
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpurity incorporation in solution crystallization: diagnosis, prevention, and control
Gerard
Capellades
 *a,
Jacob O.
Bonsu
a and
Allan S.
Myerson
*b
*a,
Jacob O.
Bonsu
a and
Allan S.
Myerson
*b
aDepartment of Chemical Engineering, Henry M. Rowan College of Engineering, Rowan University, Glassboro, New Jersey 08028, USA. E-mail: capellades@rowan.edu
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: myerson@mit.edu
First published on 7th February 2022
Abstract
Despite their widespread use for purification, our current methods for the development of solution crystallization processes lack a sufficient understanding on how impurities incorporate in growing crystals. This is, in part, due to the large number of mechanisms often encountered for impurity incorporation, and due to limitations in our methods for diagnosis of those mechanisms. These limitations propagate into largely empirical process development strategies, which are still based on trial and error and centered on solvent selection. This manuscript highlights recent developments in the diagnosis, prevention, and control of impurity incorporation during batch and continuous crystallization. The goal is to provide process development scientists with an updated toolkit for understanding how specific impurities are retained in the solid product, and to review recent prevention and control strategies that may be used to improve crystal purity in industrial crystallization processes.
1. Introduction
Crystallization as a separation process plays a critical role in the manufacturing of bulk chemicals, food products, fine chemicals, and pharmaceuticals.1,2 This unit operation is used for purification, but also as an isolation step for the separation of powders with the desired crystal size, habit, and structure. Currently, many of the products based on fine chemicals, for instance, photographic materials, explosives, and dyes, necessitate crystallization in their fabrication, and approximately 90% of small-molecule pharmaceuticals include drug substances in the crystalline solid form.1 As a result, manufacturers strive to control the crystallization process to attain products with reproducible and desired solid-state properties.In recent years, there has been a significant improvement in our understanding of solution crystallization processes. This is, in part, driven by an increasing need to design robust separation processes in pharmaceutical manufacturing, and by a push towards continuous manufacturing.3–6 Quality attributes like bioavailability and stability of pharmaceutical powders are directly related to the solid-state properties of the crystals, including crystal size, habit, and form.7–9 Those attributes can be controlled by adjusting crystallization conditions and the addition of in situ and post-crystallization mechanical processing steps.10–14 However, in contrast with the literature on crystallization for the control of crystal size, habit, and form, the amount of work relative to crystallization as a purification process is relatively scarce.
From a medical perspective, control of impurities in a manufacturing process is especially critical for products that should be fit for human consumption, and the presence of unacceptable levels of genotoxic impurities has been a reason for recent pharmaceutical recalls.15–17 Similarly, from the process perspective, traces of impurities have been shown to play a significant role in modifying the thermodynamics and kinetics of crystallization processes.18–23 The identification and control of impurity profiles through manufacturing plants is a critical step to ensure a robust operation and accurate control of the propagation of process disturbances.24,25
In the manufacturing of small-molecule pharmaceuticals, even a small number of synthesis steps can quickly lead to the generation of dozens of impurities, often with unknown composition or toxicological information.26–30 Consequently, most recent examples of end-to-end synthetic pharmaceutical manufacturing involve a number of crystallization steps where the main role is to purify an intermediate product, limiting the accumulation of impurities along the main process stream.25,26,31–38 Despite the widespread use of these purification steps, our understanding of how impurities incorporate in crystals is far behind, current models for predicting purity in solution crystallization are limited to a narrow design space,25,28 and process design is still largely based on extensive solvent screenings and trial and error.24,26,38–41 This is, in part, due to the number and complexity of mechanisms by which impurities incorporate in growing crystals, and the limited understanding of kinetic-driven interactions between impurities and solute molecules in the crystal-solution interphase.
This manuscript highlights recent advances in the diagnosis and control of impurity incorporation during solution crystallization of small organic molecules. The theory behind those mechanisms is only briefly covered to the extent that is necessary to understand the rest of this manuscript, but it is described in more detail in recent literature.42,43 The second half of the manuscript is centred on the recent publications in the context of impurity incorporation and crystallization, reviewing current efforts and novel strategies for prevention and control.
2. Impurity incorporation in solution crystallization
When it comes to impurity incorporation mechanisms, it is important to make a distinction between the manufacturing of single crystals and bulk manufacturing. For processes involving the isolation of a crystalline product, in their broadest sense, impurities can end in the solid product by incorporating inside of individual crystals, by adhering to the surface of those crystals, by entrapment within aggregates, or by forming their solid phase as a second precipitate. A summary of the individual impurity incorporation mechanisms is provided in Fig. 1.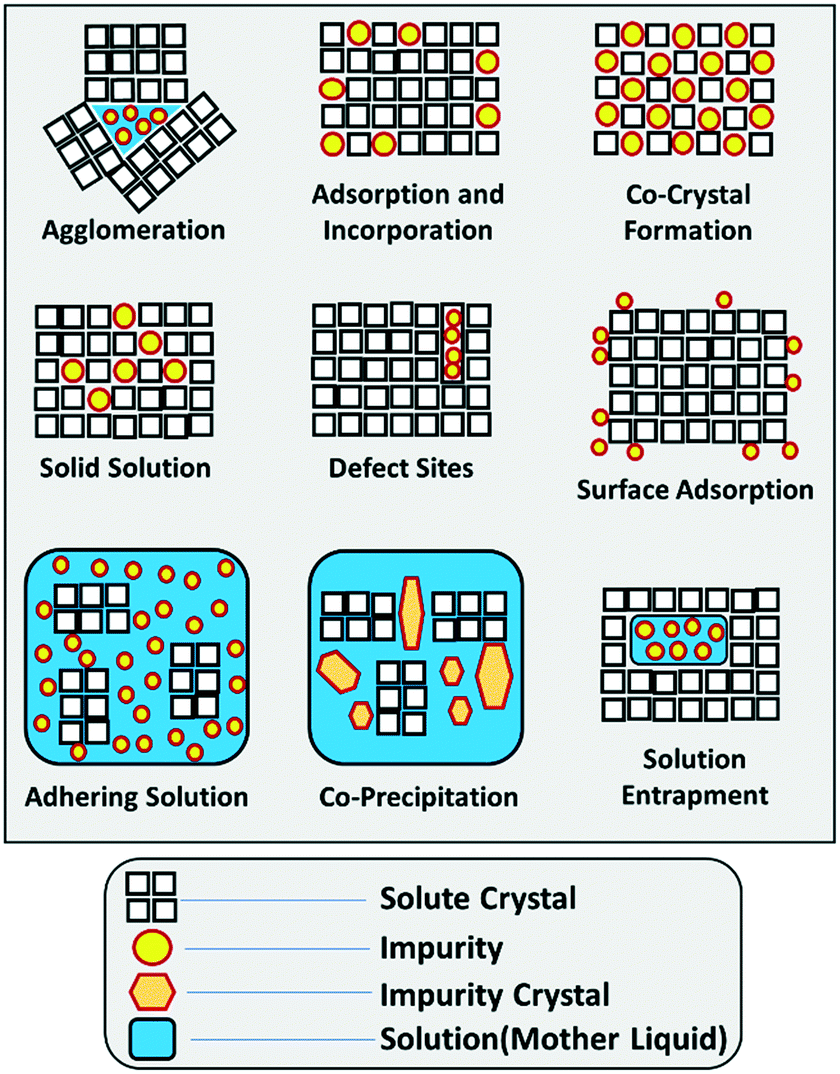 | ||
| Fig. 1 Schematic illustration of the different impurity incorporation mechanisms in solution crystallization. Inspired by Urwin et al. and expanded for this work.44 | ||
For impurity incorporation outside the crystals, i.e. by agglomeration, co-precipitation, or adhering mother liquor, the mechanisms are fairly straight-forward. The crystallization mother liquor tends to be rich in impurities that have been rejected from the crystalline phase. Thus, the formation of aggregates during crystallization has the possibility of entrapping this mother liquor, decreasing the overall purity of the solid product. Methods for preventing aggregate formation are widely available in the literature and fall out of the scope of this work.44–49 Similarly, when the mother liquor is filtered from the solids, adequate washing steps must be in place to wash out the adhering liquid while preventing the precipitation of impurities by anti-solvent effects.20,40 In this area, impurity precipitation (either during crystallization, or during washing) is driven by the generation of a supersaturated state for those impurities. If their concentration is too high, or the solubility in the crystallization solvent is too low, impurities may precipitate together with the product of interest, generating a powder that contains multiple solid phases.
A similar impurity incorporation mechanism emanates from solution entrapment, and commonly occurs for systems with porous or large crystals, where a portion of the liquid phase is entrapped in the solid product and is not easily washed away.42,44 Inclusions stem from non-equilibrium processes and may be categorized as primary (those formed during crystal growth) or secondary (those formed after crystal growth). One probable account for primary inclusion formation could be due to the disproportionate growth rates at the surface of crystals, mostly with the edges and corners growing quicker than the centre of the faces, leading to cavities formation which are ultimately sealed as growth proceeds.2,43,50
The most intricate impurity incorporation mechanisms arguably take place when impurities incorporate inside the crystal. In this area, several mechanisms have been identified in the past, and the distinction between those can be experimentally challenging with our current understanding of those mechanisms. It is important to note that, while the crystalline product is often analysed as a powder on the basis of an average purity, impurities don't necessarily incorporate evenly throughout the crystal lattice. Solute molecules follow a specific order within a crystal lattice. Thus, three-dimensional crystals presenting multiple faces will present a unique surface chemistry and a different rate of crystal growth in each face, ultimately defining crystal habit.49,51 When it comes to interactions with impurities, some impurities may preferentially interact with certain faces, either because of their growth rate or because of their surface chemistry (e.g. exposed functional groups). Similarly, differences in kinetics throughout crystal growth may lead to different impurity profiles in a grown crystal, generating a core rich in impurities and an increasing purity as we approach the crystal surface.52 These mechanisms will lead to a heterogeneous distribution of impurities within the crystal lattice (Fig. 2), with some areas of the crystal presenting impurity concentrations orders of magnitude higher than other areas.53
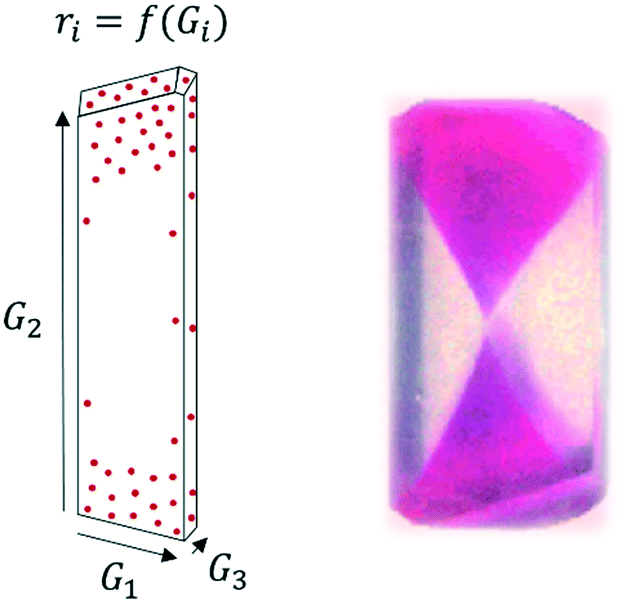 | ||
| Fig. 2 Left: Face-dependent crystal growth (Gi) and impurity incorporation (ri) in a growing crystal. Right: Example of face-dependent incorporation of sulforhodamine B in a K2SO4 crystal [adapted with permission from Kahr and Gurney.54 Copyright 2001 American Chemical Society]. | ||
A practical analysis of face-dependent impurity incorporation during industrial crystallization development is currently limited by the available diagnosis methods centred on powder analysis. However, it may be a critical step for the improved understanding of impurity incorporation in an inherently heterogeneous system. For more information and examples on face-dependent impurity incorporation, an excellent review on the topic was published by Kahr and Gurney, with focus on the incorporation of dyes in growing crystals.54
One of the main mechanisms for impurity incorporation inside a crystal is the formation of solid solutions, where impurities are uniformly distributed within the crystal lattice. These are usually presented as regions with high impurity contents within the crystal. Solid solutions can be substitutional or interstitial. In the first case, the impurity has a similar size and molecular properties as the solute molecule and incorporates by taking the solute's place in the crystal lattice. For interstitial solid solutions, the impurity enters the interstices of the crystal lattice, further disrupting the crystal order.43 True solid solutions are energetically unfavourable, and thus they are not expected to occur as a main mechanism in kinetic-driven processes. Small amounts of impurities (<1%) are often observed within the crystals, which could be indicative of incorporation in defect sites instead. However, some solid solutions have been observed in systems with structurally similar impurities.53,55–60
Similarly, it is important to note that impurities may form co-crystals with the solute system under the appropriate crystallization conditions. These are homogenous crystalline structures comprising a neutral co-former and a neutral target held together by a non-covalent interactive bond.61,62 The unique structural orientation of co-crystals is due to the non-covalent interactions between various reacting species that result in the systematic integration of both compounds (target and co-former) into the crystal lattice.62,63 For an excellent lattice substitution to take place, the incorporated impurity usually is of similar size and structure to the target compound.61 Forming co-crystals of active pharmaceutical ingredients (APIs) with a specially selected co-former is a known approach to enhance crystal quality attributes. However, the target molecule sometimes forms insoluble co-crystals with an impurity.61,64–66 For example, Ranganathan et al. observed the formation of a toxic 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-crystal of melamine and cyanuric acid, which was practically insoluble in the crystallization solvent.67
:1 co-crystal of melamine and cyanuric acid, which was practically insoluble in the crystallization solvent.67
Defects in crystals are readily formed from stresses induced by supersaturation, temperature, and mechanical action, and they can act as a hideout for impurities to incorporate in the crystal.51 Edge dislocation is a usual position of impurity uptake in crystals structure essentially because of its unique structure.68 The insertion of an extra lattice plane in edge dislocations, compresses the crystal lattice by generating several sites on the surface that can favourably concentrate impurities.
Finally, impurities may adsorb on the crystal surface if they have a sufficiently high binding energy with one or more of the crystal faces.49,51 The accumulation of adsorbed impurity molecules at the surface of the growing crystals may precede their incorporation in the crystal lattice as a new layer of solute molecules attaches in the growing crystal face. This mechanism, predominant at slow crystal growth rates, may be hard to differentiate from solid solutions that would also be expected at near-equilibrium. The adsorbed impurities have also been reported to play a role inhibiting crystal growth, and thus they may have an impact on crystallization yield, particle size, and crystal habit.69–71
3. Diagnosis of impurity incorporation mechanisms
The diagnosis of how impurities incorporate in the growing solute is a critical step for the selection of adequate control strategies that are directed to the root cause of impurity incorporation. For example, a co-precipitating impurity can be easily removed by tuning the crystallization solvent so that this species becomes undersaturated, or by the selection of a proper wash solvent in the subsequent filtration process.38 Similarly, impurity incorporation driven by agglomeration would need to be handled via mitigation of this phenomenon. Because industrial solution crystallization mostly deals with the formation of powders instead of single crystals, several methods have been described in the literature for the diagnosis of impurity incorporation mechanisms from powder analysis.3.1 Process imaging
Process imaging methods provide an advantage over the other methods described in this work, as they do not generally require separate off-line experiments for the diagnosis of impurity incorporation and they are not limited to the study of crystal purity. Several tools have been used for imaging crystallization processes in situ, including probes and integrated crystallization systems.13,72–76 While the focus is generally control of crystal size and habit, process imaging can aid in the detection of impurity incorporation mechanisms by acting as a tool to detect solution inclusions, aggregate formation, or co-precipitation. The latter can be presented as crystals with a different morphology or optical properties, but care should be taken to avoid confusing polymorphs with co-precipitating impurities. A recent example was the detection of co-precipitating phenylglycine impurities during the crystallization of cephalexin monohydrate. For this system, Salami and co-workers used a convolutional neural network (CNN) to identify the formation of phenylglycine crystals from process images taken in situ (Fig. 3).77 This detection can be used as a basis for corrective measures like temperature cycling or modifications in solvent composition.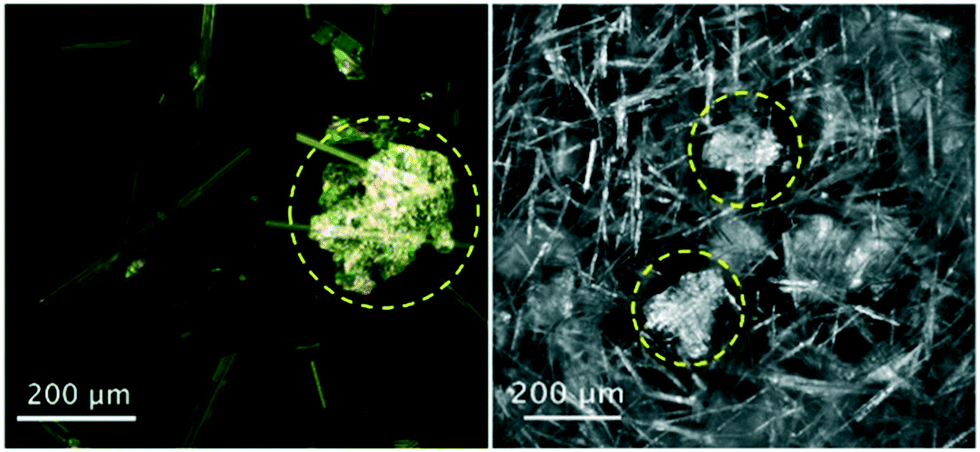 | ||
| Fig. 3 Optical microscopy (left) and in situ process images (right) of co-precipitates of phenylglycine impurities (circled) during crystallization of needle-like cephalexin monohydrate crystals [adapted with permission from Salami et al.77 Copyright 2021 American Chemical Society]. | ||
Due to its non-invasive nature, it is recommended to utilize process imaging whenever possible, as it provides valuable information about the crystallization behaviour that is hard to obtain with off-line tests. However, note that this technique is limited to the detection of co-precipitating impurities, solution entrapment, and aggregation, and it may be flawed if co-precipitating impurities present similar optical properties as the crystallizing solute. In those situations, the pairing with Raman spectroscopy could aid the detection of the new solid phase.13,14
3.2 Dry powder analysis
Similar to the use of process imaging, optical microscopy on off-line samples can aid in the detection of aggregates,25 co-precipitates (Fig. 3),77 and solution entrapment.78 For the analysis of dry powders by microscopy, it is important to mitigate the formation of aggregates or co-precipitates during washing and drying, which could induce bias in the results. Alternative methods have been used where the suspension is carefully sampled directly from the crystallizer, and diluted in a large amount of saturated solution to simultaneously quench crystallization and dilute the solids concentration to a level that facilitates imaging.10Analysis of powder mixtures by X-ray powder diffraction (XRPD) can also reveal information on crystallizing impurities, seen as the appearance of a new pattern revealing a mixture of crystalline phases,79–84 or the formation of a co-crystal.85 In addition, solid solutions have been shown to affect the diffraction peak positions by expansion or contraction of the lattice,86 as well as a reduction in peak intensities due to the loss of crystallinity.53,87 X-ray diffraction has also been used in single crystals, for specific faces showing a high degree of impurity incorporation. In those systems, X-ray diffraction can be used to identify the atomic positions of the impurities in the host crystal.43
Differential scanning calorimetry (DSC) analysis of crystallized powders can also be used to detect the incorporation of impurities. The lattice incorporation of impurities can have a small effect on the observed melting point and enthalpy of fusion,53,84,87 and the co-precipitation of impurities can be seen as peak doubling.88 However, due to the possibility of peak doubling by melting and re-crystallization of the solute crystals, care should be taken when interpreting DSC data on systems potentially presenting impurity co-precipitation and polymorphism.
3.3 Powder dissolution
Off-line tests based on powder dissolution are perhaps the most accessible method for diagnosing the location of impurities within a system. In these tests, the dry crystalline powder containing solute and impurities is slowly dissolved by gradual solvent addition, and the concentration of dissolved impurities in the washed solvent (or, alternatively, the concentration of impurities in the solids) is tracked as a function of the amount of solvent added, using analytical methods like high performance liquid chromatography (HPLC). This analysis can be based on a relative area percent (LCAP) or, if the impurities are known and available, based on their actual concentrations using a separate calibration line.89 The results are analysed using a component phase balance between the solids and the liquid phase, providing with a trend in the impurities' dissolution rate relative to the solute's dissolution rate. A high degree of impurity dissolution in the early dissolution steps is generally assumed to indicate that the impurities are located at the surface of the crystal (Fig. 4). Similarly, impurities dissolving towards the later steps would indicate that they are predominantly inside the crystal, in areas that are sometimes not accessible during the early dissolution steps.44,51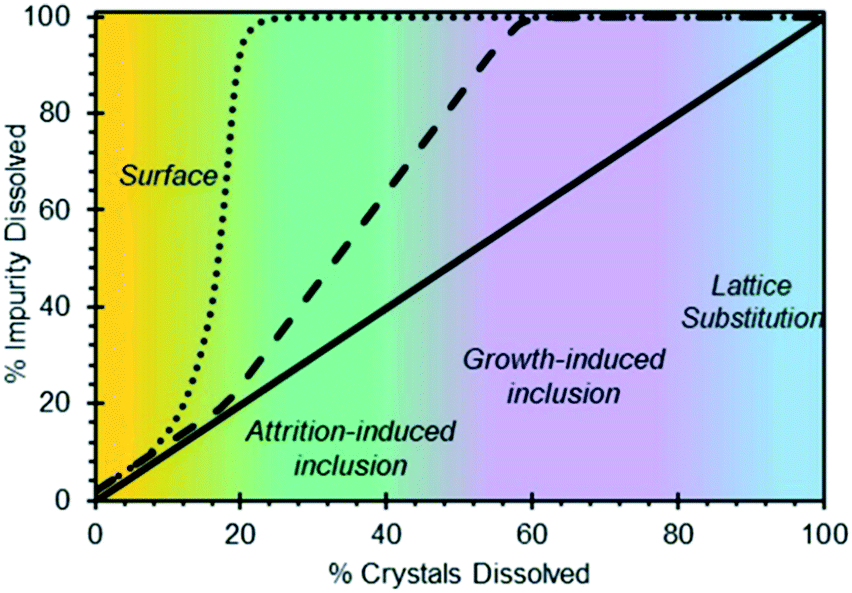 | ||
| Fig. 4 Hypothesized impurity distribution maps based on progressive powder dissolution tests [reprinted with permission from Urwin et al.44 Copyright 2020 American Chemical Society]. | ||
It is important to note that these methods rely on the assumption that crystal faces dissolve evenly and that the presence of impurities does not affect the dissolution rate of the impure crystal faces. Wang et al. conducted dissolution studies of crystallized salicylic acid, which presented elongated crystals with an impure crystal core.52 Upon the addition of a small amount of solvent, the core that was rich in impurities dissolved first, generating macrotubular crystals with a significantly higher average purity (Fig. 5).
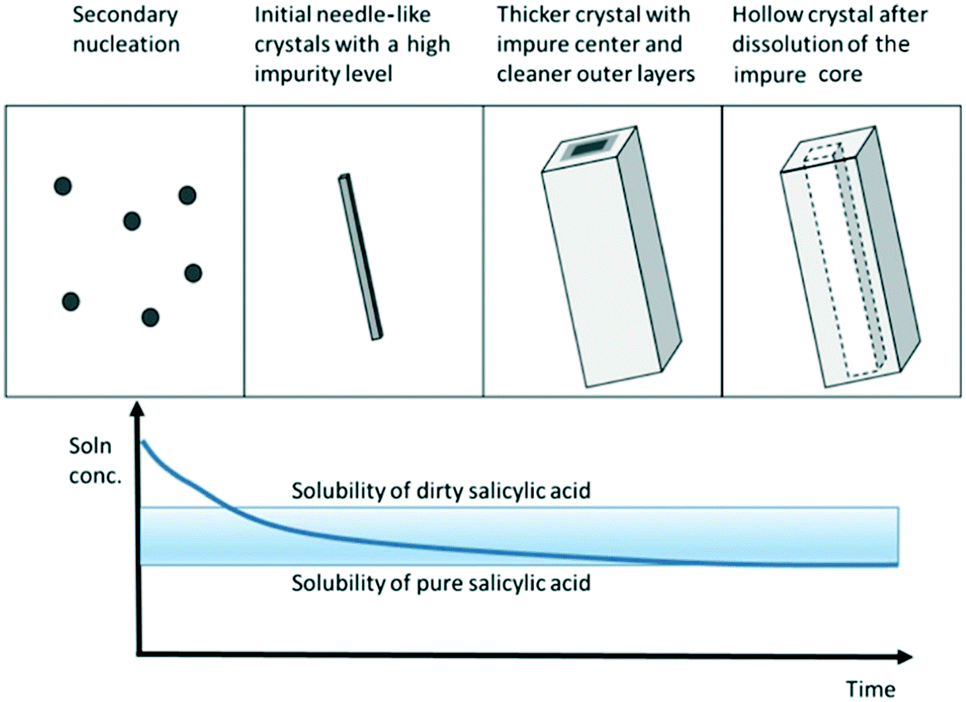 | ||
| Fig. 5 Schematic illustrating the formation of macrotubular crystals of salicylic acid by crystallization of needle-like crystals and subsequent hollowing during dissolution [reprinted with permission from Wang et al.52 Copyright 2021 American Chemical Society]. | ||
It has been widely reported that the presence of impurities in a crystallization system can have a significant impact on solubility and kinetics.18,19,21,90–92 Similarly, the incorporation of foreign species in a pure crystal can be disruptive for the crystal lattice and is energetically unfavourable. It is thus not hard to believe that, if impurities selectively incorporate at the core of the crystal or at given crystal faces,54 those areas may be the first to dissolve when presented with a solvent or undersaturated solution. Considering that progressive dissolution studies have been used for decades,49,51,93 this recent evidence implies that several systems presenting internal impurity gradients could have been misdiagnosed as surface impurities. As a preventive measure for systems exhibiting a similar behaviour, it is recommended to pair powder dissolution studies with optical microscopy, to ensure that crystal habit remains unaltered during the dissolution tests, and to verify the absence of new cavitation.
A novel type of powder dissolution test, named solubility-limited impurity purge (SLIP) test, was recently developed by Nordstrom et al., for the detection of co-precipitating impurities in powder mixtures.94 The method consists on suspending powder samples in various amounts of the crystallization solvent, centrifuging and filtering those suspensions after they have been aged for at least 24 hours. The method tracks the concentration of impurities in the solid and liquid phases using HPLC. When co-precipitating impurities are present, low amounts of solvent will generate a saturated solution of the impurities, and thus their liquid concentration will remain constant regardless of the dilution factor. This will be true until the solvent volume is enough to fully dissolve the impurities, and at this point, their concentration would drop due to dilution. The start of this drop gives the solubility of the impurity. By tracking the purity of the solid phase, it is also possible to evaluate whether the impurities or the solute are preferentially dissolved in the new solvent.
3.4 Powder-based impurity adsorption tests
Diagnosis of impurity adsorption mechanisms is difficult in kinetic-driven systems, where crystal growth can lead to the incorporation of adsorbed impurities in the growing layer. Similarly, the need to differentiate surface impurities from those in adhering mother liquor requires the careful design of a washing step that displaces the adhering mother liquor without inducing precipitation or dissolution of the surface impurities.Powder-based methods based on the use of different particle sizes are frequently used for the diagnosis of surface impurities. The powder sample can be sieved and the different sieve fractions can be analysed for crystal purity. If the impurities are located at the surface, smaller crystals with a higher surface area per unit volume will present a lower purity.43 When using this method, it is important to make sure that impurities did not co-precipitate (as a powder with likely a different size distribution) during crystallization. While lower purities in smaller crystals could be indicative of surface incorporation, lower purities for larger crystals could be a consequence of a spread in nucleation times (older crystals present at the start of crystallization, with different impurity incorporation rates at higher supersaturations), solution entrapment, or agglomeration.
An alternative approach for the simultaneous study of adsorption and adsorption-incorporation mechanisms was recently implemented by Teerakapibal et al.53 In their studies, they conducted seeded batch crystallizations with the same amount of regular and ground seeds, and investigated the purity of the final product. If adsorption plays a role in impurity incorporation, the product from the smaller seeds will present a lower purity due to the increased surface area. This approach requires a careful implementation to operate within a low, narrow range of supersaturations. This is to mitigate nucleation and to minimize bias from the different desupersaturation profiles that would be expected with two different starting surface areas.
3.5 Single crystal analysis
Several analytical methods exist for investigating the incorporation of impurities in single crystals. While industrial solution crystallization deals with powders, the analysis of single crystals is still useful to diagnose the location of impurities within a crystal, and it provides the highest level of fundamental understanding of the relationship between surface chemistry, crystal growth, and impurity incorporation.Electron microscopy and atomic force microscopy can offer information about the crystal surfaces at the nanometer scale, which can be used to detect defects in the crystals and investigate growth mechanisms in each crystal face.95–97 In combination with chemical imaging or etching studies, these techniques could reveal impurity incorporation in defect sites, as well as the relationship between the crystal growth mechanism and impurity incorporation.43
A common technique for chemical imaging is the use of Raman spectroscopy on single crystals. Industrial crystallization, especially for small molecule pharmaceutical products, often results in the attainment of crystal sizes at the order of 100 μm.98–101 With a spatial resolution of appr. 1 μm, one can use Raman microscopes to scan the surfaces of those crystals and map the impurity gradients across the crystal.52 An alternative technique, although bound to poorer spatial resolutions, is the use of infrared (IR) spectroscopy for the same purpose.43
4. Prevention and control
4.1 Solvent selection for crystallization and washing
While it may not be considered a specific strategy for impurity incorporation control, the importance of a good solvent selection for the prevention of impurity incorporation during crystallization cannot be overstated. The crystallization solvent plays a critical role in defining the attainable quality attributes of the product crystals,99,102 affecting solubility, kinetics, crystal habit, and the dominant impurity incorporation mechanism. When impurities are present, an adequate solvent selection may prevent co-precipitation of impurities entirely by keeping them undersaturated. Its direct effects on nucleation and growth kinetics will also play an indirect role in impurity incorporation, by offering control over the crystal surface area (adsorption-driven mechanisms) or minimizing crystal growth to prevent solution entrapment. Similarly, solvent plays a role in the tendency of solutes to aggregate, and recent solvent selection strategies have placed a higher emphasis on this factor.75It is thus not surprising that, when the amount of time and representative crude material are limited in process development, most of the focus is placed on the selection of crystallization and wash solvents. A recent example was reported for the last two iterations of the Pharmacy on Demand (PoD) project, where end-to-end processes for several pharmaceuticals had to be developed in parallel with synthesis steps (limited amounts of crude available for downstream) and within a very limited time frame. In the project's “Demonstration” phase, Rogers et al. presented a downstream process development framework largely based on solvent screenings with small amounts of crude material, and applied it to the development of five downstream processes for different pharmaceuticals.26 In the later “GMP” phase, Capellades et al. optimized the crystallization and wash solvent compositions for maximizing yield and purity of a recovery process for Ciprofloxacin, increasing crude purity from 78% to 97% in the first crystallization-filtration-washing cycle, and from 97% to 99% in the second cycle.38 The solvent selection process started similarly to that from the previous phase,26 with an early screening for precipitation solvents at the 10 mL scale. After selecting two promising solvent compositions for crystallization, and two compositions for washing, a factorial design was conducted to guide the final selection as soon as enough crude became available. An optimal solvent combination was chosen and scaled-up in an off-line process (Fig. 6), before being implemented in end-to-end manufacturing.27,38 While development was conducted based on a limited understanding of the mechanisms for impurity incorporation, an important part of the purification efficiency came from the selection of crystallization solvents that mitigated the co-precipitation of challenging impurities, and to the selection of wash steps to re-dissolve organic impurities and inorganic precipitates.
 | ||
| Fig. 6 Early screening of crystallization and wash solvents for the rapid development of continuous isolation processes. a) Precipitation tests in scintillation vials. b) Wash solvent screening. c) Full-scale off-line validation in an agitated filter dryer [adapted with permission from Capellades et al.38 Copyright 2021 American Chemical Society]. | ||
In a similar context, Shahid et al. presented methods for the selection of wash solvents, with focus on mitigating the co-precipitation of impurities while maximizing yield.103 The methods are suitable for small amounts of material, as they are centred on small centrifugation vials. The study, demonstrated for acetaminophen and two relevant impurities, is focused on anti-solvent effects and aids in mapping the amount and types of solvents that could be used in a series of washing steps to maximize purity and minimize API losses by dissolution.
4.2 Predictive models
While population balance models for crystallization have been available for several decades,104 the use of predictive modelling for crystallization process development has expanded significantly in recent years. In their simplest form, crystallization models relate nucleation and growth kinetics to process yield and a unidimensional crystal size distribution, assuming negligible crystal breakage, negligible aggregation, and a constant shape factor. These models largely consist on closing a mathematical loop (Fig. 7), where crystallization kinetics and a measure of time define the population of crystals in the crystallizer, n(L). With knowledge on the size, shape, and number of crystals at each size range (population), one can calculate suspension properties like the concentration of crystals at a given time (Mt), as well as their size distribution (vol(L)). Knowing the concentration of crystals and the total solute concentration, Ctot, one can calculate how much solute is left in the liquid phase (Cml) with a phase balance, and then use Cml to calculate supersaturation (σ) and the change in crystallization kinetics. The population balance equation, n(L) = f(B, G, time), depends on the type of crystallizer, but it is generally a function of the investigated crystal size, the rates of nucleation and crystal growth, and a measure of time.51,104 Simpler predictive models exist, often based in the method of moments, which sacrifices a prediction of the entire size distribution in exchange for tracking critical metrics like total solids concentration or mean particle size.104–107 In addition, variations exist to some of the equations in Fig. 7, like the use of a more accurate phase balance for high solid concentrations,108 and other simplifications to the thermodynamic definition of supersaturation that have become a standard practice in crystallization modelling, but have been shown to introduce unnecessary uncertainties in the predicted kinetics.109,110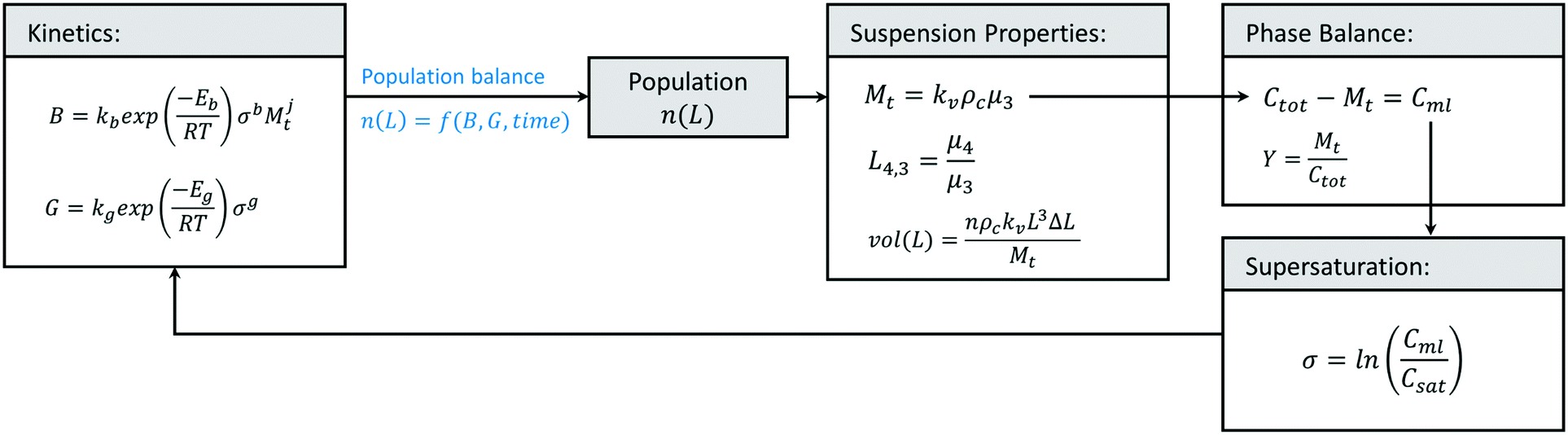 | ||
| Fig. 7 General shape of solution crystallization models for the prediction of crystallization yield (Y), mean particle size (L4,3), crystal size distribution (vol(L)), and solids concentration (Mt) from process conditions like total solute concentration (Ctot), temperature (T), and crystallization time. The models require knowledge on the system solubility (Csat), crystal shape factor (kv), crystal density (ρC), the desired distribution channels and their sizes (L, ΔL), as well as the system's kinetic parameters (Kb, Eb, kg, Eg, b, g, j) in order to calculate the evolving nucleation (B) and growth (G) rates, supersaturation (σ), mother liquor solute concentration (Cml), crystal population (n(L)), and its population moments (μi = ∫ nLidL). | ||
In the context of impurity incorporation, additional equations have been introduced to the crystallization models, often relating predictable metrics like liquid concentrations or crystal growth rates to an average partition coefficient, DCi, for the impurity (eqn (1)).
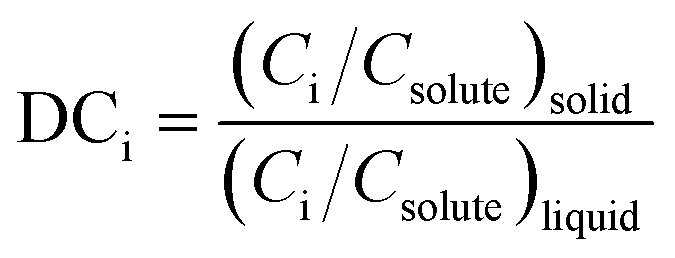 | (1) |
While impurities do incorporate at different regions within a crystal and within the powder, the modelled DCi is an average partition coefficient for the two studied phases: the solids and the liquid. Some mechanistic models do exist and are applied to crystallization from the melt,51 however, solution crystallization models for impurity incorporation in recent literature are largely empirical. A popular model was presented by Quon et al. in 2012 for the crystallization of aliskiren hemifumarate.111 The model, as shown in eqn (2), was developed from empirical observations of a linear relationship between the impurity concentration in the liquid phase of a crystallizing suspension and the distribution coefficient of that impurity.
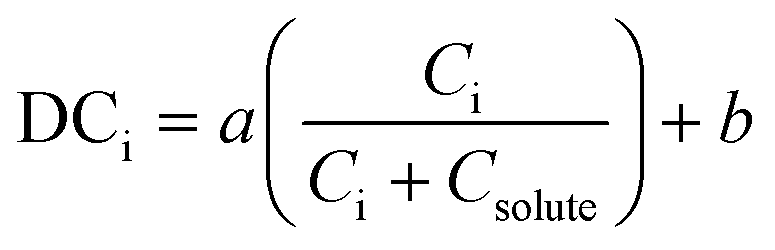 | (2) |
The model presented in eqn (2) can be introduced to the loop in Fig. 7, utilizing Ci and Csolute (as Cml) as obtained from the phase balance for the impurity and the solute, respectively. This approach has been recently implemented for the crystallizations of cyclosporine,28,112 deferasirox,112 as well as some process intermediates.25,113 In 2017, Li et al. used this model to predict the purity of cyclosporine purity in a crude system containing 20 known impurities.28 They utilized predictive models to optimize the number of crystallization stages and the crystallization conditions, maximizing purity while meeting yield constraints. In 2020, Maloney et al. implemented this model in the prediction of crystal purity for a virtual plant for a carfilzomib intermediate.25 They showed that, while the model fits for individual impurities at fixed crystallizer types and scales, a new calibration would be required for each type of crystallizer and even for varying total impurity concentrations in the crude feed (Fig. 8). This is counter-intuitive considering that the model is primarily a function of impurity concentrations, but it is not a surprising behavior for empirical models bound to a narrow design space.
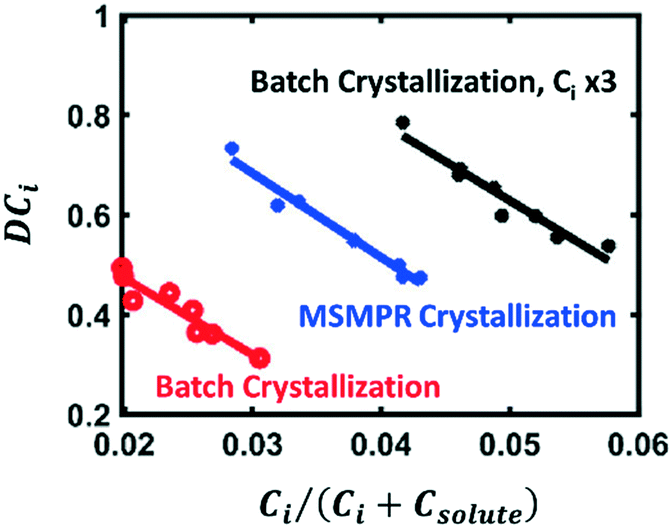 | ||
| Fig. 8 Fits to eqn (2) for the experimental distribution coefficients of an enone impurity for the nominal concentration level in batch crystallization (red), the nominal concentration level in continuous crystallization (blue), and three times the nominal concentration level in batch crystallization (black) [adapted with permission from Maloney et al.25 Copyright 2020 American Chemical Society]. | ||
A different empirical model was used in 2019 by Capellades, Wiemeyer, and Myerson, in the crystallization of Ciprofloxacin from crude solutions containing appr. 60 known impurities.18 On the observation of crystal growth-driven incorporation for the main impurities, and for the limited amount of experiments that were a consequence of limitations in the amount of crude, the empirical model shown in eqn (3) was implemented for the process.
| DCi = aG + b | (3) |
It is important to note that all the aforementioned empirical models based on an average distribution coefficient, while practical, will be flawed by definition and bound to a narrow design space. Impurities incorporate in growing crystals via a wide variety of mechanisms, and for some of these mechanisms, one crystal face can be responsible for most of the impurity incorporation.54 Overall, prediction of impurity incorporation using mechanistic models is a work in progress that requires a better understanding of impurity incorporation mechanisms, an understanding of the role of crystal faces in product purity, as well as highly reliable diagnosis methods to select mechanism-specific models. We have started seeing this modernization in the field of adsorption-driven impurity incorporation, where Borsos, Majumder, and Nagy presented a mechanistic model for the face-dependent impurity incorporation in vitamin B12. This modelling approach implemented a multidimensional population balance and considered both the effects of impurities in crystal growth and the roles of adsorption and growth in crystal purity.114
4.3 Impurity complexation
A novel method to tackle impurity incorporation during crystallization was initially demonstrated by Hsi et al. for the purification of benzamide and cinnamamide from structurally similar impurities.115 Most of the aforementioned mechanisms require impurity molecules to interact with the growing crystal so that they can be incorporated at the surface or lattice. In this context, Hsi et al. hypothesized that co-formers that can form a co-crystal with the impurities will also form a complex in solution, preventing those impurities from incorporating in the growing crystals. This work was a follow-up to prior efforts to isolate impurities by co-crystal formation, as a means to remove them from the crude solution prior to crystallizing the product of interest.116 The use of impurity complexation for control of impurity incorporation was further validated for amoxicillin trihydrate crystallization,117 showing a greater crystal purity than that obtained after two crystallizations in series without complexing agents.In 2017, this technology was also applied in continuous crystallization by Vartak and Myerson.118,119 To mitigate the common trade-off between crystallization yield and purity, their focus was to allow for solution recycling in systems with impurity complexation. Crystallization of the target molecule, filtration of the pure crystals, and nanofiltration for the removal of impurity complexes were all carried out in a continuous loop (Fig. 9). In this context, the complexing agents play two different roles: first, they prevent impurity incorporation in the crystal lattice by selectively binding with the impurities; then, by isolating the impurities in large complexes, they can be separated from the solvent and solute of interest by membrane filtration, allowing to recycle only the product of interest, and preventing loss of crystal purity due to an increasing impurity concentration in the mother liquor. This recycling approach can also be used without a complexing agent, provided that the impurities have a significantly different molecular size than the solute of interest.120
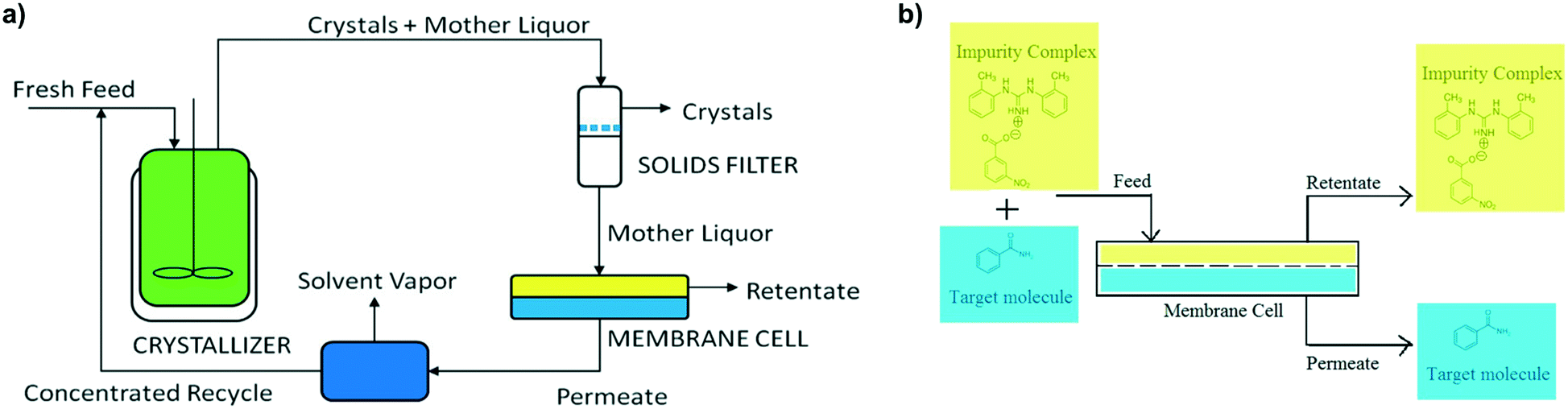 | ||
| Fig. 9 Prevention of impurity incorporation by complexation in continuous crystallizers with mother liquor recycling. a) Process diagram including continuous crystallization, solids filtration for recovery of solute crystals, nanofiltration for removal of impurity complexes, and permeate concentration prior to mother liquor recycle. b) Close-up of the nanofiltration unit, showing the retention of large impurity complexes and a permeating solution rich in the target solute molecule [adapted with permission from Vartak and Myerson.118,119 Copyright 2017 American Chemical Society]. | ||
Overall, one of the major advantages from this method is that knowledge of the impurity incorporation mechanism is not required, beyond ensuring that this mechanism is not driven by aggregation, solution entrapment, or adhering mother liquor (where the impurity incorporates as part of the solution and complexes could also be trapped). The main limitation is finding an optimal co-former for impurity complexation, that interacts with the impurities but not with the solute, and that does not incorporate in the crystal lattice.121 For known impurity systems, the Cambridge Crystallographic Data Centre (CCDC) can be used to find co-formers that would form co-crystals with the impurity, and use those as early candidates for screening.115 The measurement of solution interaction energies for potential candidates, and the use of computational models, can facilitate the selection of potential co-formers.121 However, for systems with large numbers of unknown impurities, screening for the right co-formers can take a significant amount of time and resources.
4.4 Slurry aging and temperature cycling
In those cases where prevention of impurity incorporation is not trivial or practical, an alternative to improve the purity of the final product is to implement strategies to selectively dissolve impurities post-crystallization. Some of these methods have been previously discussed, involving the choice of wash solvents to re-dissolve co-precipitating impurities.38,103 A similar method, but based on dissolving solid solutions, was used by Wang et al. for the purification of the salicylic acid crystals shown in Fig. 5.52 They suspended the impure crystals in pure water and applied mechanical agitation to promote crystal breakage and expose the impure crystal core to the wash solvent. This allowed for preferential dissolution of the impure core. Slurry aging of impure crystals in a wash solvent has been used for decades, as a means to utilize the dynamic equilibrium to remove an excess of kinetically incorporated impurities and to attain an equilibrium distribution coefficient.122–126 However, this method works best when impurities are kinetically incorporated, as the final crystal purity would be driven by thermodynamics.Temperature cycling can be used as a method to accelerate the purge of impurities in equilibrium suspensions. Instead of relying on Ostwald ripening in the dynamic dissolution-crystallization equilibrium,51 the dissolution of impure crystals and re-crystallization at low supersaturations is promoted by heating and cooling cycles, triggering small increases and drops in solubility for both the solute and the impurities. These cycles can also be implemented in supersaturated solutions during crystallization, to tackle several phenomena involved in impurity incorporation. For example, Simone et al. used temperature cycling to minimize agglomeration of piroxicam monohydrate.45 In a later contribution, this technique was used to enhance purity during crystallization of vitamin B12 using process analytical technologies (PATs) and feedback control.127 The same strategy was demonstrated for improving purity in systems presenting solution entrapment in both aggregates and single crystals.128 Presumably, the increase in solubility during the heating cycle aids in dissolving crystalline bridges within aggregates, and partially dissolves some of the formed crystals re-connecting the solution pockets with the bulk mother liquor. This was also observed by Kim and co-workers during the crystallization of cyclotrimethylene trinitramine with temperature cycling.129 As an alternative, one can use ultrasonication to break crystal aggregates and to expose entrapped solution pockets by crystal breakage.130,131
4.5 Process development workflows
Despite the wide variety of mechanisms and mechanism-dependent approaches to the design of crystallization processes for purification, some workflows do exist as systematic methodologies for the diagnosis and selection of impurity control strategies. Cheng et al. initially presented a workflow for the simultaneous development of crystallization and downstream strategies for control of crystal purity.132 The methods are based on an early screening for the understanding of impurity incorporation mechanisms, involving a solubility screening and development of phase diagrams to mitigate impurity co-crystal formation and co-precipitation. This is followed by an early precipitation screening using different solvents and crystallization conditions, and finalized with the optimization of wash solvents and deliquoring conditions.In 2020, Rogers et al. showed their crystallization development approach as part of the efforts for designing five end-to-end continuous manufacturing processes with strict time and material constraints.26 The work includes a downstream development workflow that suggests early development using purified material, and a gradual increase in the use of crude as the synthesis steps are optimized. The workflow is mostly based on solvent selection, first for crystallization and then for the washing steps.
In the same year, Urwin et al. presented a novel development workflow for diagnosis and control of impurity incorporation.44 Following the collection of preliminary knowledge, the workflow suggests conducting a set of crystallization experiments that will set the basis for development decisions. The isolated crystals are first examined for agglomerates, suggesting an anti-agglomeration strategy if this is the main mechanism for impurity retention. Then, a set of powder dissolution studies (Fig. 4) are suggested to determine if impurities are adsorbed at the surface, and to map the impurity distribution profile throughout the crystals. This can also be used to detect solvent entrapment. Finally, the workflow describes the identification of solid solutions utilizing binary phase diagrams.
In 2021, Ottoboni et al. published a different development workflow, primarily focused on solvent selection, but utilizing computational tools for the selection and ranking of crystallization and wash solvents.133 The focus is to maximize crystallization yield and purity while minimizing the overall solvent consumption. Of the aforementioned workflows, this arguably requires the smallest amount of representative crude. However, this comes at the expense of studying impurity incorporation mechanisms and the possibility of applying mechanism-specific control strategies.
5. Conclusions
Diagnosis of impurity incorporation mechanisms during solution crystallization is critical for the optimization of crystallization processes targeting high crystal purities. This is especially important in pharmaceutical processes, where products are subject to stringent quality standards, and where small amounts of certain impurities can have severe detrimental effects on the human body. Literature focused on the understanding and control of impurity incorporation is generally scarce in comparison with that focused on crystal size, habit, or polymorphism. However, in previous years, we have seen an increase in the number of crystallization papers focused on purification, development workflows, and novel approaches for the prevention of impurity incorporation. Despite these advances, there is still a gap in our understanding of impurity incorporation mechanisms, especially in modelling and predicting this impurity incorporation, and in the role of individual crystal faces on product purity. Because of this limited understanding, some of our powder-based methods for impurity detection may misdiagnose the dominant mechanism. This brings additional challenges when our prevention strategies are highly system-dependent, and when extensive screenings are not feasible within the typical time and material constraints in industrial development. This manuscript highlighted these recent efforts, in an attempt to connect new and existing process development scientists with the evolving state-of-the-art, and to facilitate the development of novel work that addresses the existing challenges in diagnosis, modelling, and prevention.Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621–629 CrossRef CAS PubMed.
- H. Lorenz and W. Beckmann, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 129–148 CAS.
- K. Plumb, Chem. Eng. Res. Des., 2005, 83, 730–738 CrossRef CAS.
- D. Zhang, S. Xu, S. Du, J. Wang and J. Gong, Engineering, 2017, 3, 354–364 CrossRef.
- Y. Ma, S. Wu, E. G. J. Macaringue, T. Zhang, J. Gong and J. Wang, Org. Process Res. Dev., 2020, 24, 1785–1801 CrossRef CAS.
- C. L. Burcham, A. J. Florence and M. D. Johnson, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 253–281 CrossRef PubMed.
- M. Fujiwara, Z. K. Nagy, J. W. Chew and R. D. Braatz, J. Process Control, 2005, 15(5), 493–504 CrossRef CAS.
- A. J. Alvarez and A. S. Myerson, Cryst. Growth Des., 2010, 10, 2219–2228 CrossRef CAS.
- J. Chen, B. Sarma, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2011, 11, 887–895 CrossRef CAS.
- G. Capellades, P. U. Joshi, K. Dam-Johansen, M. J. Mealy, T. V. Christensen and S. Kiil, Cryst. Growth Des., 2018, 18, 6455–6469 CrossRef CAS.
- Y. Yang, L. Song, Y. Zhang and Z. K. Nagy, Ind. Eng. Chem. Res., 2016, 55, 4987–4996 CrossRef CAS.
- T. C. Lai, S. Ferguson, L. Palmer, B. L. Trout and A. S. Myerson, Org. Process Res. Dev., 2014, 18, 1382–1390 CrossRef CAS.
- L. Nicoud, F. Licordari and A. S. Myerson, Org. Process Res. Dev., 2019, 23, 794–806 CrossRef CAS.
- L. Nicoud, F. Licordari and A. S. Myerson, CrystEngComm, 2019, 21, 2105–2118 RSC.
- S. Görög, TrAC, Trends Anal. Chem., 2006, 25, 755–757 CrossRef.
- Statement alerting patients and health care professionals of NDMA found in samples of ranitidine | FDA.
- Laboratory Tests | Ranitidine | FDA.
- G. Capellades, H. Wiemeyer and A. S. Myerson, Cryst. Growth Des., 2019, 19, 4008–4018 CrossRef CAS.
- C. A. Pons Siepermann and A. S. Myerson, Cryst. Growth Des., 2018, 18, 3584–3595 CrossRef CAS.
- C. A. Pons Siepermann, S. Huang and A. S. Myerson, Cryst. Growth Des., 2017, 17, 2646–2653 CrossRef CAS.
- N. Kubota, Cryst. Res. Technol., 2001, 36, 749–769 CrossRef CAS.
- Z. Berkovitch-Yellin, L. Addadi, M. Idelson, M. Lahav and L. Leiserowitz, Angew. Chem., Int. Ed. Engl., 1982, 21, 1336–1345 CrossRef.
- K. Sangwal and E. Mielniczek-Brzóska, Cryst. Res. Technol., 2003, 38, 113–124 CrossRef CAS.
- H. A. Moynihan and D. E. Horgan, Org. Process Res. Dev., 2017, 21, 689–704 CrossRef CAS.
- A. J. Maloney, E. Içten, G. Capellades, M. G. Beaver, X. Zhu, L. R. Graham, D. B. Brown, D. J. Griffin, R. Sangodkar, A. Allian, S. Huggins, R. Hart, P. Rolandi, S. D. Walker and R. D. Braatz, Org. Process Res. Dev., 2020, 24, 1891–1908 CrossRef CAS.
- L. Rogers, N. Briggs, R. Achermann, A. Adamo, M. Azad, D. Brancazio, G. Capellades, G. Hammersmith, T. Hart, J. Imbrogno, L. P. Kelly, G. Liang, C. Neurohr, K. Rapp, M. G. Russell, C. Salz, D. A. Thomas, L. Weimann, T. F. Jamison, A. S. Myerson and K. F. Jensen, Org. Process Res. Dev., 2020, 24, 2183–2196 CrossRef CAS.
- C. Armstrong, Y. Miyai, A. Formosa, D. Thomas, E. Chen, T. Hart, V. Schultz, B. K. Desai, A. Y. Cai, A. Almasy, K. Jensen, L. Rogers and T. Roper, Org. Process Res. Dev., 2021, 25, 1524–1533 CrossRef CAS.
- J. Li, T. C. Lai, B. L. Trout and A. S. Myerson, Cryst. Growth Des., 2017, 17, 1000–1007 CrossRef CAS.
- K. P. Cole, J. M. Groh, M. D. Johnson, C. L. Burcham, B. M. Campbell, W. D. Diseroad, M. R. Heller, J. R. Howell, N. J. Kallman, T. M. Koenig, S. A. May, R. D. Miller, D. Mitchell, D. P. Myers, S. S. Myers, J. L. Phillips, C. S. Polster, T. D. White, J. Cashman, D. Hurley, R. Moylan, P. Sheehan, R. D. Spencer, K. Desmond, P. Desmond and O. Gowran, Science, 2017, 356, 1144–1150 CrossRef CAS PubMed.
- S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef CAS PubMed.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead and T. Stelzer, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- P. Zhang, N. Weeranoppanant, D. A. Thomas, K. Tahara, T. Stelzer, M. G. Russell, M. O'Mahony, A. S. Myerson, H. Lin, L. P. Kelly, K. F. Jensen, T. F. Jamison, C. Dai, Y. Cui, N. Briggs, R. L. Beingessner and A. Adamo, Chem. – Eur. J., 2018, 24, 2776–2784 CrossRef CAS PubMed.
- E. Içten, A. J. Maloney, M. G. Beaver, D. E. Shen, X. Zhu, L. R. Graham, J. A. Robinson, S. Huggins, A. Allian, R. Hart, S. D. Walker, P. Rolandi and R. D. Braatz, Org. Process Res. Dev., 2020, 24, 1861–1875 CrossRef.
- E. Içten, A. J. Maloney, M. G. Beaver, X. Zhu, D. E. Shen, J. A. Robinson, A. T. Parsons, A. Allian, S. Huggins, R. Hart, P. Rolandi, S. D. Walker and R. D. Braatz, Org. Process Res. Dev., 2020, 24, 1876–1890 CrossRef.
- C. Hu, C. J. Testa, W. Wu, K. Shvedova, D. E. Shen, R. Sayin, B. S. Halkude, F. Casati, P. Hermant, A. Ramnath, S. C. Born, B. Takizawa, T. F. O'Connor, X. Yang, S. Ramanujam and S. Mascia, Chem. Commun., 2020, 56, 1026–1029 RSC.
- K. P. Cole, B. J. Reizman, M. Hess, J. M. Groh, M. E. Laurila, R. F. Cope, B. M. Campbell, M. B. Forst, J. L. Burt, T. D. Maloney, M. D. Johnson, D. Mitchell, C. S. Polster, A. W. Mitra, M. Boukerche, E. W. Conder, T. M. Braden, R. D. Miller, M. R. Heller, J. L. Phillips and J. R. Howell, Org. Process Res. Dev., 2019, 23, 858–869 CrossRef CAS.
- B. J. Reizman, K. P. Cole, M. Hess, J. L. Burt, T. D. Maloney, M. D. Johnson, M. E. Laurila, R. F. Cope, C. V. Luciani, J. Y. Buser, B. M. Campbell, M. B. Forst, D. Mitchell, T. M. Braden, C. K. Lippelt, M. Boukerche, D. R. Starkey, R. D. Miller, J. Chen, B. Sun, M. Kwok, X. Zhang, S. Tadayon and P. Huang, Org. Process Res. Dev., 2019, 23, 870–881 CrossRef CAS.
- G. Capellades, C. Neurohr, N. Briggs, K. Rapp, G. Hammersmith, D. Brancazio, B. Derksen and A. S. Myerson, Org. Process Res. Dev., 2021, 25, 1534–1546 CrossRef CAS.
- S. Ottoboni, C. J. Price, C. Steven, E. Meehan, A. Barton, P. Firth, A. Mitchell and F. Tahir, J. Pharm. Sci., 2019, 108, 372–381 CrossRef CAS PubMed.
- N. Estime, S. Teychené, J. M. Autret and B. Biscans, Powder Technol., 2011, 208, 337–342 CrossRef CAS.
- M. Müller, U. Meier, D. Wieckhusen, R. Beck, S. Pfeffer-Hennig and R. Schneeberger, Cryst. Growth Des., 2006, 6, 946–954 CrossRef.
- C. Darmali, S. Mansouri, N. Yazdanpanah and M. W. Woo, Ind. Eng. Chem. Res., 2019, 58, 1463–1479 CrossRef CAS.
- A. S. Myerson, D. Edemir and A. Y. Lee, Handbook of Industrial Crystallization, Cambridge University Press, Third Edit., 2019 Search PubMed.
- S. J. Urwin, G. Levilain, I. Marziano, J. M. Merritt, I. Houson and J. H. Ter Horst, Org. Process Res. Dev., 2020, 24, 1443–1456 CrossRef CAS PubMed.
- E. Simone, R. Othman, G. T. Vladisavljević and Z. K. Nagy, Pharmaceutics, 2018, 10(1), 17 CrossRef PubMed.
- E. M. Ålander, M. S. Uusi-Penttilä and Å. C. Rasmuson, Ind. Eng. Chem. Res., 2004, 43, 629–637 CrossRef.
- R. David, P. Marchal, J. P. Klein and J. Villermaux, Chem. Eng. Sci., 1991, 46, 205–213 CrossRef CAS.
- R. David, P. Marchal and B. Marcant, Chem. Eng. Technol., 1995, 18, 302–309 CrossRef CAS.
- J. W. Mullin, Crystallization, Butterworth-Heinemann, Fourth., 2001 Search PubMed.
- K. G. Denbigh and E. T. White, Nature, 1963, 199, 799–800 CrossRef CAS.
- A. S. Myerson, Handbook of Industrial Crystallization, Butterworth-Heinemann, 2nd edn, 2002 Search PubMed.
- Y. Wang, H. Li, M. Raikes, B. Linehan, J. Robson and F. L. Nordstrom, Cryst. Growth Des., 2021, 21, 4100–4110 CrossRef CAS.
- R. Teerakapibal, H. Li, B. Linehan and F. L. Nordstrom, Cryst. Growth Des., 2020, 20, 1716–1728 CrossRef CAS.
- B. Kahr and R. W. Gurney, Chem. Rev., 2001, 101, 893–951 CrossRef CAS PubMed.
- A. J. Florence, C. K. Leech, N. Shankland, K. Shankland and A. Johnston, CrystEngComm, 2006, 8, 746–747 RSC.
- A. J. Cruz Cabeza, G. M. Day, W. D. S. Motherwell and W. Jones, Chem. Commun., 2007, 1600–1602 RSC.
- F. P. A. Fabbiani, L. T. Byrne, J. J. McKinnon and M. A. Spackman, CrystEngComm, 2007, 9, 728–731 RSC.
- D. H. Case, V. K. Srirambhatla, R. Guo, R. E. Watson, L. S. Price, H. Polyzois, J. K. Cockcroft, A. J. Florence, D. A. Tocher and S. L. Price, Cryst. Growth Des., 2018, 18, 5322–5331 CrossRef CAS.
- A. Burel, N. Couvrat, S. Tisse, Y. Cartigny, P. Cardinael and G. Coquerel, Eur. Phys. J.: Spec. Top., 2017, 226, 869–880 CAS.
- D. E. Horgan, L. M. Crowley, S. P. Stokes, S. E. Lawrence and H. A. Moynihan, in Advanced Topics in Crystallization, ed. Y. Mastai, InTechOpen, 2015 Search PubMed.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CrossRef CAS PubMed.
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC.
- M. C. Etter, J. Phys. Chem., 1991, 95, 4601–4610 CrossRef CAS.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2015, 52, 640–655 RSC.
- M. Karimi-Jafari, L. Padrela, G. M. Walker and D. M. Croker, Cryst. Growth Des., 2018, 18, 6370–6387 CrossRef CAS.
- A. Karagianni, M. Malamatari and K. Kachrimanis, Pharmaceutics, 2018, 10(1), 18 CrossRef PubMed.
- A. Ranganathan, V. R. Pedireddi and C. N. R. Rao, J. Am. Chem. Soc., 1999, 121, 1752–1753 CrossRef CAS.
- J. D. Wright, Molecular crystals, Cambridge University Press, 2nd edn, 1995 Search PubMed.
- R. J. Davey, J. Cryst. Growth, 1976, 34, 109–119 CrossRef CAS.
- S. Dobberschütz, M. R. Nielsen, K. K. Sand, R. Civioc, N. Bovet, S. L. S. Stipp and M. P. Andersson, Nat. Commun., 2018, 9, 1–6 CrossRef PubMed.
- D. D. Patel and B. D. Anderson, J. Pharm. Sci., 2015, 104, 2923–2933 CrossRef CAS PubMed.
- M. Jiang, X. Zhu, M. C. Molaro, M. L. Rasche, H. Zhang, K. Chadwick, D. M. Raimondo, K. K. Kim, L. Zhou, Z. Zhu, M. H. Wong, D. O'Grady, D. Hebrault, J. Tedesco and R. D. Braatz, Ind. Eng. Chem. Res., 2014, 53, 5325–5336 CrossRef CAS.
- Y. H. Luo, G. G. Wu and B. W. Sun, J. Chem. Eng. Data, 2013, 58, 588–597 CrossRef CAS.
- J. Schöll, D. Bonalumi, L. Vicum, M. Mazzotti and M. Müller, Cryst. Growth Des., 2006, 6, 881–891 CrossRef.
- C. J. Brown, T. Mcglone, S. Yerdelen, V. Srirambhatla, F. Mabbott, R. Gurung, M. L. Briuglia, B. Ahmed, H. Polyzois, J. Mcginty, F. Perciballi, D. Fysikopoulos, P. MacFhionnghaile, H. Siddique, V. Raval, T. S. Harrington, A. D. Vassileiou, M. Robertson, E. Prasad, A. Johnston, B. Johnston, A. Nordon, J. S. Srai, G. Halbert, J. H. ter Horst, C. J. Price, C. D. Rielly, J. Sefcik and A. J. Florence, Mol. Syst. Des. Eng., 2018, 3, 518–549 RSC.
- B. O'Sullivan, P. Barrett, G. Hsiao, A. Carr and B. Glennon, Org. Process Res. Dev., 2003, 7, 977–982 CrossRef.
- H. Salami, M. A. Mcdonald, A. S. Bommarius, R. W. Rousseau and M. A. Grover, Org. Process Res. Dev., 2021, 25, 1670–1679 CrossRef CAS.
- A. Waldschmidt, V. Dupray, B. Berton, N. Couvrat, S. Petit and G. Coquerel, J. Cryst. Growth, 2012, 342, 72–79 CrossRef CAS.
- G. Shete, V. Puri, L. Kumar and A. K. Bansal, AAPS PharmSciTech, 2010, 11, 598–609 CrossRef CAS PubMed.
- M. Mirmehrabi, S. Rohani, K. S. K. Murthy and B. Radatus, Cryst. Growth Des., 2006, 6, 141–149 CrossRef CAS.
- M. Sun, X. Hu, X. Zhou and J. Gu, Powder Diffr., 2017, 32, 78–85 CrossRef CAS.
- M. Tiwari, G. Chawla and A. K. Bansal, J. Pharm. Biomed. Anal., 2007, 43, 865–872 CrossRef CAS PubMed.
- M. Sundaram, S. Natarajan, A. G. Dikundwar and H. Bhutani, Powder Diffr., 2020, 35, 226–232 CrossRef CAS.
- W. Beckmann and H. Lorenz, Chem. Eng. Technol., 2006, 29, 226–232 CrossRef CAS.
- S. Ishihara, Y. Hattori and M. Otsuka, Spectrochim. Acta, Part A, 2019, 221, 117142 CrossRef CAS PubMed.
- G. Descamps, Y. Cartigny, M. Sanselme, M. N. Petit, S. Petit, E. Aubin and G. Coquerel, Cryst. Growth Des., 2009, 9, 3910–3917 CrossRef CAS.
- S. P. Duddu and D. J. W. Grant, Thermochim. Acta, 1995, 248, 131–145 CrossRef CAS.
- M. S. Rabello and J. R. White, Polymer, 1997, 38, 6389–6399 CrossRef CAS.
- D. D. Caspi and F. L. Nordstrom, Org. Process Res. Dev., 2018, 22, 856–861 CrossRef CAS.
- A. B. Herhold, D. Ertaş, A. J. Levine and H. E. King, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 59, 6946–6955 CrossRef CAS PubMed.
- K. Sangwal, K. Wójcik and J. Borc, Cryst. Res. Technol., 2007, 42, 1243–1251 CrossRef CAS.
- K. Sangwal, Prog. Cryst. Growth Charact. Mater., 1996, 32, 3–43 CrossRef CAS.
- L. Addadi, S. Weinstein, E. Gati, I. Weissbuch and M. Lahav, J. Am. Chem. Soc., 1982, 104, 4610–4617 CrossRef CAS.
- F. L. Nordstrom, B. Linehan, R. Teerakapibal and H. Li, Cryst. Growth Des., 2019, 19(2), 1336–1346 CrossRef CAS.
- A. McPherson, A. J. Malkin and Y. G. Kuznetsov, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 361–410 CrossRef CAS PubMed.
- P. Pantaraks and A. E. Flood, Cryst. Growth Des., 2005, 5, 365–371 CrossRef CAS.
- A. G. Shtukenberg, L. N. Poloni, Z. Zhu, Z. An, M. Bhandari, P. Song, A. L. Rohl, B. Kahr and M. D. Ward, Cryst. Growth Des., 2015, 15, 921–934 CrossRef CAS.
- G. Capellades, A. Duso, K. Dam-Johansen, M. J. Mealy, T. V. Christensen and S. Kiil, Org. Process Res. Dev., 2019, 23, 252–262 CrossRef CAS.
- J. M. Schall, J. S. Mandur, R. D. Braatz and A. S. Myerson, Cryst. Growth Des., 2018, 18, 1560–1570 CrossRef CAS.
- G. Morris, G. Power, S. Ferguson, M. Barrett, G. Hou and B. Glennon, Org. Process Res. Dev., 2015, 19, 1891–1902 CrossRef CAS.
- T. Vetter, C. L. Burcham and M. F. Doherty, Chem. Eng. Sci., 2014, 106, 167–180 CrossRef CAS.
- J. M. Schall, G. Capellades, J. S. Mandur, R. D. Braatz and A. S. Myerson, Org. Process Res. Dev., 2019, 23, 1960–1969 CrossRef CAS.
- M. Shahid, G. Sanxaridou, S. Ottoboni, L. Lue and C. Price, Org. Process Res. Dev., 2021, 25, 969–981 CrossRef CAS PubMed.
- A. D. Randolph and M. A. Larson, Theory of particulate processes - Analysis and Techniques of Continuous Crystallization, Academic Press, Inc., 1971 Search PubMed.
- N. Bajcinca, S. Qamar, D. Flockerzi and K. Sundmacher, Chem. Eng. Sci., 2011, 66, 3711–3720 CrossRef CAS.
- C. T. Óciardhá, K. W. Hutton, N. A. Mitchell and P. J. Frawley, Cryst. Growth Des., 2012, 12, 5247–5261 CrossRef.
- P. J. Frawley, N. A. Mitchell, C. T. Ó'Ciardhá and K. W. Hutton, Chem. Eng. Sci., 2012, 75, 183–197 CrossRef CAS.
- J. Li, B. L. Trout and A. S. Myerson, Org. Process Res. Dev., 2016, 20, 510–516 CrossRef CAS.
- J. M. Schall, G. Capellades and A. S. Myerson, CrystEngComm, 2019, 21, 5811–5817 RSC.
- G. Lunardon Quilló, S. Bhonsale, B. Gielen, J. F. Van Impe, A. Collas and C. Xiouras, Cryst. Growth Des., 2021, 21, 5403–5420 CrossRef.
- J. L. Quon, H. Zhang, A. Alvarez, J. Evans, A. S. Myerson and B. L. Trout, Cryst. Growth Des., 2012, 12, 3036–3044 CrossRef CAS.
- S. Y. Wong, A. P. Tatusko, B. L. Trout and A. S. Myerson, Cryst. Growth Des., 2012, 12, 5701–5707 CrossRef CAS.
- H. Zhang, J. Quon, A. J. Alvarez, J. Evans, A. S. Myerson and B. Trout, Org. Process Res. Dev., 2012, 16, 915–924 CrossRef CAS.
- A. Borsos, A. Majumder and Z. K. Nagy, Cryst. Growth Des., 2016, 16, 555–568 CrossRef CAS.
- K. H. Hsi, M. Kenny, A. Simi and A. S. Myerson, Cryst. Growth Des., 2013, 13, 1577–1582 CrossRef CAS.
- K. H. Y. Hsi, K. Chadwick, A. Fried, M. Kenny and A. S. Myerson, CrystEngComm, 2012, 14, 2386–2388 RSC.
- K. H. Y. Hsi, A. J. Concepcion, M. Kenny, A. A. Magzoub and A. S. Myerson, CrystEngComm, 2013, 15, 6776–6781 RSC.
- S. Vartak and A. S. Myerson, Org. Process Res. Dev., 2017, 21, 253–261 CrossRef CAS.
- S. Vartak and A. S. Myerson, Cryst. Growth Des., 2017, 17, 5506–5516 CrossRef CAS.
- S. Ferguson, F. Ortner, J. Quon, L. Peeva, A. Livingston, B. L. Trout and A. S. Myerson, Cryst. Growth Des., 2014, 14, 617–627 CrossRef CAS.
- C. C. Weber, G. P. F. Wood, A. J. Kunov-Kruse, D. E. Nmagu, B. L. Trout and A. S. Myerson, Cryst. Growth Des., 2014, 14, 3649–3657 CrossRef CAS.
- I. M. Kolthoff and G. E. Noponen, J. Am. Chem. Soc., 1938, 60, 197–201 CrossRef CAS.
- S. Gaines and A. S. Myerson, AIChE Symp. Ser., 1982, 215, 42–45 Search PubMed.
- P. M. Brown and A. S. Myerson, AIChE J., 1989, 35, 1749–1752 CrossRef CAS.
- M. Saska and A. S. Myerson, AIChE J., 1987, 33, 848–852 CrossRef CAS.
- P. M. Brown, M. Marquering and A. S. Myerson, Ind. Eng. Chem. Res., 1990, 29, 2089–2093 CrossRef CAS.
- E. Simone, W. Zhang and Z. K. Nagy, Cryst. Growth Des., 2015, 15, 2908–2919 CrossRef CAS.
- A. N. Saleemi, G. Steele, N. I. Pedge, A. Freeman and Z. K. Nagy, Int. J. Pharm., 2012, 430, 56–64 CrossRef CAS PubMed.
- J. W. Kim, J. K. Kim, H. S. Kim and K. K. Koo, Org. Process Res. Dev., 2011, 15, 602–609 CrossRef CAS.
- T. T. H. Nguyen, A. Khan, L. M. Bruce, C. Forbes, R. L. O'Leary and C. J. Price, Crystals, 2017, 7(10), 294 CrossRef.
- C. Forbes, T. T. H. Nguyen, R. L. O'Leary and C. J. Price, Proc. Meet. Acoust., 2018, 32, 045017 Search PubMed.
- Y. S. Cheng, K. W. Lam, K. M. Ng and C. Wibowo, AIChE J., 2010, 56, 633–649 CAS.
- S. Ottoboni, B. Wareham, A. Vassileiou, M. Robertson, C. J. Brown, B. Johnston and C. J. Price, Org. Process Res. Dev., 2021, 25, 1143–1159 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |