
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diverse reactivity of a magnesium silanide toward ketones†
Bibian
Okokhere-Edeghoghon
 ,
Michael S.
Hill
*,
Mary F.
Mahon
and
Claire L.
McMullin
*
,
Michael S.
Hill
*,
Mary F.
Mahon
and
Claire L.
McMullin
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: msh27@bath.ac.uk; cm2025@bath.ac.uk
First published on 30th August 2022
Abstract
The reactivity of a molecular magnesium silanide toward ketones displays a significant variability of outcome, resulting in adduct formation, deprotonation, dearomatisation or deoxygenation, that is dependent on the structure and electronic character of the carbonyl-containing reagent.
Addition of a silicon-centred nucleophile, primarily in the form of a stabilised triorganosilyllithium reagent, to a carbonyl-containing molecule has long been known to provide the kinetic product of attack at the electrophilic C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbon centre.1 Such transformations, however, are also prone to be quite ill-defined and bedevilled by a variety of subsequent thermodynamically-driven rearrangement processes.2 Although, for example, addition of t-butyldiphenylsilyllithium to acetophenone affords the expected α-hydroxysilane at −20 °C, the initial C–Si bonded species rapidly undergoes decomposition via a Brook-type rearrangement at more elevated temperatures (Scheme 1a).3 These latter processes, in particular, are also strongly reminiscent of our recent use of the β-diketiminato (BDI) magnesium silanide (1) as an easily generated source of the triorganosilyl nucleophile,4,5 and its reactivity with a range of organic isocyanates (Scheme 1b).6
O carbon centre.1 Such transformations, however, are also prone to be quite ill-defined and bedevilled by a variety of subsequent thermodynamically-driven rearrangement processes.2 Although, for example, addition of t-butyldiphenylsilyllithium to acetophenone affords the expected α-hydroxysilane at −20 °C, the initial C–Si bonded species rapidly undergoes decomposition via a Brook-type rearrangement at more elevated temperatures (Scheme 1a).3 These latter processes, in particular, are also strongly reminiscent of our recent use of the β-diketiminato (BDI) magnesium silanide (1) as an easily generated source of the triorganosilyl nucleophile,4,5 and its reactivity with a range of organic isocyanates (Scheme 1b).6
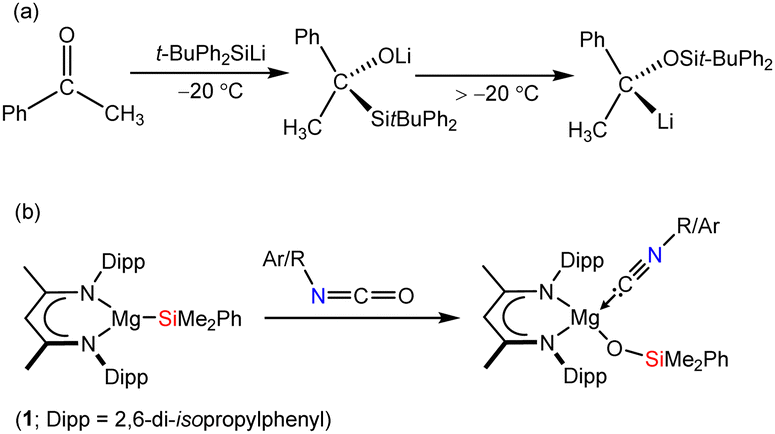 | ||
| Scheme 1 (a) Addition of t-butyldiphenylsilyllithium to acetophenone;3 (b) silanative deoxygenation of organic isocyanates by compound 1.6 | ||
Although some dependence was observed to result from variation of the R/ArNCO alkyl or aryl substituent, these reactions typically resulted in deoxygenation of the heterocumulene through the generation of a magnesium siloxide and the corresponding isonitrile. In a process reminiscent of that depicted in Scheme 1a, this reactivity was computed to ensue via initial nucleophilic attack of silicon at the sp-hybridised carbon of the magnesium-coordinated isocyanate and rearrangement of the resultant C-silylated amidate to the ultimately observed siloxymagnesium isonitrile adduct.6 Prompted by the potential for elaboration of such silanative deoxygenation to a more expansive palette of C–O-bonded small molecules, in this contribution we report the reactivity of 1 with the more diverse electronic and steric variability presented by the commercially available ketones, acetophenone, benzophenone, 4-fluorobenzophenone and 9-fluorenone.
An equimolar reaction of 1 with acetophenone performed on a NMR (ca. 50 mg) scale in C6D6 resulted in the generation of a pale pink solution at room temperature and complete consumption of the starting material was evidenced within ten minutes by 1H NMR spectroscopy (Scheme 2). Well-defined and mutually coupled doublet (δ 0.23 ppm) and septet (4.63 ppm) resonances were assigned as the respective methyl and Si–H signals of dimethylphenylsilane, while the generation of a predominant β-diketiminate-containing species (2) was apparent from a BDI γ-methine signal at δ 5.01 ppm that emerged in unison with a broadened resonance integrating to 2H at δ 4.04 ppm. This latter signal was found to correlate by HSQC and HMBC NMR experiments with resonances at δ 91.6 and 160.4 ppm in the corresponding 13C{1H} NMR spectrum. These chemical shift data are reminiscent of those arising from the {OC![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH2} unit of the handful of enolate anions provided by deprotonation of the bulkier 2,4,6-Me3C6H2COMe with mono- or heterobimetallic magnesium alkyl and amide bases,7 an inference that was confirmed by X-ray diffraction analysis of a single crystal of 2 isolated from hexane solution at −30 °C (Fig. 1).
CH2} unit of the handful of enolate anions provided by deprotonation of the bulkier 2,4,6-Me3C6H2COMe with mono- or heterobimetallic magnesium alkyl and amide bases,7 an inference that was confirmed by X-ray diffraction analysis of a single crystal of 2 isolated from hexane solution at −30 °C (Fig. 1).
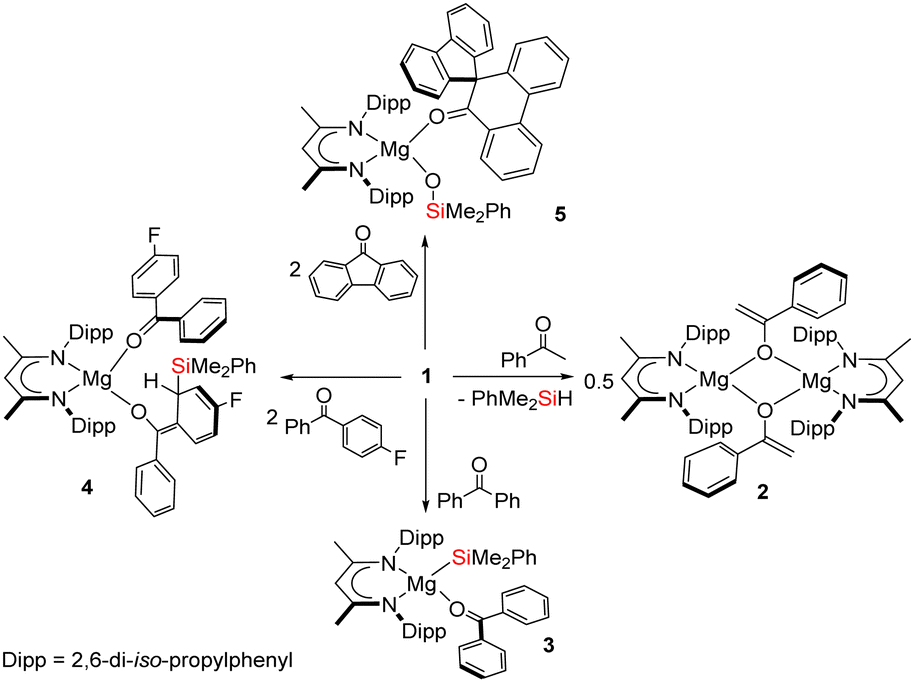 | ||
| Scheme 2 Synthesis of compounds 2–5. | ||
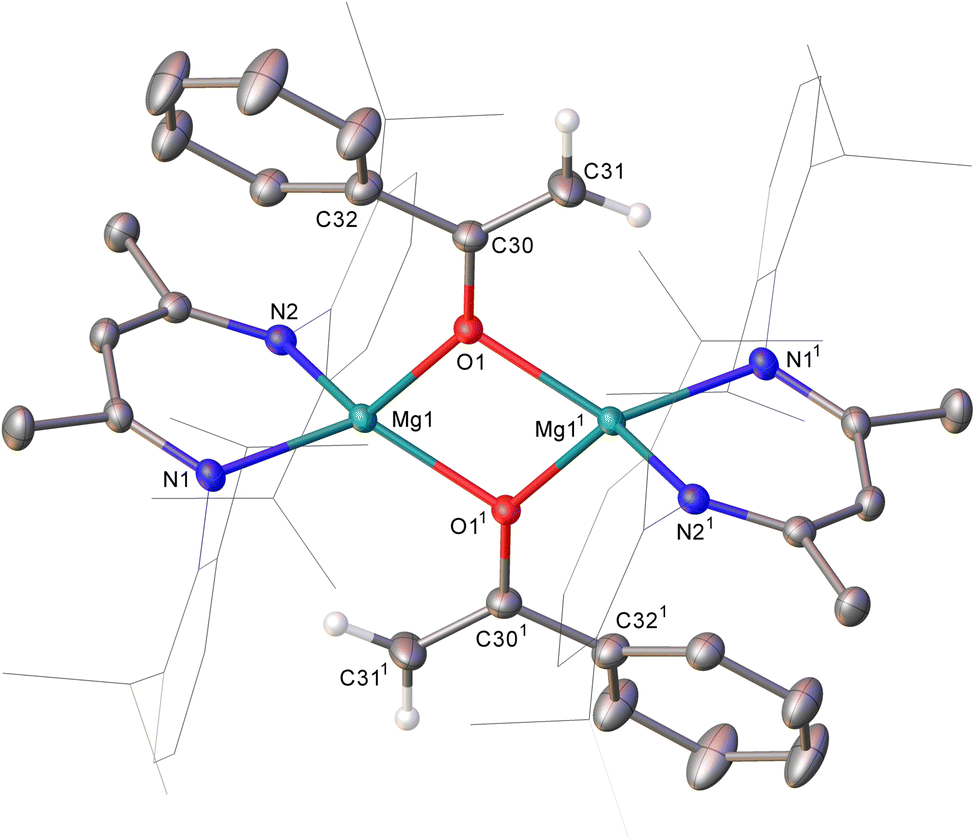 | ||
| Fig. 1 Displacement ellipsoid (30% probability) plot of compound 2. BDI Dipp substituents are shown in wireframe view for visual simplicity and hydrogen atoms, apart from those attached to C31/C311 are also omitted for clarity. Selected bond lengths (Å) and angles (°): Mg1–N1 2.0992(12), Mg1–N2 2.106412, Mg1–O1 2.0052(10), Mg1–O11 2.0498(10), O1–C30 1.3522(17), C30–C31 1.330(2); O1–Mg1–O11 78.81(4), O11–Mg1–N2 124.56(5), O1–Mg1–N2 119.22(4), O11–Mg1–N1 125.89(5), O1–Mg1–N1 121.13(5), N1–Mg1–N2 90.98(5), C30–O1–Mg1 140.38(9), C30–O1–Mg11 118.43(9). Atoms with superscripted labels are related to those in the asymmetric unit by the 1 − x, 1 − y, 1 − z symmetry operator. | ||
The structure of compound 2 comprises a centrosymmetric dimer in which the BDI-coordinated magnesium centres are connected via two μ2-O bridging acetophenolate ligands. Although the resultant enolate structure is unremarkable and provides metric data that are broadly commensurate with those arising from the aforementioned Mg-μ2-O bridged derivatives of the {OC(= CH2)2,4,6-Me3C6H2} anion,7 the structure of 2, to the best of our knowledge, provides the first documented evidence of the Brønsted basicity of a magnesium silanide toward a C–H acidic reagent.
To preclude deprotonation, a subsequent reaction was performed between compound 1 and the non-enolisible ketone, benzophenone. Addition of benzene to an equimolar mixture of the two solid reagents at room temperature resulted in the immediate formation of a brown-green solution, analysis of which by 1H NMR spectroscopy revealed the emergence of a major new BDI-containing species (3) characterised by a γ-methine resonance at δ 5.11 ppm (Scheme 2). Single crystals suitable for X-ray diffraction analysis were obtained by recrystallisation from hexane at room temperature, enabling the identification of 3 as a monomeric benzophenone adduct of the magnesium silanide (Fig. 2a). Consistent with this formulation, the Mg1–O1 distance [2.0443(13) Å] in 3 is effectively identical to the terminal Mg–O interactions observed in the only previous structurally characterised benzophenone adduct of magnesium, the amide/alkoxide complex, [{(Me3Si)2NMg[μ-OC(H)Ph2]·(OCPh2)}2], and in which the magnesium centres were similarly 4-coordinate [2.046(3) Å].8 Although structural characterisation of s-block benzophenone ketyl complexes has revealed that the C–O and C–Cipso [1.233(2) and 1.478(2) Å] distances are only marginally perturbed in comparison to the corresponding bonds in free benzophenone [1.222 and 1.496 Å mean],9 confidence in the assignment of the {OCPh2} unit of 3 as a neutral two electron donor, is provided by its diamagnetism and the essentially planar geometry of the oxygen-bonded C30 atom (Σangles about C(30) = 359.99°). Similarly, the Mg1–Si1 bond length [2.6488(6) Å] demonstrates a significant elongation in comparison to that of the 3-coordinate starting material, compound 1 [2.5897(8) Å],4a and is commensurate with the Mg–Si distances of previously described terminal silanides of 4-coordinate magnesium.4b,10
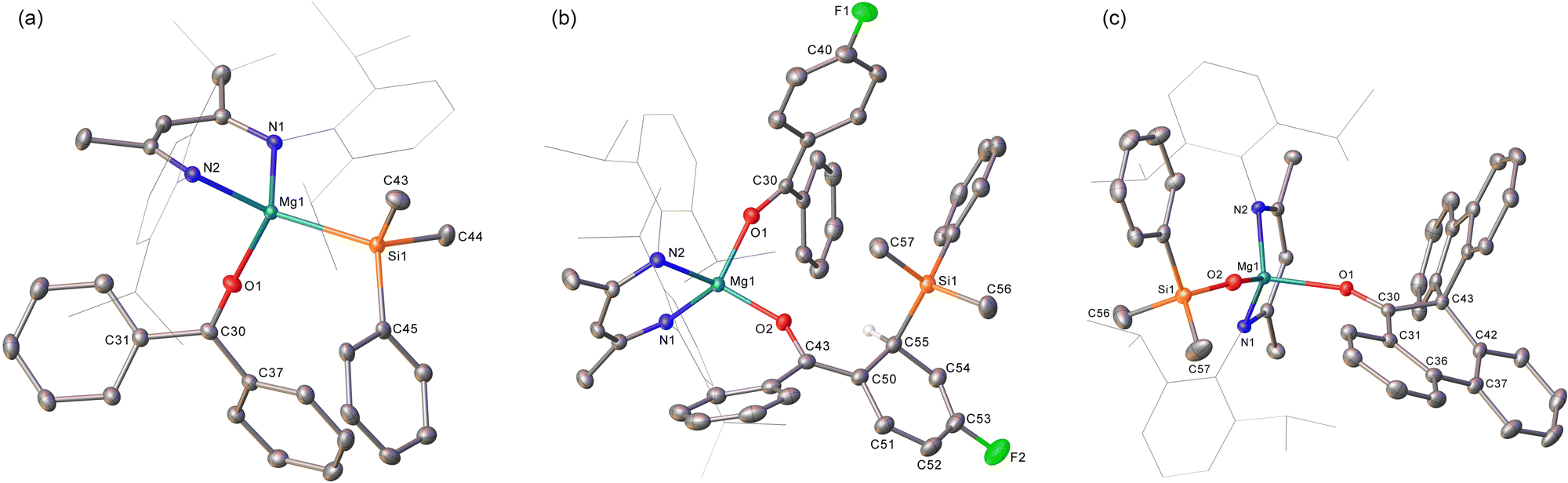 | ||
| Fig. 2 Displacement ellipsoid (30% probability) plots of (a) compound 3, (b) compound 4 and (c) compound 5. BDI Dipp substituents are shown in wireframe view for visual simplicity. Hydrogen atoms, apart from that attached to C55 of 4 (plus the disorder in 4) have also been omitted for clarity. Selected bond lengths (Å) and angles (°): (3) Mg1–O1 2.0443(13), O1–C30 1.233(2), Mg1–Si1 2.6488(6), O1–Mg1–Si1 112.02(4), N1–Mg1–Si1 117.52(4); (4) Mg1–O1 1.971(2), Mg1–O2 1.888(2), O1–C30 1.239(3), O2–C43 1.325(3); O2–Mg1–O1 96.19(9), O1–Mg1–N1 116.69(10), O1–Mg1–N2 103.96(9), O2–Mg1–N1 114.74(9), O2–Mg1–N2 131.50(10); (5) Mg1–O1 2.0567(10), Mg1–O2 1.8505(10), O1–C30 1.2332(15), Si1–O2 1.5931(10); O2–Mg1–O1 110.67(4), O2–Mg1–N2 119.62(5), N2–Mg1–O1 101.56(4), N2–Mg1–N1 94.41(4), N1–Mg1–O1 101.39(4). C30–O1–Mg1 153.57(9), Si1–O2–Mg1 150.50(7). | ||
We reasoned that the installation of an electron-withdrawing arylfluorine substituent would enhance the electrophilicity of coordinated benzophenone ligand and promote intramolecular nucleophilic attack. Accordingly, an equimolar quantity of 1 and 4-fluorobenzophenone was reacted in benzene (Scheme 2). Initial scrutiny of the resultant dark brown solution by 1H NMR spectroscopy was consistent with the onset of a more complex reaction. A predominant new compound (4), however, could be identified in its 1H NMR spectrum after 48 hours at room temperature, while removal of volatiles and crystallisation from a saturated hexane solution yielded brown single crystals at room temperature. Compound 4 was identified by X-ray diffraction analysis as an asymmetric monomer with two C–O donor ligands arising from the interaction of 4-fluorobenzophenone and the magnesium centre. One unperturbed ketone molecule is again coordinated to magnesium, while the other may be identified as an alkoxide-like anion resulting from silanide ion migration (Fig. 2b). Although, in mitigation of our initial hypothesis, intramolecular attack is facilitated by the presence of the strongly electronegative para-positioned fluorine atom, this process results in ortho-dearomatisation of the 4-fluorobenzophenone substituent rather than C–Si bond formation at the former carbonyl carbon atom. The Mg1–O1 [1.971(2) Å] and Mg1–O2 [1.888(2) Å] bond lengths in 4 reflect the assignment of the two O-donor units as neutral and alkoxide-like ligands, respectively. The dearomatisation at the 2-position of the 4-fluorophenyl substituent by addition of the {SiPh2Me} unit at C55 is apparent from the alternating long [C50–C51, 1.441(4), C52–C53, 1.430(5) Å] and short [C51–C52, 1.357(4), C53–C54, 1.326(4) Å] C–C bonds that clearly denote the C6 carbocycle as a cyclic diene. Although, as a result of the dearomatisation process, the former carbonyl carbon centre retains its planar sp2 hybridisation state (Σangles about C45 = 360.0°), the C43–C50 [1.380(4) Å] and C43–C44 [1.489(4) Å] bond lengths indicate that the resultant anion is best considered as a triorganosilicon-substituted, magnesium enolate (Scheme 2).
We then turned to DFT calculations to shed light on the divergent reactivity leading to compounds 3 and 4 (see ESI† for methodology details). The generation of adduct species equivalent to compound 3 was found to be comparably exergonic with both benzophenone (−10.2 kcal mol−1) and 4-fluorobenzophenone (A = −11.7 kcal mol−1). We anticipated that any transformation of these adduct species would occur through silanide addition at the carbon atom of the coordinated carbonyl. This process, however, was precluded by the steric demands of the SiPhMe2 unit. Rather, the onward progression of the reactions was calculated to proceed via direct silanide-to-ortho-carbon transfer to the coordinated diaryl ketones. These processes were calculated to be only marginally more viable for the transformation of compound 1 to compound 4 [ΔG = −37.3 kcal mol−1] (Fig. S12, ESI†) than for silylation of a phenyl substituent of benzophenone [(4′′) ΔG = −33.5 kcal mol−1] (Fig. S13, ESI†). On this basis, therefore, the room temperature formation of compound 4 may be judged to result from a marginally enhanced kinetic preference for the fluoroaromatic substituent to undergo intramolecular attack by the coordinated triorganosilyl residue. The evidently subtle, but influential, impact of the fluoride substituent during the formation of 4 was underscored by the location of a transition state leading to intramolecular attack of the silyl nucleophile at the 2-position of the non-fluorinated ring of 4-fluorobenzophenone and the generation of 4′ [ΔG = −36.3 kcal mol−1] (Fig. S13, ESI†). This invokes a barrier height [+15.9 kcal mol−1] that is only 1.5 kcal mol−1 higher than the observed process of addition providing dearomatisation of the fluorinated phenyl substituent. In mitigation of these theoretical deductions, and consistent with competitive kinetic access to these alternate reaction pathways, analogous reactions to those providing 3 and 4 performed at elevated (>40 °C) temperatures provided more complex and intractable mixtures of products.
We have previously observed that similar ortho-dearomatisation of benzophenone itself is facilitated by reaction with the diboranate derivative, [(BDI)Mg{pinB-B(n-Bu)pin}] (pin = pinacolato), which comprises the identical β-diketiminato magnesium platform as compound 1 and behaves as a surrogate for the [Bpin]− nucleophile.11 In contrast to this boron-derived reactivity, however, the enolato component of compound 4 displayed no evidence for onward aldol-type C–C coupling with further equivalents of the 4-fluorobenzophenone substrate. [(BDI)Mg{pinB-B(n-Bu)pin}] was also found to induce aldol-type C–C coupling of 9-fluorenone but without any observable generation of the putative dearomatised intermediate. The spontaneous nature of this latter process was, thus, ascribed to the higher energetic cost associated with the disruption of the more extended π-system of the fused ring ketone.11 With this rationale in mind, compound 1 was treated with two molar equivalents of 9-fluorenone, resulting in the immediate development of a brownish-green colouration of the reaction mixture. Monitoring by 1H NMR spectroscopy indicated that complete conversion to compound 5 was achieved after 48 hours at 60 °C. The 1H NMR spectrum of 5 displayed resonances between δ 7.59–7.32 ppm and a series of mutually coupled quartet, two triplet and doublet resonances between δ 6.95–6.56 ppm, which were assigned to the CH environments of an intact fluorenyl unit. A 1H-13C-HMBC NMR experiment also inferred that a carbon atom resonating at δ 144.6 ppm corresponded to the OC carbon of a fluorenone-like residue. The origin of these observations was resolved by an X-ray diffraction analysis of compound 5, which was performed on a single crystal isolated from a saturated hexane solution (Fig. 2c). Compound 5 is a 4-coordinate β-diketiminato magnesium siloxide, comprising an analogous {OSiMe2Ph} anion to that observed in our previous study of the reactivity of 1 toward organic isocyanates (Scheme 1b).6 In this instance, however, the magnesium coordination sphere is completed by a molecule of spiro[fluorene-9,9′-phenanthren-10′-one]. Although the synthesis of this spirocyclic ketone has previously been achieved by reductive coupling of 9-fluorenone and subsequent acid-catalysed pinacol rearrangement,12 the simultaneous generation of the siloxide unit in 5 argues for a mechanism that is better precedented by our earlier observations of isocyanate reduction (Scheme 1b). Although the generation of 5 has not been further investigated, and we cannot discount possible radical-based pathways,13 the generation of nucleophilic carbene intermediates by photochemical or thermally initiated 1,2-Brook-type rearrangement of an acyl or amidoylsilane is well precedented.14 We suggest, therefore, that the formation of the spirocyclic unit of 5 is best rationalised as resulting from insertion of the carbene product of 9-fluorenone deoxygenation into a C–C(O) bond of a further molecule of magnesium-coordinated 9-fluorenone. Under this regime carbene generation occurs via the formation of a C-silylated alkoxide intermediate that undergoes Brook-type rearrangement in a similar manner to our previously proposed pathway for isocyanate deoxygenation.6 In contrast to the stable isonitrile products, however, we hypothesise that the resultant highly reactive di-organocarbene can initiate C–C activation of a further molecule of magnesium-coordinated 9-fluorenone. Commensurate with its identity as a formally charge neutral donor, the O1–C30 bond length of the resultant spirocycle [1.2332(15) Å] is closely comparable to the respective O1–C30 distances observed in compounds 3 and 4 [(3) 1.233(2); (4) 1.239(3) Å], while the Si1–O2 bond length [1.5931(10) Å] lies in the range established for the previously reported products of isocyanate deoxygenation.
In conclusion, the magnesium silanide 1 offers a variety of substrate-dependent reactivity when reacted with ketone small molecules. Whilst the reaction of 1 with acetophenone yields an enolate species by silane protonation, benzophenone forms a magnesium adduct species. Installation of a 4-fluorophenyl substituent, however, induces a sufficiently enhanced kinetic facility for intramolecular nucleophilic attack and the isolation of a further enolato species by arene dearomatisation. In further contrast to these behaviours, treatment of 9-fluorenone with 1 yields a more complex outcome and the generation of a magnesium siloxide resulting from ketone deoxygenation and the inferred insertion of the resultant carbenic species into a (O)C–C bond of a further molecule of the ketone reagent.
BOE performed the synthesis and characterisation of the new compounds reported. MSH and CLM conceptualised the study and finalised the manuscript for submission. CLM performed the computational study and MFM finalised the X-ray diffraction analyses for publication.
We acknowledge financial support from the EPSRC (research grant EP/R020752/1) and the Petroleum Technology Development Fund (PTDF) of Nigeria for the provision of a PhD scholarship for BO-E. This research made use of the Anatra High Throughput Computing (HTC) Cluster at the University of Bath.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Gilman and G. D. Lichtenwalter, J. Am. Chem. Soc., 1958, 80, 2680–2682 CrossRef CAS.
- A. G. Brook, J. Am. Chem. Soc., 1958, 80, 1886–1889 CrossRef CAS.
- P. Cuadrado, A. M. González, B. González and F. J. Pulido, Synth. Commun., 1989, 19, 275–283 CrossRef CAS.
- (a) D. J. Liptrot, M. Arrowsmith, A. L. Colebatch, T. J. Hadlington, M. S. Hill, G. Kociok-Kohn and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 15280–15283 CrossRef CAS ; see also ; (b) G. Coates, H. Y. Tan, C. Kalff, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2019, 58, 12514–12518 CrossRef CAS.
- B. Okokhere-Edeghoghon, M. Dehmel, M. S. Hill, R. Kretschmer, M. F. Mahon, C. L. McMullin, L. J. Morris and N. A. Rajabi, Inorg. Chem., 2020, 59, 13679–13689 CrossRef CAS PubMed.
- B. Okokhere-Edeghoghon, S. E. Neale, M. S. Hill, M. F. Mahon and C. L. McMullin, Dalton Trans., 2022, 51, 136–144 RSC.
- (a) J. F. Allan, K. W. Henderson, A. R. Kennedy and S. J. Teat, Chem. Commun., 2000, 1059–1060 RSC; (b) E. Hevia, K. W. Henderson, A. R. Kennedy and R. E. Mulvey, Organometallics, 2006, 25, 1778–1785 CrossRef CAS; (c) M.-L. Hsueh, B.-T. Ko, T. Athar, C.-C. Lin, T.-M. Wu and S.-F. Hsu, Organometallics, 2006, 25, 4144–4149 CrossRef CAS; (d) Z. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, C. M. Thomas, T. Roisnel, O. Casagrande Jr. and J.-F. Carpentier, New J. Chem., 2008, 32, 2279–2291 RSC.
- K. W. Henderson, J. F. Allan and A. R. Kennedy, Chem. Commun., 1997, 1149–1150 RSC.
- (a) C. Jones, L. McDyre, D. M. Murphy and A. Stasch, Chem. Commun., 2010, 46, 1511–1513 RSC; (b) T. A. Scott, B. A. Ooro, D. J. Collins, M. Shatruk, A. Yakovenko, K. R. Dunbar and H.-C. Zhou, Chem. Commun., 2009, 65–67 RSC; (c) Z. Hou, X. Jia, A. Fujita, H. Tezuka, H. Yamazaki and Y. Wakatsuki, Chem. – Eur. J., 2000, 6, 2994–3005 CrossRef CAS.
- (a) A. R. Claggett, W. H. Isley, T. J. Anderson, M. D. Glick and J. P. Oliver, J. Am. Chem. Soc., 1977, 99, 1797–1801 CrossRef CAS; (b) R. Goddard, C. Kruger, N. A. Ramadan and A. Ritter, Angew. Chem., Int. Ed. Engl., 1995, 34, 1030–1032 CrossRef CAS; (c) D. W. Goebel Jr., J. L. Hencher and J. P. Oliver, Organometallics, 1983, 2, 746–750 CrossRef; (d) L. Rösch, J. Pickardt, S. Imme and U. Börner, Z. Naturforsch., B: J. Chem. Sci., 1986, 41, 1523–1526 CrossRef; (e) J. D. Farwell, M. F. Lappert, C. Marschner, C. Strissel and T. D. Tilley, J. Organomet. Chem., 2000, 185, 185–188 CrossRef; (f) H.-W. Lerner, S. Scholz, M. Bolte, N. Wiberg, H. Nöth and I. Krossing, Eur. J. Inorg. Chem., 2003, 666–670 CrossRef CAS; (g) W. Gaderbauer, M. Zirngast, J. Baumgartner, C. Marschner and T. D. Tilley, Organometallics, 2006, 25, 2599–2606 CrossRef CAS; (h) H. Wagner, J. Baumgartner and C. Marschner, Organometallics, 2007, 26, 1762–1770 CrossRef CAS; (i) K. Yan, B. M. Upton, J. Zhu, A. Ellern and A. D. Sadow, Organometallics, 2013, 32, 6834–6843 CrossRef CAS; (j) J. Luo, E.-H. Yan, H. Zhao, X.-Q. Xiao and Z. Li, Polyhedron, 2015, 102, 233–238 CrossRef CAS; (k) L. E. Lemmerz, V. Leich, D. Martin, T. P. Spaniol and J. Okuda, Inorg. Chem., 2017, 56, 14979–14990 CrossRef CAS PubMed.
- A. F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Organometallics, 2018, 37, 4457–4464 CrossRef.
- Y.-C. Chen, S.-K. Huang, S.-S. Li, Y.-Y. Tsai, C.-P. Chen, C.-W. Chen and Y. J. Chang, ChemSusChem, 2018, 11, 3225–3233 CrossRef CAS.
- See, for example; (a) Z. Hou and Y. Wakatsuki, Chem. – Eur. J., 1997, 3, 1005–1008 CrossRef CAS; (b) Z. Hou, A. Fujita, T.-A. Koizumi, H. Yamazaki and Y. Wakatsuki, Organometallics, 1999, 18, 1979–1985 CrossRef CAS; (c) Z. Hou, A. Fujita, Y. Zhang, T. Miyano, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 1998, 120, 754–766 CrossRef CAS; (d) Z. Hou, X. Jia, M. Hoshino and Y. Wakatsuki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1292–1294 CrossRef CAS; (e) W. Clegg, K. Izod, P. O’Shaughnessy, C. Eaborn and J. D. Smith, Angew. Chem., Int. Ed. Engl., 1997, 36, 2815–2817 CrossRef CAS; (f) Z. Hou, A. Fujita, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 1996, 118, 7843–7844 CrossRef CAS; (g) Z. Hou, A. Fujita, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 1996, 118, 2503–2504 CrossRef CAS; (h) Z. Hou, T. Miyano, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 1995, 117, 4421–4422 CrossRef CAS.
- For a comprehensive review, see; D. L. Priebbenow, Adv. Synth. Catal., 2020, 362, 1927–1946 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray analysis of compounds 2–5 (CCDC 2167201–2167204), details of the computational analysis and atomic coordinates. of the DFT computed structures. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc03966d |
| This journal is © The Royal Society of Chemistry 2022 |