
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Prebiotic triose glycolysis promoted by co-catalytic proline and phosphate in neutral water†
Álvaro F.
Magalhães
 and
Matthew W.
Powner
*
and
Matthew W.
Powner
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: matthew.powner@ucl.ac.uk
First published on 14th November 2022
Abstract
Proline and phosphate promote a near-quantitative aldol reaction between glycolaldehyde phosphate and formaldehyde at neutral pH in water. Our results demonstrate the important role of general acid-base catalysis in water and underscore the essential role that amino acid catalysis may have played in early evolution of life's core metabolic pathways.
Metabolism is orchestrated by highly evolved, genetically encoded enzymes. However, at the origins of life, before the advent of enzyme catalysis, the chemical reactions that sustained life must have been organized by the innate reactivity of simple abiotic molecules.1a–o Therefore, the boundary between intrinsically and enzymatically controlled reactions lies at the heart of elucidating the origins of life. The triose glycolysis pathway (TGP) serves as a central pillar of metabolism. It is one of life's most highly conserved and densely connected metabolic pathways.2a,b This underscores the TGP's deep-seated antiquity,3a,b but also raises the fascinating possibility that biological triose glycolysis was foreshadowed by a prebiotic reaction pathway, and that its central role in metabolism was predisposed by the inherent reactivity of simple sugars.4a,b
In the TGP, glyceraldehyde-3-phosphate (1) is first oxidised to glyceric acid-3-phosphate (2), the phosphate must then be enzymatically migrated to yield glyceric acid-2-phosphate (3), before enzymatic elimination yields phosphoenol pyruvate (4) (Fig. 1a). We have previously identified the constitutional relationship between glyceraldehyde-2-phosphate (7) and the carbon framework of 4;4a demonstrating phosphorylation of glycolaldehyde 5 – which yields glycolaldehyde phosphate (6) – controls a network of reactions that delivered not only 4, but also the other components of the TGP. Rerouting the metabolic pathway – i.e., switching the order of oxidation and elimination – yielded a chemically predisposed sequence of reactions in which elimination occurs at the aldehyde oxidation level from 7 (Fig. 1b).
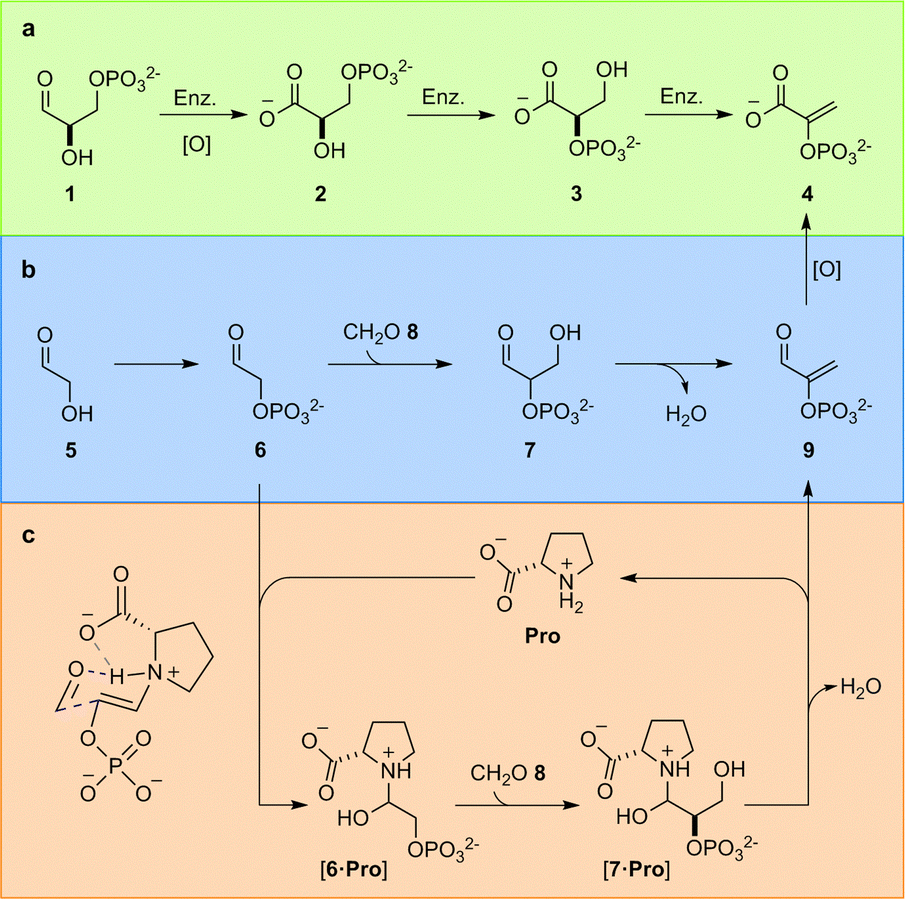 | ||
| Fig. 1 Triose glycolysis. (a) Biological enzyme (Enz.) catalysed triose glycolysis pathway (TGP); (b) proposed prebiotic triose glycolysis. (c) Proline-catalysed triose glycolysis. | ||
This juxtaposition between predisposed and enzymatically activated pathways, opens the intriguing (general) question of how predisposed chemical reactions would first become catalytically controlled, en route to becoming embedded within an enzyme-gated biochemical network. We therefore set out to investigate the relationship between simple, prebiotically, and biologically relevant, catalysts and the key aldol reaction required to transition between sugar 5 and phosphoenol 4.
Aldolase enzymes greatly enhance the rate and stereoselectivity of (biochemical) aldol reactions,5a,b and can be divided into two mechanistic classes. Type I aldolases have a catalytic amine in their active site and form an enamine intermediate that accelerates their cognate aldol reaction, whilst Type II aldolases coordinate a Lewis acid cofactor, such as Zn2+, to accelerate an aldol reaction.6 Aldolase enzymes are clearly the product of evolution, and too structurally complex to have been available to control energy metabolism at the origins of life. However, small molecule catalysts must have been prebiotically available. If these small molecules endowed enhanced reactivity, they could then have been acted upon by evolution to provide an evolutionary trajectory to modern enzyme catalyzed metabolism.
The remarkable (type I) aldol activity of proline (Pro) in organic solvents,7a–d and the high intracellular concentrations of amino acids, have suggested to some that in vivo natural products synthesis may be influenced by amino acid catalysis and, by extension, that organocatalysis may lie at the root of biological (enzyme) catalysis and even biological homochirality.1e,8a–i However, despite the magnitude of these questions, and a large body of work attempting to address organocatalysis in water,8a–i,9a–d this concept remains elusive. Most organocatalytic reactions employ heterogeneous media (rather than water) exploiting e.g. non-aqueous additives, co-solvents, extreme (>molar) substrate concentrations, emulsions or amphiphiles due to the challenges imposed by implementing organocatalysis in dilute water.8i,9a–d,10a–c Although Clarke and co-workers have reported Pro is incapable of catalyzing the dimerization of glycolaldehyde (5) in water,8gPro does catalyze the aldol reaction of acetone and p-nitrobenzaldehyde in aqueous solution [with organic co-solvent, i.e. 10% DMSO].11a,bL-Pro has also been reported to yield an unsubstantiated (small) quantity of enantioenriched glyceraldehyde from the reaction of glycolaldehyde (5) and formaldehyde (8).12a–e Together these results highlight the ineffective nature of Pro catalysis in water, but hint at the potential for Pro, or a related small molecule catalyst, to enhance the rate of the key aldol reaction in prebiotic triose glycolysis (Fig. 1c) and provide a (small molecule) chemical link towards the evolution of enzyme-catalyzed metabolism.
The key aldol reaction of the prebiotic TGP, between glycolaldehyde phosphate (6, 100 mM) and formaldehyde (8, 10 equiv.) yields 7 (66%) in alkaline solution (pH 10.7) at room temperature (rt) after 6 days.13 However, with no catalyst, no reaction was observed at neutral pH (Fig. 2a and ESI,† Fig. S15, S16). We suspected neutral pH reactivity would be established with the appropriate catalyst, so we set out to study the effect of amino acids on this reaction.10a–c,14a,b
 | ||
Fig. 2 Proline/phosphate co-catalysed reaction of glycolaldehyde phosphate and formaldehyde. 1H NMR [400 MHz, noesygpprld, H2O/D2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)] spectra to show the reaction of glycolaldehyde phosphate (6, 100 mM) with formaldehyde (8, 100 mM) after 48 h at pH 7 and 40 °C: (a) in water; (b) with 50 mol% L-Pro in water; (c) with 50 mol% L-Pro in 500 mM phosphate. :1)] spectra to show the reaction of glycolaldehyde phosphate (6, 100 mM) with formaldehyde (8, 100 mM) after 48 h at pH 7 and 40 °C: (a) in water; (b) with 50 mol% L-Pro in water; (c) with 50 mol% L-Pro in 500 mM phosphate. | ||
We initially investigated glycine (Gly) and Pro (50 mol%, pH 7, rt; ESI,† Fig. S15). In water only Pro was observed to have significant reactivity, yielding 7 as a proline-hemiaminal [7·Pro] (10%) (Fig. 1), alongside trace amounts of the elimination product 9 (ESI,† Fig. S17), but the conversion was low with the remaining mass balance (89% 6) unreacted after 14 days.
We suspected that [7·Pro] was not equilibrating with 7. This was confirmed by studying authentically synthesized 7.15 Upon incubation with Pro, 7 was observed to directly dehydrate to 9, but only yield small amounts of [7·Pro] (Fig. 3 and ESI,† Fig. S33–S35). This low concentration of [7·Pro] (8%) stands in stark contrast to the observation that [7·Pro] (27%) was a more major compound (at intermediate time points) in the Pro-catalysed reaction of 6 and 8, where only trace quantities of aldehyde 7 (<1%) were observed. Furthermore, the dehydration of 7 was not notably enhanced by Pro; 40% and 43% conversion of 7 (25 mM) to 9 was observed with and without Pro (50 mol%) after 4.5 days at 40 °C and pH 7, respectively (ESI,† Fig. S33 and S35). It is likely the highly electron-withdrawing α-phosphate retards the rate of hemiaminal-to-aldehyde equilibration, and trapping Pro in hemiaminal [7·Pro] would clearly block turnover and suppress catalytic activity.16 This suggested enhancing the turnover and release of Pro would be highly beneficial.
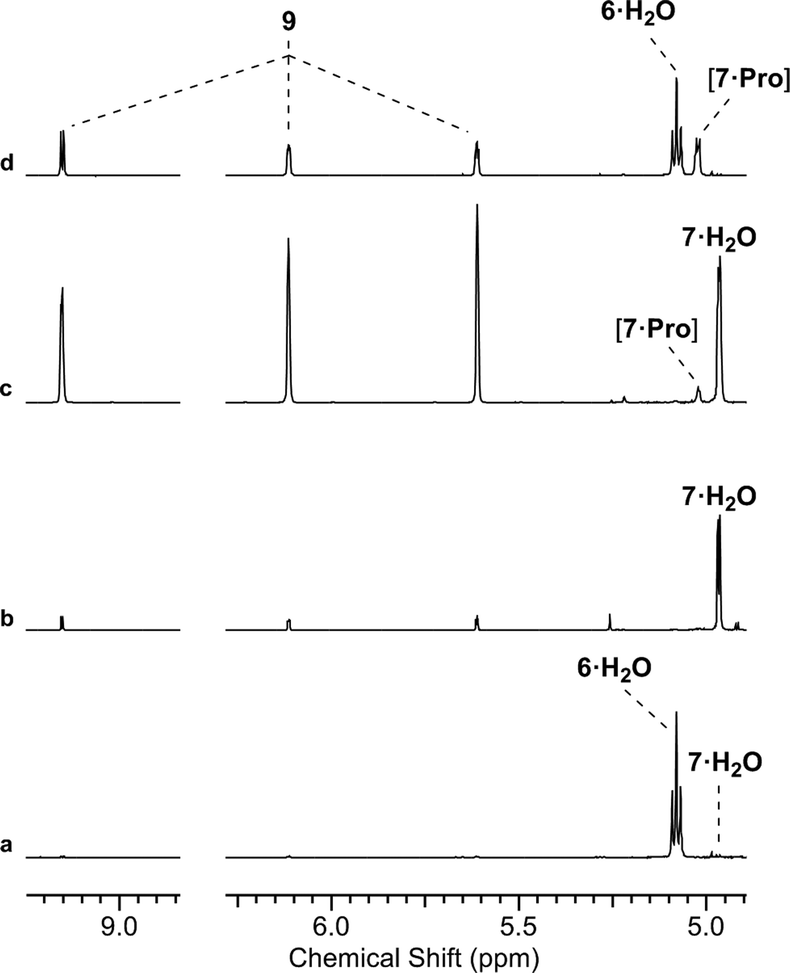 | ||
| Fig. 3 Proline-catalysed aldol-elimination cascade. (a) 1H NMR [400 MHz, noesygpprld, H2O/D2O (9:1)] spectrum to show the reaction of glycolaldehyde phosphate (6, 100 mM) with formaldehyde (8, 100 mM) in phosphate buffer (500 mM, pH 7) at 40 °C after 48 h; (b and c) 1H NMR [700 MHz, noesygpprld, H2O/D2O (9:1)] spectra to show the reaction of glyceraldehyde-2-phosphate (7, 100 mM) in phosphate buffer (500 mM, pH 7) with 50 mol% L-Pro at 40 °C after: (b) 0.5 h; (c) 48 h; (d) 1H NMR [400 MHz, noesygpprld, H2O/D2O (9:1)] spectrum to show the reaction of 6 (100 mM) with 8 (100 mM) in phosphate buffer (500 mM, pH 7) with 50 mol% L-Pro at 40 °C after 7 h. | ||
Exploring other amino acids, amino amides and simple peptides did not lead to improved activity (ESI,† Fig. S15).8h,17a,b Indeed, the activity of Pro was depressed by transformation into its methyl ester and nitrile derivatives (ESI,† Fig. S15), and so we continued to focus on Pro-catalysis.
Disappointed, but not surprised, by the poor organocatalytic activity observed in neutral water, we reflected on the distribution of charged moieties (e.g. in catalytic triads) in enzyme active sites.18a–d We postulated a combination of enamine catalysis and general acid-base (GAB) catalysed proton transfer would be the key to effective aldol activity at neutral pH. Within enzyme active sites GAB catalysis is mediated by the structural proximity of acidic and basic moieties,19a,b however, without the structured scaffold of a complex, coded polypeptide, we recognized this activity could be facilely replaced by an appropriate buffer. Pleasingly, when co-catalytic phosphate (Pi)14a,20a,b was introduced the conversion to the desired products markedly (3-fold) increased, such that co-catalytic Pro (50 mol%) and Pi (500 mM, pH 7) yielded [7·Pro] (17%) and phosphoenol 9 (31%) after 14 days at rt (ESI,† Fig. S20). Pi was observed to enhance the performance of all amine catalysts tested (ESI,† Fig. S18). To confirm Pi was acting as a GAB catalyst we investigated the effect of Pi concentration and, as expected, increased Pi concentration enhanced the reaction (Fig. 4a).
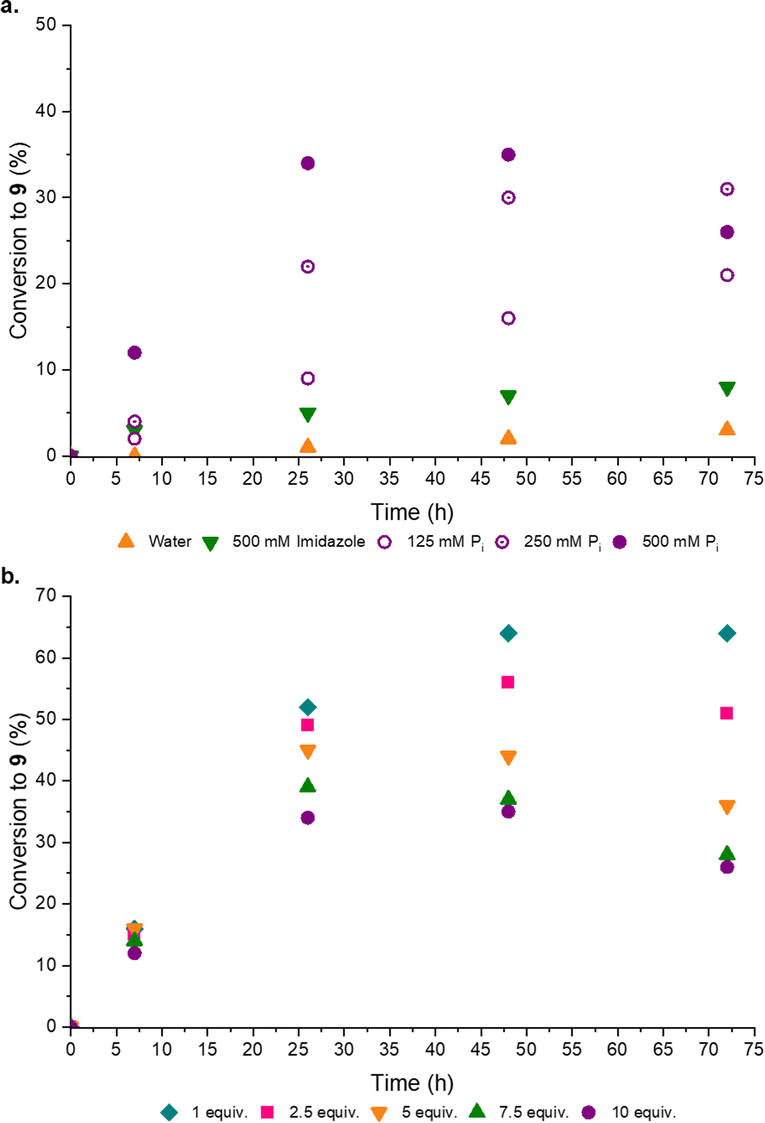 | ||
| Fig. 4 Proline-catalysed synthesis of phosphoenol pyruvaldehyde. Graphs to show the effect of buffer and stoichiometry. (a) Reaction of 6 (100 mM) and 8 (10 equiv.) in neutral water or specified buffer (125–500 mM, pH 7) with L-Pro (50 mol%) at 40 °C; (b) reactions of 6 (100 mM) and 8 (1–10 equiv.) in Pi buffer (500 mM, pH 7) with L-Pro (50 mol%) at 40 °C. | ||
Previously a high concentration and large excess of 8 (10 equiv.) has been employed in these aldol reactions,4a,13 however, in the Pro catalysed reaction we observed a dramatic improvement in the conversion of 6 to 9 when the initial concentration of 8 was decreased. Indeed, the maximum yield of 9 (Fig. 4b) was observed with 1 equiv. of formaldehyde 8. This stoichiometric (1:1) reaction, catalyzed by Pro/Pi, thus afforded the highest reported yield of 9. It seemed likely 8 could suppress the aldol reaction through competitive sequestration of Pro (into e.g. [8·Pro], ESI,† Fig. S32), but it was perhaps more noteworthy that higher initial concentrations of 8 resulted in more rapid degradation of 9, suggesting 8 accelerates the hydrolysis of 9. In line with these observations, we next tested setting 8 as the limiting reagent, and pleasingly we found that substoichiometric 8 (0.5 equiv.) led to a near-quantitative combined yield of [7·Pro] (18%) and 9 (76%) (ESI,† Table S6).
Finally, L-Pro was observed to induce enantioenrichment of 7, furnishing more D-7 (ee = 50%; ESI,† Fig. S36–S38). This reaction pairs L-amino acid (i.e.L-Pro) catalysis with D-sugar-phosphates, mirroring life's symmetry relationship between amino acids and sugar-phosphates. It is therefore of note that 7 is the first (simplest) chiral sugar-phosphate that can be synthesized by an aldol reaction from two achiral precursors.
The co-catalytic action of phosphate and amino acids, but especially Pro, markedly accelerate phosphoenol synthesis in the reported prebiotic TGP.4a These results highlight, the often overlooked, importance of GAB catalysis for promoting proton transfer in neutral water, and how GAB catalysts can be readily paired with a complementary co-catalyst to enhance the co-catalysts activity and promote otherwise ineffective reaction. GAB catalysis is an essential element of enzyme activity because enzymes have evolved to operate in neutral water, and so GAB catalysis must also be essential to understanding the transition from chemical to biochemical reactivity.
It is also of note, with respect to aldol catalysis, that even in water a simple small molecule catalyst, like Pro, can remain substrate-bound following aldol addition. The GAB activity of Pi likely facilitates proton transfer during enamine catalysis, but also, importantly, promotes the elimination of [7·Pro] to complete the catalytic cycle (Fig. 1c). Geochemical models suggest high concentrations of Pi (>1 M) would accumulate coincidently with high concentrations of carbonate in natural (prebiotic) environments.21 Interestingly, we observe carbonate has no detrimental effect on the observed Pro/Pi catalysed aldol (ESI,† Fig. S39 and S40). Further investigation of the co-catalytic action of chiral amines and phosphates, as well as the potential to enhance chiroselectivity through (hemi-)aminal dynamic resolution,12e,22 in the context of (synthetic) aldol reactions and the origins of sugar-phosphate homochirality are warranted.
M. W. P. conceived the research. M. W. P. and Á. F. M. designed and analysed the experiments and wrote the manuscript. Á. F. M. conducted the experiments.
The European Union Horizon 2020 (Marie Skłodowska-Curie grant 813873), EPSRC (EP/P020410/1) and Simons Foundation (318881FY19) supported this work. We thank K. Karu (Mass spectrometry) and A. E. Aliev (NMR spectroscopy) for assistance, and D. Whitaker and J. Singh for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. J. Morowitz, J. D. Kostelnik, J. Yang and G. D. Cody, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7704–7708 CrossRef CAS PubMed; (b) N. R. Pace, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 805–808 CrossRef CAS PubMed; (c) A. Eschenmoser, Tetrahedron, 2007, 63, 12821–12844 CrossRef CAS; (d) L. E. Orgel, PLOS Biol., 2008, 6, e18 CrossRef PubMed; (e) A. Eschenmoser, Angew. Chem., Int. Ed., 2011, 50, 12412–12472 CrossRef CAS PubMed; (f) M. W. Powner and J. D. Sutherland, Philos. Trans. R. Soc., B, 2011, 366, 2870–2877 CrossRef CAS PubMed; (g) B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS PubMed; (h) S. Islam and M. W. Powner, Chem, 2017, 2, 470–501 CrossRef CAS; (i) M. Ralser, Biochem. J., 2018, 475, 2577–2592 CrossRef CAS PubMed; (j) J. S. Teichert, F. M. Kruse and O. Trapp, Angew. Chem., Int. Ed., 2019, 58, 9944–9947 CrossRef CAS PubMed; (k) K. B. Muchowska, S. J. Varma and J. Moran, Nature, 2019, 569, 104–107 CrossRef CAS PubMed; (l) R. T. Stubbs, M. Yadav, R. Krishnamurthy and G. Springsteen, Nat. Chem., 2020, 12, 1016–1022 CrossRef CAS PubMed; (m) D. J. Ritson, Sci. Adv., 2021, 7, eabh3981 CrossRef CAS PubMed; (n) Z. Liu, L.-F. Wu, C. L. Kufner, D. D. Sasselov, W. W. Fischer and J. D. Sutherland, Nat. Chem., 2021, 13, 1126–1132 CrossRef CAS PubMed; (o) M. Yadav, S. Pulletikurti, J. R. Yerabolu and R. Krishnamurthy, Nat. Chem., 2022, 14, 170–178 CrossRef CAS PubMed.
- (a) C. T. Walsh, T. E. Benson, D. H. Kim and W. J. Lees, Chem. Biol., 1996, 3, 83–91 CrossRef CAS PubMed; (b) D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, 2004 Search PubMed.
- (a) S. Potter and L. A. Fothergill-Gilmore, Biochem. Educ., 1993, 21, 45–48 CrossRef CAS; (b) R. A. Bender, in Brenner's Encyclopedia of Genetics, ed. S. Maloy and K. Hughes, Academic Press, San Diego, 2nd edn, Glycolysis, 2013, pp. 346–349 Search PubMed.
- (a) A. J. Coggins and M. W. Powner, Nat. Chem., 2017, 9, 310–317 CrossRef CAS PubMed; (b) A. J. Wagner and D. G. Blackmond, ACS Cent. Sci., 2016, 2, 775–777 CrossRef CAS PubMed.
- (a) H. P. Meloche and J. P. Glusker, Science, 1973, 181, 350–352 CrossRef CAS PubMed; (b) J. K. Lassila, D. Baker and D. Herschlag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4937–4942 CrossRef CAS PubMed.
- W.-D. Fessner, A. Schneider, H. Held, G. Sinerius, C. Walter, M. Hixon and J. V. Schloss, Angew. Chem., Int. Ed. Engl., 1996, 35, 2219–2221 CrossRef CAS.
- (a) B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS; (b) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798–6799 CrossRef CAS PubMed; (c) L. Hoang, S. Bahmanyar, K. N. Houk and B. List, J. Am. Chem. Soc., 2003, 125, 16–17 CrossRef CAS PubMed; (d) S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475–2479 CrossRef CAS PubMed.
- (a) S. Pizzarello and A. L. Weber, Science, 2004, 303, 1151 CrossRef CAS PubMed; (b) R. Breslow and M. S. Levine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12979–12980 CrossRef CAS PubMed; (c) Y. Hayashi, M. Matsuzawa, J. Yamaguchi, S. Yonehara, Y. Matsumoto, M. Shoji, D. Hashizume and H. Koshino, Angew. Chem., Int. Ed., 2006, 45, 4593–4597 CrossRef CAS PubMed; (d) M. Klussmann, H. Iwamura, S. P. Mathew, D. H. Wells, U. Pandya, A. Armstrong and D. G. Blackmond, Nature, 2006, 441, 621–623 CrossRef CAS PubMed; (e) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638–4660 CrossRef CAS PubMed; (f) C. F. Barbas III, Angew. Chem., Int. Ed., 2008, 47, 42–47 CrossRef PubMed; (g) L. Burroughs, M. E. Vale, J. A. R. Gilks, H. Forintos, C. J. Hayes and P. A. Clarke, Chem. Commun., 2010, 46, 4776–4778 RSC; (h) H. Wennemers, Chem. Commun., 2011, 47, 12036–12041 RSC; (i) M. P. van der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491–508 CrossRef CAS.
- (a) B. List, Tetrahedron, 2002, 58, 5573–5590 CrossRef CAS; (b) Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Chem. Soc. Rev., 2018, 47, 4388–4480 RSC; (c) C. S. Foden, S. Islam, C. Fernández-García, L. Maugeri, T. D. Sheppard and M. W. Powner, Science, 2020, 370, 865–869 CrossRef CAS PubMed; (d) J. Singh, D. Whitaker, B. Thoma, S. Islam, C. S. Foden, A. E. Aliev, T. D. Sheppard and M. W. Powner, J. Am. Chem. Soc., 2022, 144, 10151–10155 CrossRef CAS PubMed.
- (a) N. Zotova, A. Franzke, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2007, 129, 15100–15101 CrossRef CAS PubMed; (b) M. Raj and V. K. Singh, Chem. Commun., 2009, 6687–6703 RSC; (c) F. Cruz-Acosta, P. de Armas and F. García-Tellado, Chem. – Eur. J., 2013, 19, 16550–16554 CrossRef CAS PubMed.
- The aldehyde must be pre-dissolved in the organic solvent, i.e., DMSO: (a) T. J. Dickerson and K. D. Janda, J. Am. Chem. Soc., 2002, 124, 3220–3221 CrossRef CAS PubMed; (b) T. J. Dickerson, T. Lovell, M. M. Meijler, L. Noodleman and K. D. Janda, J. Org. Chem., 2004, 69, 6603–6609 CrossRef CAS PubMed.
- Breslow and co-workers reported L-Pro enriches L- not D-glyceraldehyde (20–42%ee) under acidic conditions (pH 3), and because this reaction enriched the non-biological enantiomer of glyceraldehyde, they cast doubt on whether Pro could have played a role on the prebiotic Earth. Hein and Blackmond confirmed that 8%ee L-glyceraldehyde was observed under similar conditions, but they observed 13%ee D-glyceraldehyde in the presence of an organic base (i.e. tetrabutylammonium acetate). Breslow and co-workers re-examined Pro catalysis at pH 7.6, where negligible chiral enrichment (2%ee) for D-glyceraldehyde was observed, but again no yield was reported. Upon repeating this reaction we were unable to detect glyceraldehyde by NMR, though it may be below the NMR detection limit. Interestingly, whilst studying a related kinetic peptide mediated resolution of glyceraldehyde, Blackmond and co-workers have also recently reported Pro is unreactive: (a) R. Breslow and Z.-L. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5723–5725 CrossRef CAS PubMed; (b) J. E. Hein and D. G. Blackmond, Acc. Chem. Res., 2012, 45, 2045–2054 CrossRef CAS PubMed; (c) A. M. Steer, N. Bia, D. K. Smith and P. A. Clarke, Chem. Commun., 2017, 53, 10362–10365 RSC; (d) R. Breslow, V. Ramalingam and C. Appayee, Origins Life Evol. Biospheres, 2013, 43, 323–329 CrossRef PubMed; (e) J. Yu, A. X. Jones, L. Legnani and D. G. Blackmond, Chem. Sci., 2021, 12, 6350–6354 RSC.
- D. Müller, S. Pitsch, A. Kittaka, E. Wagner, C. E. Wintner, A. Eschenmoser and G. Ohlofjgewidmet, Helv. Chim. Acta, 1990, 73, 1410–1468 CrossRef.
- (a) B. List, L. Hoang and H. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5839–5842 CrossRef CAS PubMed; (b) M. B. Schmid, K. Zeitler and R. M. Gschwind, Angew. Chem., Int. Ed., 2010, 49, 4997–5003 CrossRef CAS PubMed.
- C. Anastasi, F. F. Buchet, M. A. Crowe, M. Helliwell, J. Raftery and J. D. Sutherland, Chem. – Eur. J., 2008, 14, 2375–2388 CrossRef CAS PubMed.
- D. Seebach, X. Sun, M.-O. Ebert, W. B. Schweizer, N. Purkayastha, A. K. Beck, J. Duschmalé, H. Wennemers, T. Mukaiyama, M. Benohoud, Y. Hayashi and M. Reiher, Helv. Chim. Acta, 2013, 96, 799–852 CrossRef CAS.
- (a) P. Krattiger, R. Kovasy, J. D. Revell, S. Ivan and H. Wennemers, Org. Lett., 2005, 7, 1101–1103 CrossRef CAS PubMed; (b) J. D. Revell and H. Wennemers, Adv. Synth. Catal., 2008, 350, 1046–1052 CrossRef CAS.
- (a) W. J. Albery and J. R. Knowles, Angew. Chem., Int. Ed. Engl., 1977, 16, 285–293 CrossRef CAS PubMed; (b) W. P. Jencks, Catalysis in Chemistry and Enzymology, Dover, 1987, pp. 163–242 Search PubMed; (c) J. Kraut, Science, 1988, 242, 533–540 CrossRef CAS PubMed; (d) J. R. Knowles, Science, 1987, 236, 1252–1258 CrossRef CAS PubMed.
- (a) C. Y. Lai, N. Nakai and D. Chang, Science, 1974, 183, 1204–1206 CrossRef CAS PubMed; (b) W. J. Lees and G. M. Whitesides, J. Org. Chem., 1993, 58, 1887–1894 CrossRef CAS.
- (a) M. W. Powner, B. Gerland and J. D. Sutherland, Nature, 2009, 459, 239–242 CrossRef CAS PubMed; (b) M. W. Powner and J. D. Sutherland, Angew. Chem., Int. Ed., 2010, 49, 4641–4643 CrossRef CAS PubMed.
- (a) J. D. Toner and D. C. Catling, Proc. Natl. Acad. Sci. U. S. A., 2019, 117, 883–888 CrossRef PubMed; (b) M. P. Brady, R. Tostevin and N. J. Tosca, Nat. Commun., 2022, 13, 5162 CrossRef CAS PubMed.
- S. Islam, D.-K. Bučar and M. W. Powner, Nat. Chem., 2017, 9, 584–589 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: https://doi.org/10.1039/d2cc05466c |
| This journal is © The Royal Society of Chemistry 2022 |