron reductions of the ancillary phen or TAP moieties (Table S2 and Fig. S18–S23, ESI†). From the estimated reduction potentials at the excited state, complexes 2a–c exhibit stronger photo-oxidizing potentials than compounds 1a–c. Therefore, the photoreactivity of the complexes towards 2′-deoxyguanosine 5′-monophosphate (dGMP) has been investigated thanks to luminescence quenching experiments. As expected, the oxidation potential of dGMP being close to 1.10 V vs. Ag/AgCl,31 no luminescence quenching was observed with complexes 1a–c. In contrast, the addition of dGMP to a solution of complexes 2a–c leads to efficient luminescence quenching (Fig. S24, ESI†), with a high quenching rate constant (3.1 × 109 M−1 s−1, 1.4 × 109 M−1 s−1 and 1.1 × 109 M−1 s−1 for 2a–c, respectively). In agreement with the electrochemical data and the literature, this luminescence quenching can safely be ascribed to the photo-reduction of the complexes 2a–c by a guanine moiety, leading to the generation of guanine radical cations (eqn (1)).
| [Ru*–Ru]4+ + G → [Ru–Ru]3+ + G˙+ | (1) |
Then, the capacity of complexes 1a–c and 2a–c to interact with G4 model structures G1 (3′TT(GGGATT)3GGG5′) and G2T1 (5′TAGGG(TTAGGG)73′) (Fig. 1(B)) was investigated in sodium and potassium buffer by circular dichroism (CD) and bio-layer interferometry (BLI) analysis. Upon addition of complexes 1a–c and 2a–c, no major change was observed in the CD spectra at room temperature, neither with G1 nor G2T1, in sodium or potassium buffer, suggesting that the complexes did not induce any major structural changes at 20 °C (Fig. S25–S28, ESI†). CD melting assays were then performed in both Na+ and K+ containing buffers to assess the thermal stabilization of the quadruplex structures G1 and G2T1 when interacting with complexes 1a–c and 2a–c. The melting temperature of G1 and G2T1 was recorded upon addition of the complexes 1a–c and 2a–c with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Ru(II) metal centre with respect to each G-quadruplex structure (Table S3 and Fig. S29–S32, ESI†). The addition of 1a–c and 2a–c induced a significant thermal stabilization of the dimeric G2T1 that was more pronounced in Na+ containing buffer. A very low stabilization – or even a destabilization – was also observed with the monomeric structure G1. However, it should be mentioned that the stabilization imparted by the ligand would be naturally more pronounced in the intrinsically less stable oligonucleotides (i.e. the dimeric G2T1), the direct comparison of ΔTm values cannot provide straightforward information on the binding affinity.32
:1 ratio of Ru(II) metal centre with respect to each G-quadruplex structure (Table S3 and Fig. S29–S32, ESI†). The addition of 1a–c and 2a–c induced a significant thermal stabilization of the dimeric G2T1 that was more pronounced in Na+ containing buffer. A very low stabilization – or even a destabilization – was also observed with the monomeric structure G1. However, it should be mentioned that the stabilization imparted by the ligand would be naturally more pronounced in the intrinsically less stable oligonucleotides (i.e. the dimeric G2T1), the direct comparison of ΔTm values cannot provide straightforward information on the binding affinity.32
Thus, to gain insight into the binding affinity of the complexes for the different DNA G4s, bio-layer interferometry (BLI) experiments were carried out. This technique indeed allows the study of the affinity independently of the stability of the oligonucleotides. The kinetic and thermodynamic parameters of the interaction towards G2T1, G1 and duplex hairpin DNA HP GC (3′(GC)4TTTT(GC)45′) as a control, both in sodium and potassium buffer (Table 1, Table S4 and Fig. S33–S44) were determined. All complexes display high affinity in the nanomolar range (KD = 100–650 nM) for the dimeric and monomeric G4 structures, in both buffers. In comparison with the previously reported mononuclear Ru(II) complexes, for which the equilibrium dissociation constants values were in the micromolar range against the G1 structure (KD = 1.0–22 μM), the new dinuclear complexes show a much higher affinity towards G-quadruplex DNA.25 For all complexes, the affinity proved to be slightly stronger for the dimeric quadruplex G2T1 than for the monomeric structure G1. We also noticed that the length of the polyether linkers mildly impacts the binding affinity with dimeric and monomeric G-quadruplex DNAs.
| DNA structures | Buffer | 1a | 1b | 1c | 2a | 2b | 2c |
|---|---|---|---|---|---|---|---|
| a Equilibrium dissociation constants deduced from the kinetic rate constants. The provided errors are standard deviations from the mean values. The running buffer was Tris–HCl 10 mM, NaCl or KCl 100 mM (pH 7.04) and 0.5% v/v surfactant. | |||||||
| G2T1 | Na+ | 0.33 ± 0.14 | 0.17 ± 0.01 | 0.12 ± 0.02 | 0.10 ± 0.01 | 0.22 ± 0.10 | 0.21 ± 0.05 |
| K+ | 0.32 ± 0.14 | 0.36 ± 0.17 | 0.52 ± 0.30 | 0.33 ± 0.15 | 0.37 ± 0.13 | 0.48 ± 0.17 | |
| G1 | Na+ | 0.43 ± 0.04 | 0.26 ± 0.04 | 0.23 ± 0.13 | 0.17 ± 0.04 | 0.25 ± 0.05 | 0.65 ± 0.18 |
| K+ | 0.55 ± 0.22 | 0.45 ± 0.30 | 0.40 ± 0.19 | 0.52 ± 0.29 | 0.50 ± 0.09 | 1.65 ± 0.89 | |
| HP GC | Na+ | 1.60 ± 0.05 | 1.20 ± 0.25 | 0.48 ± 0.02 | 0.37 ± 0.05 | 0.38 ± 0.04 | 0.96 ± 0.14 |
| K+ | 1.15 ± 0.20 | 2.66 ± 0.28 | 3.09 ± 0.83 | 7.61 ± 3.64 | 1.41 ± 0.95 | 0.94 ± 0.28 | |
Confocal microscopy was implemented to investigate the cellular uptake of the complexes in U2OS human osteosarcoma cells. As depicted in Fig. 2 (see also Fig. S45, ESI†), all complexes showed effective cell penetration. In the studied concentration range, differences in intracellular distribution were observed: while complex 1a seems to localize mostly to the nucleus, complexes 1b–c and 2a–c localized to both the cytoplasm and the nucleus which was already noticed for the previously reported mononuclear equivalents.25
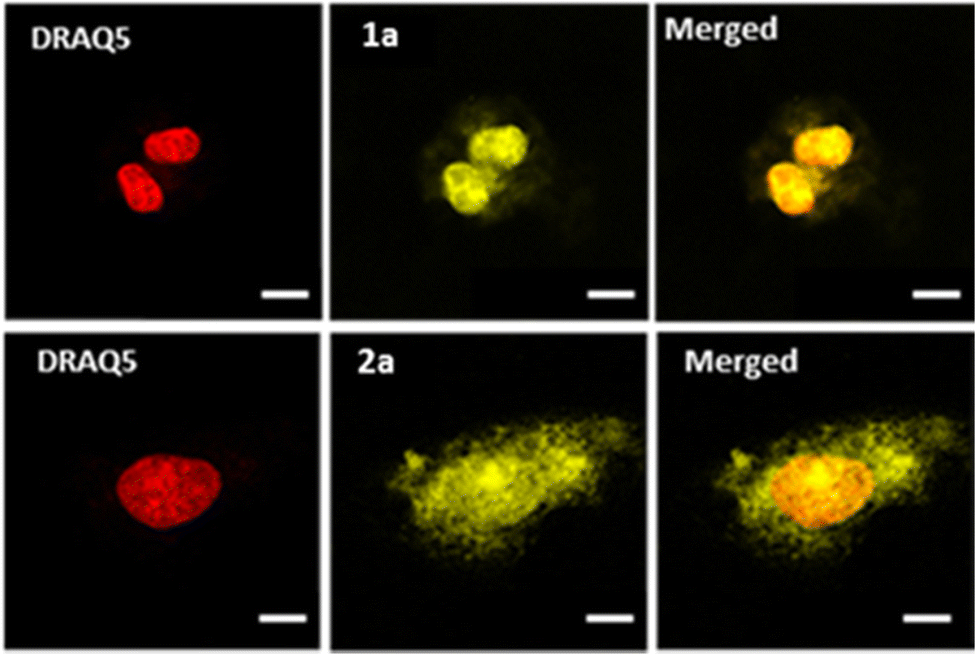 | ||
| Fig. 2 Fluorescence microscopy images of U2OS cells after incubation (1 h 30) with 1a (10 μM) or 2a (50 μM)* complex in DMEM buffer. From left to right: the nucleus in red, stained by DRAQ5; 1a and 2a luminescent complexes in yellow; merged images. Scale, 10 μM. *Due to the inherent quenching associated with TAP-containing complexes, a higher concentration was used for 2a. | ||
The capacity of the complexes to photo-induce cellular toxicity towards the U2OS cell line was then tested. Importantly, no cellular toxicity was observed when different complexes were added to the medium in the dark as previously evidenced for the mononuclear complexes (IC50 > 10 μM).25 In contrast, light irradiation led to a dramatic decrease of the survival rate of U2OS cells. The IC50 values for each complex were within the submicromolar range which was not reported for the mononuclear equivalents (Table 2 and Fig. S46, ESI†).25 The strong photo-cytotoxicity of reference complexes 1a–c could be explained by the photo-sensitization of singlet oxygen, namely type II photo-oxidation as those types of Ru(II) compounds are studied for photodynamic therapy applications.33,34 The singlet oxygen photo-sensitization yields were found to reach 30% and 40% for TAP (2a) and phen (1a) complexes, respectively (see Fig. S49, ESI†), suggesting a high efficiency of cell mortality via ROS formation for each complex. Due to the singlet oxygen diffusion, this process should lead to delocalised damage. However, 2a–c complexes are likely to induce cell mortality also by type I photo-oxidation (i.e. photo-induced electron transfer) as mentioned above, allowing for direct oxidation at the binding site.
| 1a | 1b | 1c | 2a | 2b | 2c | |
|---|---|---|---|---|---|---|
| Cell viability studies. U2OS cells were incubated for 1 h 30 min with complexes 1a–c and 2a–c before 30 minutes irradiation with blue LED light (405 nm at 15.7 W m−2). Tetrazolium salt-based cellular viability assays were performed 24h post-irradiation. Negative control: blue light irradiation without any Ru(II) complex shows no effect on cell viability. | ||||||
| Light | 0.83 | 1.02 | 0.59 | 1.71 | 0.84 | 2.27 |
| Dark | >10 | >10 | >10 | >10 | >10 | >10 |
In addition to the photo-toxicity experiments, the ability of the compounds to target and damage telomeric DNA was studied in U2OS cells. Telomere dysfunction-induced foci (TIF) assays allow monitoring damaged telomeres in cells thanks to the combination of fluorescent in situ hybridization (FISH) – to detect telomeres – and immunofluorescence (IF) against a DNA damage marker, in this case 53BP1.35 We selected two lead compounds for TIF analyses, namely 1a and 2a.36 The bar chart of Fig. 3 displays the frequency of nuclei with at least two damaged telomeres (TIF) under various depicted conditions. We found that, while light irradiation of U2OS cells pre-incubated with 1a did not increase the TIF frequency, a significant three-fold increase in the frequency of nuclei with >2 TIF was observed upon light irradiation for cells incubated with 2a. Note that the presence of a low frequency of TIF in non-treated cells was expected as U2OS cells maintain their telomeres with a telomerase-independent mechanism, dubbed ALT, characterized by replicative stress and low level of DNA damage at telomeres.37 Importantly, TIF were not increased when 2a was added in the dark. It can therefore be stated that complex 2a is able to drastically increase the amount of telomeric damage thanks to its ability to photo-oxidize guanine residues. Light irradiation also resulted in the appearance of 53BP1 foci outside of telomeres (see Fig. S47, ESI†). This is consistent with the fact that although G4 structures are prevalent at telomeres, they are also detected at other genomic loci, including oncogene promoters, gene bodies or 5′ untranslated transcribed regions.38
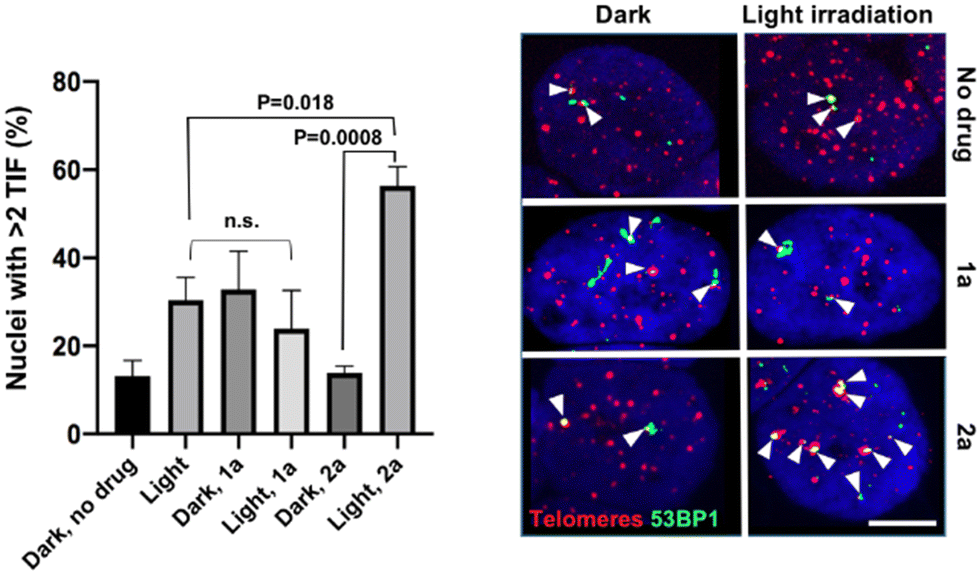 | ||
| Fig. 3 TIF experiments in U2OS cells. Left panel, bar chart of the frequency of nuclei showing at least two TIF (at least 50 nuclei per experiment). Mean ± SEM. Unpaired Student's t tests were applied; n.s.: non-significant. Right panel, representative pictures from the FISH/IF experiments. Telomeres, detected by FISH, appear in red. 53BP1, a marker of DNA damage, detected by IF, appears in green. Arrowheads indicate TIF. Scale, 10 μM. | ||
In conclusion, a series of new ruthenium(II) dinuclear complexes bearing 1,10-phenanthroline (phen) and 1,4,5,8-tetraazaphenthrene (TAP) ligands have been synthesized as new telomeric photo-reactive agents. Complexes 2a–c can (i) strongly interact with telomeric DNA and (ii) photo-induce electron transfer with dGMP. All complexes were found to efficiently penetrate the cells and to induce dramatic damage upon light irradiation, whereas no toxicity was observed in the dark. Crucially, we evidenced the presence of photo-induced DNA lesions at the telomeres of U2OS osteosarcoma cells incubated with complex 2a. These results emphasize the importance of direct PET with guanine and represent, to the best of our knowledge, the first in cellulo evidence of G-driven photo-oxidative damage at telomeric DNA of cancer cells. In the future, the synthesis of related photo-redox active structures with higher selectivity towards duplex DNA may provide new powerful approaches to target telomeres. This is currently under investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fonds de la Recherche Scientifique – FNRS under Grant no. J.0091.18. J. W. thanks the “Fonds pour la Formation à la Recherche dans l’Industrie et dans l’Agriculture” (FRIA) and the Erasmus programme for financial support. This work was partially supported by the French “Agence National de la Recherche” (ANR-16-CE11-0006-01) and CBH-EUR-GS (ANR-17-EURE-0003) agencies, the région Auvergne-Rhône-Alpes and the Fondation Belge Contre le Cancer (convention 2018-072) agency. The NanoBio-ICMG platforms (UAR 2607) are acknowledged for their support.References
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential, Nat. Rev. Mol. Cell Biol., 2017, 18(5), 279–284 CrossRef.
- M. Di Antonio, A. Ponjavic, A. Radzevičius, R. T. Ranasinghe, M. Catalano and X. Zhang, et al., Single-molecule visualization of DNA G-quadruplex formation in live cells, Nat. Chem., 2020, 12(9), 832–837 CrossRef CAS PubMed.
- D. Sen and W. Gilbert, Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis, Nature, 1988, 334(6180), 364–366 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner and A. Parry, et al., G-quadruplex structures mark human regulatory chromatin, Nat. Genet., 2016, 48(10), 1267–1272 CrossRef PubMed.
- J. W. Shay and W. E. Wright, Telomeres and telomerase in normal and cancer stem cells, FEBS Lett., 2010, 584(17), 3819–3825 CrossRef CAS PubMed.
- E. H. Blackburn, E. S. Epel and J. Lin, Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection, Science, 2015, 350(6265), 1193–1198 CrossRef CAS PubMed.
- A. L. Moye, K. C. Porter, S. B. Cohen, T. Phan, K. G. Zyner and N. Sasaki, et al., Telomeric G-quadruplexes are a substrate and site of localization for human telomerase, Nat. Commun., 2015, 6, 7643 CrossRef PubMed.
- J. W. Shay and W. E. Wright, Telomeres and telomerase: Three decades of progress, Nat. Rev. Genet., 2019, 20(5), 299–309 CrossRef CAS PubMed.
- S. Asamitsu, T. Bando and H. Sugiyama, Ligand design to acquire specificity to intended G-quadruplex structures, Chem. – Eur. J., 2019, 25(2), 417–430 CrossRef CAS.
- N. Kosiol, S. Juranek, P. Brossart, A. Heine and K. Paeschke, G-quadruplexes: A promising target for cancer therapy, Mol. Cancer, 2021, 20(1), 40 CrossRef CAS PubMed.
- S. Balasubramanian and S. Neidle, G-quadruplex nucleic acids as therapeutic targets, Curr. Opin. Chem. Biol., 2009, 13(3), 345–353 CrossRef CAS PubMed.
- A. Łęczkowska, J. Gonzalez-Garcia, C. Perez-Arnaiz, B. Garcia, A. J. P. White and R. Vilar, Binding studies of metal–salphen and metal–bipyridine complexes towards G-quadruplex DNA, Chem. – Eur. J., 2018, 24(45), 11785–11794 CrossRef.
- M. Gillard, J. Weynand, H. Bonnet, F. Loiseau, A. Decottignies and J. Dejeu, et al., Flexible Ru(II) Schiff base complexes: G-quadruplex DNA binding and photo-induced cancer cell death, Chem. – Eur. J., 2020, 26(61), 13849–13860 CrossRef CAS PubMed.
- G. Piraux, L. Bar, M. Abraham, T. Lavergne, H. Jamet and J. Dejeu, et al., New ruthenium-based probes for selective G-quadruplex targeting, Chem. – Eur. J., 2017, 23, 11872–11880 CrossRef CAS.
- H. Bertrand, S. Bombard, D. Monchaud, E. Talbot, A. Guédin and J.-L. Mergny, et al., Exclusive platination of loop adenines in the human telomeric G-quadruplex, Org. Biomol. Chem., 2009, 7(14), 2864–2871 RSC.
- E. Wachter, D. Moyá, S. Parkin and E. C. Glazer, Ruthenium complex “Light Switches” that are selective for different G-quadruplex structures, Chem. – Eur. J., 2016, 22(2), 550–559 CrossRef CAS PubMed.
- M. Nadai, F. Doria, M. Di Antonio, G. Sattin, L. Germani and C. Percivalle, et al., Naphthalene diimide scaffolds with dual reversible and covalent interaction properties towards G-quadruplex, Biochimie, 2011, 93(8), 1328–1340 CrossRef CAS PubMed.
- Z. Yu, M. Han and J. A. Cowan, Toward the design of a catalytic metallodrug: Selective cleavage of G-quadruplex telomeric DNA by an anticancer copper–acridine–ATCUN complex, Angew. Chem., Int. Ed., 2015, 54(6), 1901–1905 CrossRef CAS PubMed.
- M. Nadai, F. Doria, M. Scalabrin, V. Pirota, V. Grande and G. Bergamaschi, et al., A catalytic and selective scissoring molecular tool for quadruplex nucleic acids, J. Am. Chem. Soc., 2018, 140(44), 14528–14532 CrossRef CAS PubMed.
- A. D. Beniaminov, R. A. Novikov, O. K. Mamaeva, V. A. Mitkevich, I. P. Smirnov and M. A. Livshits, et al., Light-induced oxidation of the telomeric G4 DNA in complex with Zn(II) tetracarboxymethyl porphyrin, Nucleic Acids Res., 2016, 44(21), 10031–10041 CAS.
- C. Vialas, G. Pratviel and B. Meunier, Oxidative damage generated by an oxo-metalloporphyrin onto the human telomeric sequence, Biochemistry, 2000, 39, 9514 CrossRef CAS.
- M. Nadai, F. Doria, L. Germani, S. N. Richter and M. Freccero, A photoreactive G-quadruplex ligand triggered by green light, Chem. – Eur. J., 2015, 21(6), 2330–2334 CrossRef CAS PubMed.
- S. A. Archer, A. Raza, F. Dröge, C. Robertson, A. J. Auty and D. Chekulaev, et al., A dinuclear ruthenium(II) phototherapeutic that targets duplex and quadruplex DNA, Chem. Sci., 2019, 10, 3502–3513 RSC.
- J. Weynand, H. Bonnet, F. Loiseau, J.-L. Ravanat, J. Dejeu and E. Defrancq, et al., Targeting G-rich DNA structures with photoreactive bis-cyclometallated iridium(III) complexes, Chem. – Eur. J., 2019, 25(55), 12730–12739 CrossRef CAS PubMed.
- J. Weynand, A. Diman, M. Abraham, L. Marcélis, H. Jamet and A. Decottignies, et al., Towards the development of photo-reactive ruthenium(II) complexes targeting telomeric G-quadruplex DNA, Chem. – Eur. J., 2018, 24(72), 19216–19227 CrossRef CAS PubMed.
- M. Gillard, G. Piraux, M. Daenen, M. Abraham, L. Troian-Gautier and L. Bar, et al., Photo-oxidizing ruthenium(II) complexes with enhanced visible light absorption and G-quadruplex DNA binding abilities, Chem. – Eur. J., 2022, e202202251 Search PubMed.
- C.-Q. Zhou, T.-C. Liao, Z.-Q. Li, J. Gonzalez-Garcia, M. Reynolds and M. Zou, et al., Dinickel–Salphen complexes as binders of human telomeric dimeric G-quadruplexes, Chem. – Eur. J., 2017, 23(19), 4713–4722 CrossRef CAS PubMed.
- B. Elias and A. Kirsch-De Mesmaeker, Photo-reduction of polyazaaromatic Ru(II) complexes by biomolecules and possible applications, Coord. Chem. Rev., 2006, 250(13–14), 1627–1641 CrossRef CAS.
- S. Rickling, L. Ghisdavu, F. Pierard, P. Gerbaux, M. Surin and P. Murat, et al., A Rigid dinuclear ruthenium(II) complex as an efficient photoactive agent for bridging two guanine bases of a duplex or quadruplex oligonucleotide, Chem. – Eur. J., 2010, 16(13), 3951–3961 CrossRef CAS PubMed.
- A. Raza, S. A. Archer, S. D. Fairbanks, K. L. Smitten, S. W. Botchway and J. A. Thomas, et al., A dinuclear ruthenium(II) complex excited by near-infrared light through two-photon absorption induces phototoxicity deep within hypoxic regions of melanoma cancer spheroids, J. Am. Chem. Soc., 2020, 142(10), 4639–4647 CrossRef CAS PubMed.
- S. Steenken and S. V. Jovanovic, How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution, J. Am. Chem. Soc., 1997, 119(3), 617–618 CrossRef CAS.
- R. Rocca, F. Moraca, G. Costa, M. Nadai, M. Scalabrin and C. Talarico, et al., Identification of G-quadruplex DNA/RNA binders: Structure-based virtual screening and biophysical characterization, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861(5, Part B), 1329–1340 CrossRef CAS PubMed.
- J. Karges, S. Kuang, F. Maschietto, O. Blacque, I. Ciofini and H. Chao, et al., Rationally designed ruthenium complexes for 1- and 2-photon photodynamic therapy, Nat. Commun., 2020, 11(1), 3262 CrossRef CAS PubMed.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Light-activated ruthenium complexes photobind DNA and are cytotoxic in the photodynamic therapy window, Chem. Commun., 2012, 48(77), 9649–9651 RSC.
- I. Mender and J. W. Shay, Telomere dysfunction induced foci (TIF) ANalysis, Bio-Protoc., 2015, 5(22), e1656 Search PubMed.
- These complexes were chosen due to strong interaction with dimeric G-quadruplexes DNA and their major localization in the nucleus of the cancer cells.
- M. Raghunandan, D. Geelen, E. Majerova and A. Decottignies, NHP2 downregulation counteracts hTR-mediated activation of the DNA damage response at ALT telomeres, EMBO J., 2021, 40(6), e106336 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, J. Spiegel, G. Marsico, D. Tannahill and S. Balasubramanian, Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing, Nat. Protoc., 2018, 13(3), 551–564 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00192f |
| This journal is © The Royal Society of Chemistry 2022 |