
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A synthesis-enabled relative configurational assignment of the C31–C46 region of hemicalide†
Tegan P.
Stockdale
,
Nelson Y. S.
Lam
 * and
Ian
Paterson
*
* and
Ian
Paterson
*
Yusuf Hamied Department of Chemistry, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 14th April 2022
Abstract
With 21 unknown stereocentres embedded in spatially separated stereoclusters, the cytotoxic polyketide hemicalide represents a seemingly intractible structural assignment problem. Herein, through the targeted synthesis of configurationally defined fragments, as well as “encoded” mixtures of diastereomers, the stereochemical elucidation of the C31–C46 region of hemicalide is achieved. Detailed NMR spectroscopic analysis of candidate fragments and comparison with the related hemicalide data strongly supported a 31,32-syn, 32,36-anti and 42,46-anti relationship. In combination with previous work on hemicalide, this reduces the number of possible structural permutations down to a more manageable eight diastereomers.
Hemicalide (1, Fig. 1) is a complex polyketide isolated from the marine sponge hemimycale sp., exhibiting extraordinary picomolar IC50 values against a panel of human cancer cell lines through a putative antimitotic mode of action.1 First reported in the patent literature in 2011, all 21 stereocentres within hemicalide were initially unassigned, leaving over two million possible permutations.
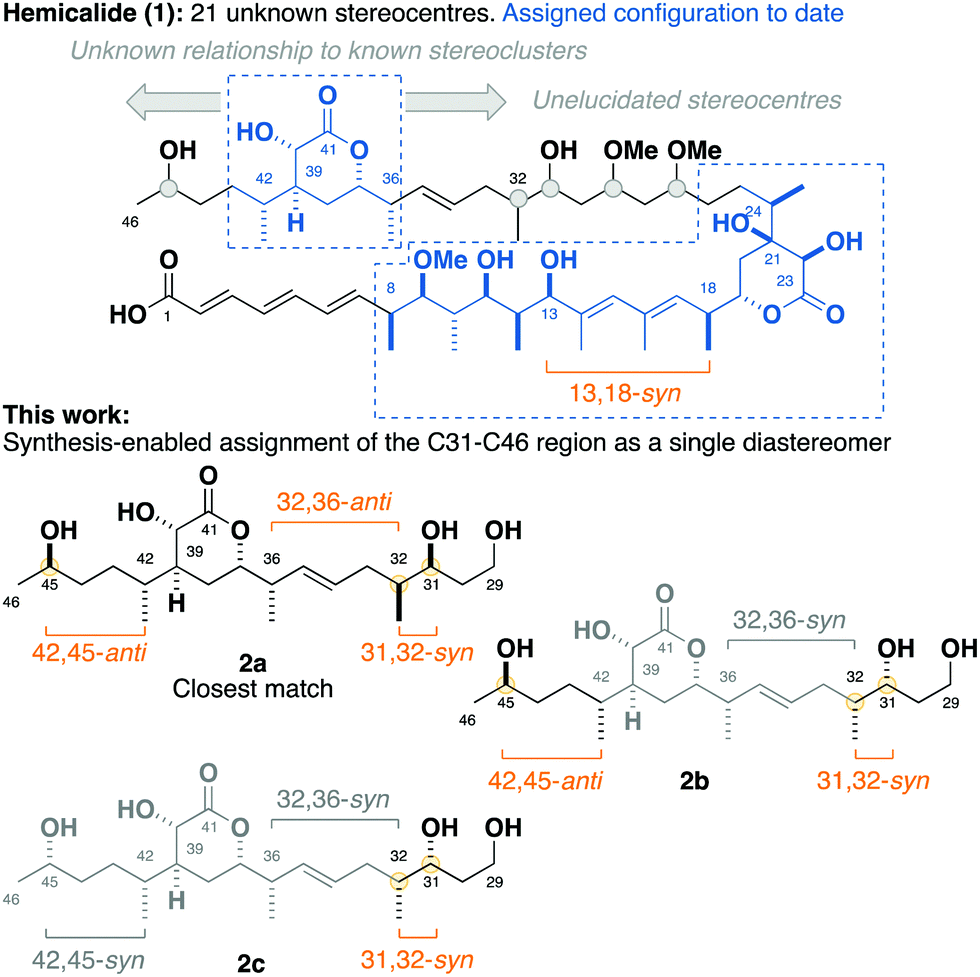 | ||
| Fig. 1 Structure of hemicalide (1) and the assigned relative configurations of each region determined to date. Through the stereocontrolled synthesis of the candidate truncate fragments 2a, 2b and 2c, followed by detailed 1H and 13C NMR correlations, this work assigns the C31–C46 region as a single diastereomer as in 2a. | ||
By interrogation of the available 1H and 13C NMR data for hemicalide, our group along with that of Ardisson–Meyer–Cossy have focused on solving this challenging stereochemical conundrum through the synergistic combination of synthetic and computational approaches. To date, this has enabled the confident assignment of the C8–C13 stereohexad,2,3 C18–C24 dihydroxylactone4 and C36–C42 hydroxylactone regions.5,6 In addition, a synthesis-enabled investigation established the relative configuration between the C8–C13 and C18–C24 regions.7 These cumulative efforts served to narrow the possible structural permutations down to 128 stereoisomers, leaving the C27–C32 stereotetrad, the isolated C45 stereo centre, as well as their relationship to the other stereoclusters, unassigned.8–10
Herein, we report a targeted synthesis of the C35–C43 region of hemicalide and its elaboration to generate the candidate fragments 2a, 2b and 2c. Detailed NMR spectroscopic comparison with the natural product then served to assign the previously unknown C31, C32 and C45 stereocentres relative to the C35–C43 region. Through this evolutionary approach,11,12 the C31–C46 region is narrowed down to a single diastereomer, translating to only eight possible diastereomers remaining for hemicalide.
Seeking flexibility in the installation of the C45 hydroxyl-bearing stereocentre, as well as modularity in appending candidate C29–C34 fragments, 2 was disconnected across the internal C34–C35 alkene to afford the terminal alkenes 3 and 4 (Fig. 2).13 Given the isolated nature of C45, we planned to perform a stereoselective carbonyl reduction of the corresponding enone under reagent control followed by hydrogenation. Recognition of the 1,4-syn relationship between C37 and C42 alluded to the execution of a boron-mediated aldol reaction between the Roche ester-derived building blocks 5 and 6. Critically, the antipode of 3 could then be readily obtained by using the enantiomeric components.
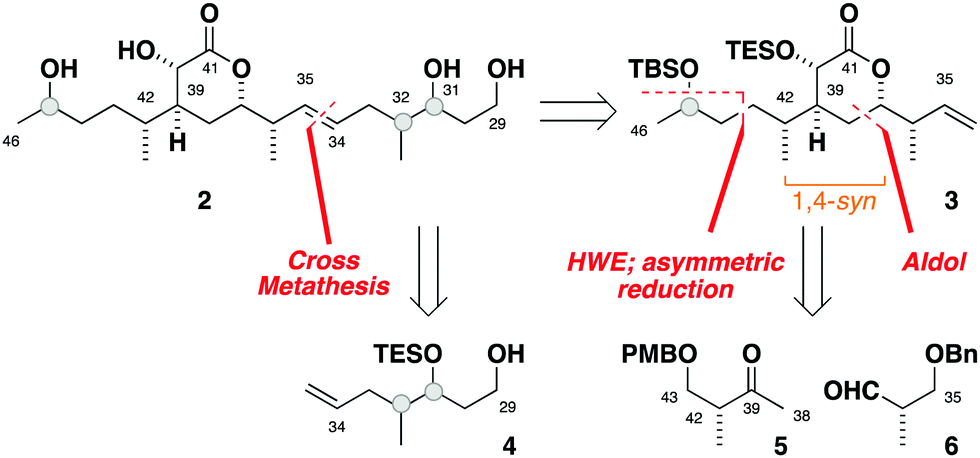 | ||
| Fig. 2 Retrosynthesis for the C29–C46 truncate 2. | ||
Synthesis of the alkene 3 commenced with a (+)-Ipc2BCl mediated aldol reaction between the known ketone 54 and the aldehyde 614 to afford 7 (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr),15 following silyl ether formation (Scheme 1). Preliminary studies towards installing the C39 stereocentre via the conjugate reduction of a cyclic enoate derivative afforded the undesired configuration,16 indicating that a suitable acyclic enoate might be sought. To this end, a tandem aldol reaction on the ketone 7 using the lithium enolate of 8, followed by in situ Peterson olefination gave the Z-enoate 9. Extensive experimentation revealed that the desired C39 configuration was best installed through a hydroxyl-directed reduction under optimised conditions. Thus, following silyl ether cleavage of 9, hydrogenation of the resulting alkene 10 mediated by Crabtree's catalyst ([Ir(cod)py(PCy3)][PF6], 13 mol%, H2, 1 atm) at low temperature (−23 °C) and acidic workup delivered the δ-lactone 11 (5:1 dr) now favouring the desired C39 configuration.17 From 11, α-hydroxylation via reaction of the derived potassium enolate with Davis oxaziridine18 cleanly gave the alcohol 12 (>20:1 dr) bearing the required C40 configuration.5 Silyl ether formation then enabled the chromatographic separation of the C39 epimers to obtain the desired C35–C43 fragment 13.
:1 dr),15 following silyl ether formation (Scheme 1). Preliminary studies towards installing the C39 stereocentre via the conjugate reduction of a cyclic enoate derivative afforded the undesired configuration,16 indicating that a suitable acyclic enoate might be sought. To this end, a tandem aldol reaction on the ketone 7 using the lithium enolate of 8, followed by in situ Peterson olefination gave the Z-enoate 9. Extensive experimentation revealed that the desired C39 configuration was best installed through a hydroxyl-directed reduction under optimised conditions. Thus, following silyl ether cleavage of 9, hydrogenation of the resulting alkene 10 mediated by Crabtree's catalyst ([Ir(cod)py(PCy3)][PF6], 13 mol%, H2, 1 atm) at low temperature (−23 °C) and acidic workup delivered the δ-lactone 11 (5:1 dr) now favouring the desired C39 configuration.17 From 11, α-hydroxylation via reaction of the derived potassium enolate with Davis oxaziridine18 cleanly gave the alcohol 12 (>20:1 dr) bearing the required C40 configuration.5 Silyl ether formation then enabled the chromatographic separation of the C39 epimers to obtain the desired C35–C43 fragment 13.
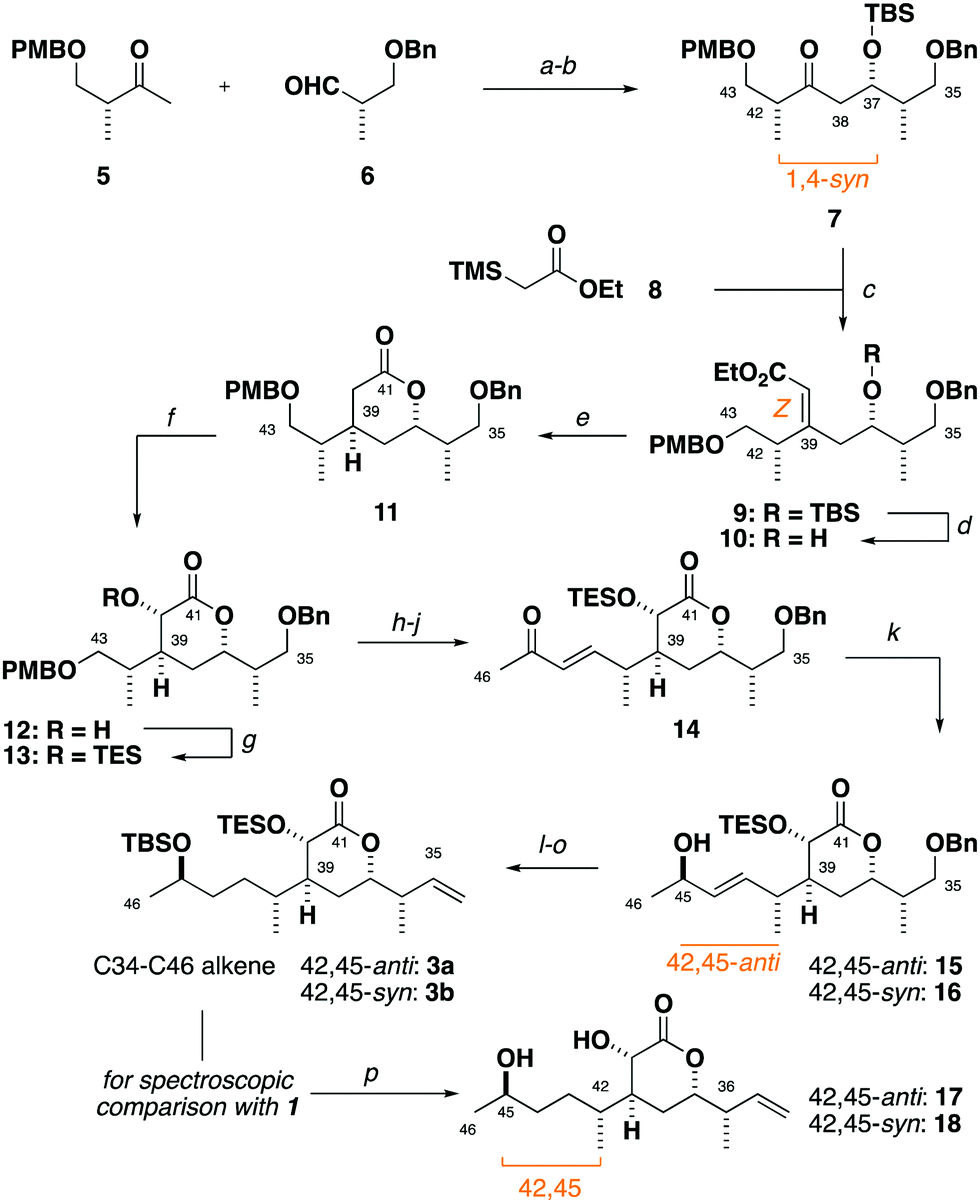 | ||
| Scheme 1 Synthesis of hemicalide C34–C46 alkenes 3a and 3b, and fragments 17 and 18 for the spectroscopic determination of the C45 relative configuration. Reagents and conditions: (a) 5, (+)-Ipc2BCl, Et3N, Et2O, 0 °C; 6 −78 → −20 °C, 91%, >20:1 dr; (b) TBSOTf, 2,6-lutidine, CH2Cl2, −78 → 0 °C, 99%; (c) 8, LDA, THF, −78 °C; 7, THF, −78 °C → rt, 80%, >19:1 Z:E; (d) TsOH·H2O, MeOH, rt, 97%; (e) [Ir(cod)py(PCy3)][PF6] (13 mol%), H2 (1 atm), CH2Cl2, −20 °C → rt; TsOH·H2O, 93%, 5:1 dr; (f) 11, KHMDS, THF, −78 °C; 2-phenylsulfonyl-3-phenyloxaziridine, THF, −78 °C, 81%, >20:1 dr; (g) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C, >99%; isolated 37,39-anti diastereomer: 85%; (h) DDQ, CH2Cl2/pH 9.2 buffer (4:1), rt, 97%; (i) (COCl)2, DMSO, CH2Cl2; Et3N, −78 → −20 °C; (j) dimethyl 2-oxopropylphosphonate, Ba(OH)2, THF/H2O (40:1), 0 °C → rt, 74% over two steps, >20:1 E:Z; (k) For 15: LiAlH4, (+)-N-methylephedrine, N-ethylaniline, Et2O; −94 °C, 62%, 6:1 dr; (l) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 92%; (m) RANEY® Ni, H2, EtOAc, rt, 99%; (n) Dess–Martin Periodinane, NaHCO3, CH2Cl2, rt; (o) MePPh3Br, nBuLi, THF, 0 °C → rt, 81% over two steps; (p) HF·py, py, THF, 0 °C, 99%. | ||
From 13, a sequence of PMB ether cleavage, followed by oxidation of the resulting alcohol to the aldehyde and Horner–Wadsworth–Emmons (HWE) olefination cleanly delivered the requisite E-enone 14.19 A preliminary screen revealed that the flexible installation of the C45 stereocentre was achievable under Terashima asymmetric reduction conditions, with either C45-configuration (42,45-anti15 and 42,45-syn16) selectively obtained (6:1 dr) through use of the appropriate antipode of the N-methylephedrine ligand.20
Preliminary reconnaissance next revealed that the C45 alcohol was best derivatised as its TBS ether to avoid any desilylation under subsequent hydrogenation conditions. In a parallel sequence, concomitant alkene hydrogenation and benzyl ether cleavage with RANEY® nickel, oxidation and Wittig methylenation afforded both candidate fragments of the full C34–C46 alkene (42,45-anti3a, 42,45-syn3b) in readiness for the downstream cross metathesis for chain extension.
At this stage, global desilylation permitted a head-to-head NMR spectroscopic comparison with hemicalide, enabled by the timely provision of the original FID files by the isolation team (Table 1). Interrogation of the 1H and 13C chemical shift data for the epimeric fragments 17 and 18 indicated that 17 containing a 42,45-anti relationship (entry 1: Σ|ΔH| = 0.05 ppm; Σ|ΔC| = 0.38 ppm) was a closer fit to the natural product than the alternative 42,45-syn configuration present in 18 (entry 2: Σ|ΔH| = 0.13 ppm; Σ|ΔC| = 0.67 ppm).
At this juncture, we undertook the detailed spectroscopic analysis of a reported full skeletal structure of hemicalide bearing a 31,32-anti configuration,9 noting significant 1H and 13C NMR chemical shift deviations in this region vis-à-vis the natural product (Fig. 3). This provided strong evidence against a 31,32-anti relationship and gave us confidence to concentrate on the alternative and unconsidered 31,32-syn configuration.
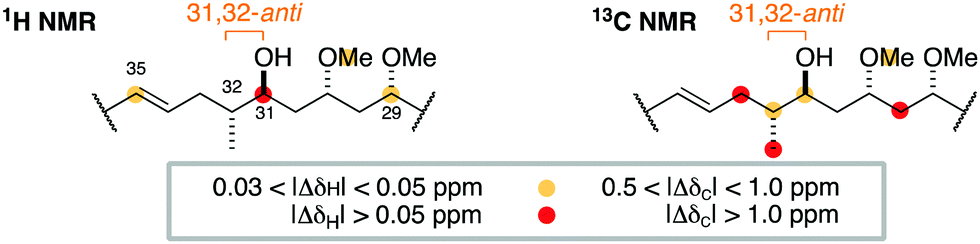 | ||
| Fig. 3 Comparison of the 1H and 13C NMR chemical shifts for the C29–C35 region of a previously reported diastereomer (ref. 9) with the corresponding data for hemicalide does not support the 31,32-anti configuration. | ||
Synthesis of the C29–C35 alkene fragment commenced with a Brown syn-crotylation of the aldehyde 1921 and silylation to afford the TBS ether 20 and set the 31,32-syn configuration (Scheme 2).22 Alkene hydroboration, alcohol oxidation and Wittig methylenation then provided the homologated alkene 4a. An analogous sequence using the enantiomeric crotylation reagent delivered ent-4a as required to establish the relationship between the stereoclusters. From here, a parallel cross-metathesis with 3a (containing the 42,45-anti configuration) mediated by Hoveyda–Grubbs II catalyst (13 mol%), followed by PMB ether cleavage, gave the separate C29–C46 fragments 32,36-anti21 and 32,36-syn22.23 Global desilylation under fluorous conditions then gave the truncated tetraols 2a and 2b for detailed NMR spectroscopic comparison. An analogous sequence with 3b (42,45-syn configuration) and ent-4a provided the 32,36-syn, 42,45-syn diastereomer 2c.
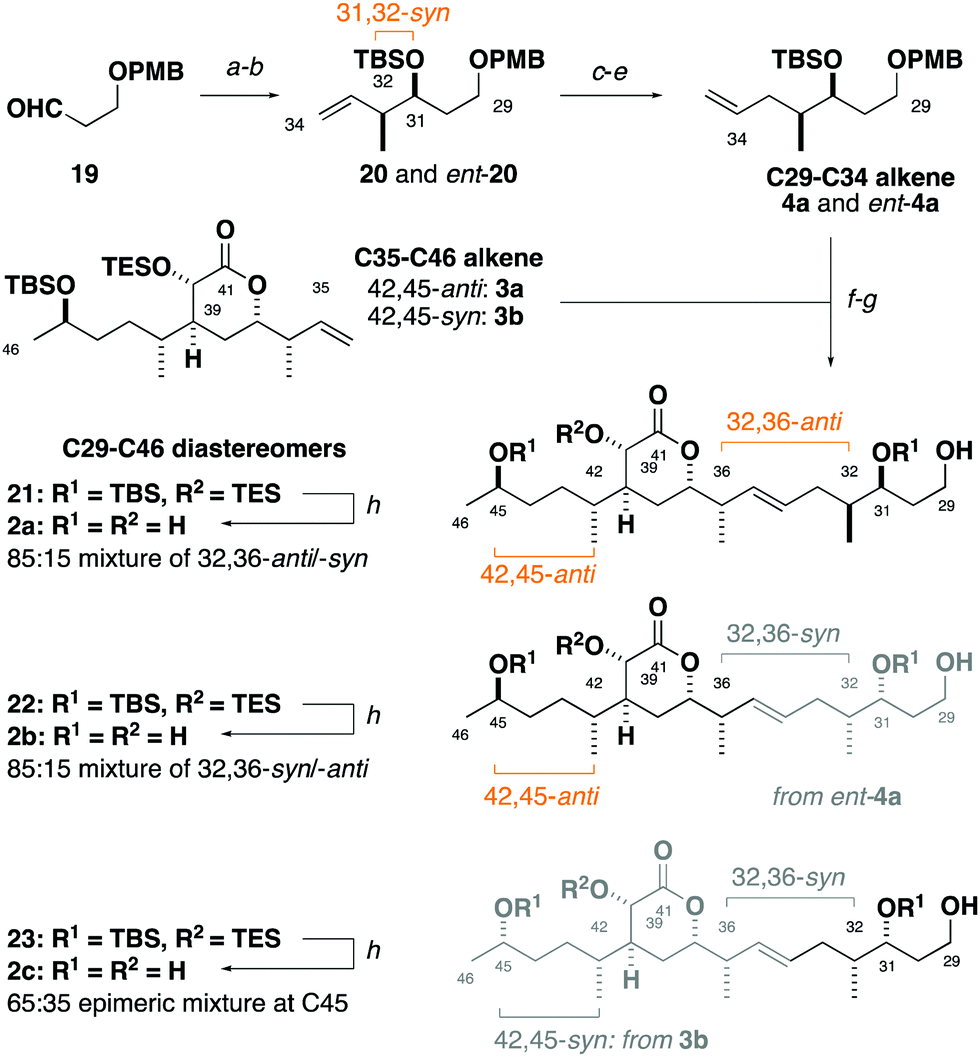 | ||
| Scheme 2 Synthesis of the C29–C34 alkene and fragment union. Reagents and conditions: (a) cis-but-2-ene, tBuOK, nBuLi, THF, −78 → −45 °C; (+)-Ipc2BOMe, −78 °C; BF3·OEt2; 19, THF, −78 °C; H2O2, NaOH, −78 °C → rt; (b) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 88% over two steps, >20:1 dr; (c) BH3·SMe2, THF, 0 °C → rt; H2O2, NaOH, MeOH, 0 °C → rt, 80%; (d) (COCl)2, DMSO; Et3N, CH2Cl2, –78 °C → rt; (e) MePPh3Br, nBuLi, THF, 0 °C → rt, 60% over two steps; (f) Hoveyda–Grubbs II Catalyst (13 mol%), p-benzoquinone, PhMe, 80 °C; (g) DDQ, CH2Cl2/pH 9.2 buffer (4:1), 0 °C → rt, 59% over two steps; (h) aq. HF/MeCN (10%), −20 °C; Et3N, 99%. | ||
At the outset, we were cognisant that subtle and minute chemical shift differences resulting from 1,4- and 1,5-related stereoclusters separated by flexible acyclic linkers could confound conclusions in this study. This was anticipated for the as yet unassigned stereocentres at C31, C32 and C45 in the sidechains appended to the established δ-lactone region. To ameliorate this, we prepared fragment 2c bearing the 32,36-syn, 42,45-syn configuration containing an “encoded” 65:35 epimeric mixture at C45, as well as a configurationally pure C35–C46 region coupled with an “encoded” 85:15 mixture of the C29–C35 alkene enantiomers to generate 2b as an 85:15 mixture of 32,36-syn and -anti diastereomers. Analogously, 2a was synthesised containing an 85:15 mixture of 32,36-anti and -syn diastereomers with 2b as the minor component (see the ESI†).24 With these fragments in hand, detailed 1H and 13C NMR spectroscopic comparison with the corresponding region for the hemicalide spectra (CD3OD) processed in-house could now be conducted (Table 2 and Fig. 4). Initial inspection revealed that all the 31,32-syn diastereomers (entries 1 to 3, Σ|ΔH| < 0.11 ppm; Σ|ΔC| < 2.86 ppm) possessed a much lower chemical shift deviation relative to hemicalide compared with the 31,32-anti configuration previously reported (entry 4: Σ|ΔH| = 0.33 ppm; Σ|ΔC| = 8.48 ppm).9 This strongly supported the assignment of a 31,32-syn configuration in the natural product. Next, a comparison of the 13C NMR data for fragments 2c (42,45-syn) and 2b (42,45-anti) with hemicalide supported the 42,45-anti configuration assigned above (entry 2: Σ|ΔC| = 2.72 ppm) over the alternative 42,45-syn isomer (entry 3: Σ|ΔC| = 2.86 ppm). A final comparison of 32,36-syn2b (entry 2), 32,36-anti2a (entry 1) and the hemicalide 1H and 13C NMR data conclusively gave lower chemical shift deviations for the 32,36-anti diastereomer (entry 1: Σ|ΔH| = 0.06 ppm; Σ|ΔC| = 1.57 ppm) over the alternative 32,36-syn isomer (entry 2: Σ|ΔH| = 0.11 ppm; Σ|ΔC| = 2.72 ppm), representing the best fit candidate for future targeted synthetic efforts. Importantly, this provides strong evidence for the relative configurational assignment of the C31, C32 and C45 stereocentres.
| Entry | Sum |Δ| 1H | Max |Δ| 1H | Sum |Δ| 13C | Max |Δ| 13C |
|---|---|---|---|---|
| a Absolute errors taken for 1H and 13C NMR chemical shifts between H/C31-H/C46. b |Δ| = δ(experimental shift) – δ(reported shift), errors in ppm. | ||||
| 1. 31,32-syn, 32,36-anti, 42,45-anti 2a | 0.06 | 0.04 | 1.57 | 0.81 |
| 2. 31,32-syn, 32,36-syn, 42,45-anti2b | 0.11 | 0.04 | 2.72 | 0.72 |
| 3. 31,32-syn, 32,36-syn, 42,45-syn2c | 0.10 | 0.03 | 2.86 | 0.73 |
| 4. Lecourt et al. diastereomer (ref. 9), 31,32-anti, 32,36-syn, 42,45-anti | 0.33 | 0.14 | 8.48 | 1.32 |
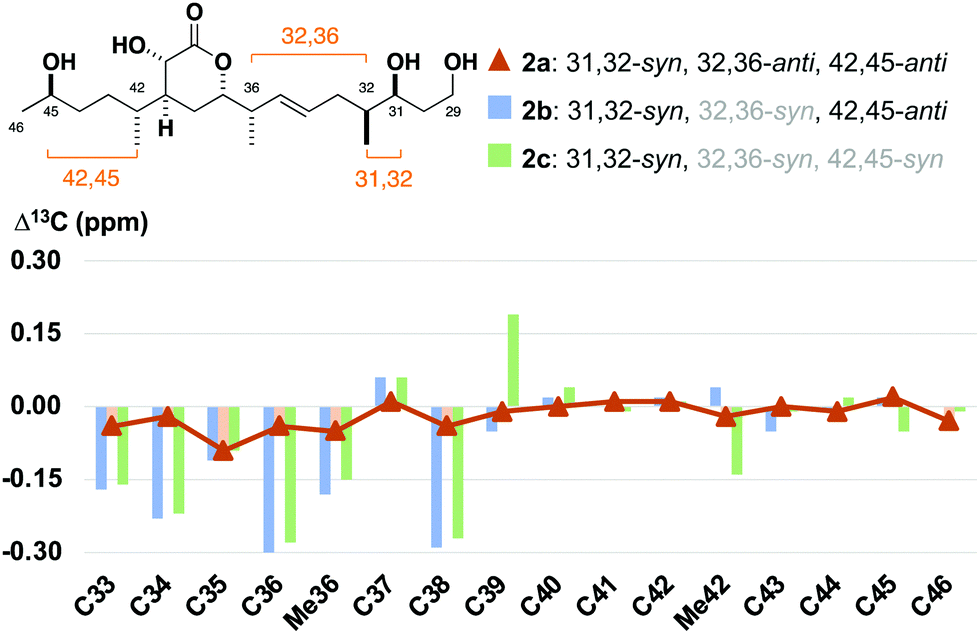 | ||
| Fig. 4 Bar graph highlighting the 13C NMR chemical shift differences between the C33–C46 diastereomers 2a, 2b and 2c relative to hemicalide (1), overlaid with a line graph for the best match in 2a. See the ESI† for expanded bar graphs and detailed tabulated data. | ||
In summary, we have developed a streamlined synthesis of the C29–C46 region of hemicalide, enabling the flexible installation of both the C45 hydroxyl and the C31–C32 stereocluster to give candidate structural permutations.
Spectroscopic comparison with each diastereomer then provided firm evidence in support of the (i) 31,32-syn, (ii) 32,36-anti and (iii) 42,45-anti configuration. In conjunction with the prior assignment of the C1–C24 region,7 this serves to reduce the stereochemical conundrum to only eight possible candidate diastereomers (out of >1 million). In addition to reaffirming the undisputed power of chemical synthesis as the arbiter of stereochemical determination,25–27 the current work should enable future access to advanced intermediates towards interrogating a now-tractable set of possible diastereomers within the vast stereochemical space occupied by hemicalide.
We thank the Herchel Smith Fund (TPS), Woolf Fisher Trust and Trinity Hall, Cambridge (NYSL) for financial support, Dr Georges Massiot for the kind supply of the FID NMR data for hemicalide and, together with Prof Janine Cossy, for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) I. Carletti, C. Debitus and G. Massiot, Molécules polykétides comme agents anticancéreux, Pat. Appl. Pub., WO2011051380A1 (FR), 2011; Chem. Abstr., 2011, 154, 5130950 Search PubMed; (b) L. Marcourt and G. Massiot, Eur. J. Org. Chem., 2022, e22020208 Search PubMed.
- E. Fleury, M.-I. Lannou, O. Bistri, F. Sautel, G. Massiot, A. Pancrazi and J. Ardisson, J. Org. Chem., 2009, 74, 7034 CrossRef CAS PubMed.
- S. G. Smith and J. M. Goodman, J. Am. Chem. Soc., 2010, 132, 12946 CrossRef CAS PubMed.
- C. I. MacGregor, B. Y. Han, J. M. Goodman and I. Paterson, Chem. Commun., 2016, 52, 4632 RSC.
- S. Specklin, G. Boissonnat, C. Lecourt, G. Sorin, M.-I. Lannou, J. Ardisson, F. Sautel, G. Massiot, C. Meyer and J. Cossy, Org. Lett., 2015, 17, 2446 CrossRef CAS PubMed.
- E. De Gussem, W. Herrebout, S. Specklin, C. Meyer, J. Cossy and P. Bultinck, Chem. – Eur. J., 2014, 20, 17385 CrossRef CAS PubMed.
- B. Y. Han, N. Y. S. Lam, C. I. MacGregor, J. M. Goodman and I. Paterson, Chem. Commun., 2018, 54, 3247 RSC.
- C. Lecourt, S. Boinapally, S. Dhambri, G. Boissonnat, C. Meyer, J. Cossy, F. Sautel, G. Massiot, J. Ardisson, G. Sorin and M.-I. Lannou, J. Org. Chem., 2016, 81, 12275 CrossRef CAS PubMed.
- C. Lecourt, S. Dhambri, K. Yamani, G. Boissonnat, S. Specklin, E. Fleury, K. Hammad, E. Auclair, S. Sablé, A. Grondin, P. B. Arimondo, F. Sautel, G. Massiot, C. Meyer, J. Cossy, G. Sorin, M. I. Lannou and J. Ardisson, Chem. – Eur. J., 2019, 25, 2745 CrossRef CAS PubMed.
- G. Sorin, E. Fleury, C. Tran, E. Prost, N. Molinier, F. Sautel, G. Massiot, S. Specklin, C. Meyer, J. Cossy, M. I. Lannou and J. Ardisson, Org. Lett., 2013, 15, 4734 CrossRef CAS PubMed.
- N. Y. S. Lam and I. Paterson, Eur. J. Org. Chem., 2020, 2310 CrossRef CAS.
- K. C. Nicolaou and S. A. Snyder, Angew. Chem., Int. Ed., 2005, 44, 1012 CrossRef CAS PubMed.
- M. J. Anketell, T. M. Sharrock and I. Paterson, Angew. Chem., Int. Ed., 2020, 59, 1572 CrossRef CAS PubMed.
- I. Paterson and K.-S. Yeung, Tetrahedron Lett., 1993, 34, 5347 CrossRef CAS.
- I. Paterson, J. M. Goodman, M. A. Lister, R. C. Schumann, C. K. McClure and R. D. Norcross, Tetrahedron, 1990, 46, 4663 CrossRef CAS.
-
Copper hydride-mediated conjugate reduction of the corresponding α,β-unsaturated δ-lactone gave exclusively the 39-epi lactone 11a. (a) (BDP)CuH (10 mol%), PMHS, PPh3, PhMe, 50 °C, 60%. B. A. Baker, Z. V. Bošković and B. H. Lipschutz, Org. Lett., 2008, 10, 289 CrossRef CAS PubMed. - I. Wang, T. J. Chen, K. J. Chaa, T. C. J. Tsai, G. Lohaus, L. Friedman, H. Shechter, J. M. Brown, R. G. Naik, S. A. Hall, R. H. Crabtree, M. W. Davis, G. Stork and D. E. Kahne, J. Org. Chem., 2002, 51, 2655 Search PubMed.
- F. A. Davis, L. C. Vishwakarma, J. M. Billmers and J. Finn, J. Org. Chem., 1984, 49, 3241 CrossRef CAS.
- I. Paterson, K.-S. Yeung and J. B. Smaill, Synlett, 1993, 774 CrossRef CAS.
- S. Terashima, N. Tanno and K. Koga, J. Chem. Soc., Chem. Commun., 1980, 1026 RSC.
- I. Paterson and M. Tudge, Angew. Chem., Int. Ed., 2003, 42, 343 CrossRef CAS PubMed.
- H. C. Brown, P. K. Jadhav and K. S. Bhat, J. Am. Chem. Soc., 1988, 110, 1535 CrossRef CAS.
- M. J. Anketell, T. M. Sharrock and I. Paterson, Org. Biomol. Chem., 2020, 18, 8109 RSC.
- J. Wu, P. Lorenzo, S. Zhong, M. Ali, C. P. Butts, E. L. Myers and V. K. Aggarwal, Nature, 2017, 547, 436 CrossRef CAS PubMed.
- T. P. Stockdale, N. Y. S. Lam, M. J. Anketell and I. Paterson, Bull. Chem. Soc. Jpn., 2021, 94, 713 CrossRef CAS.
- N. Y. S. Lam, T. P. Stockdale, M. J. Anketell and I. Paterson, Chem. Commun., 2021, 57, 3171 RSC.
- N. Y. S. Lam, G. Muir, V. R. Challa, R. Britton and I. Paterson, Chem. Commun., 2019, 55, 9717 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and characterisation details; NMR correlation tables and bar graphs. See DOI: https://doi.org/10.1039/d2cc01802k |
| This journal is © The Royal Society of Chemistry 2022 |