
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A reversible pressure-induced bond rearrangement of flexible lanthanide 2,5-bis(allyloxy)terephthalate coordination polymer networks†
L. R.
Hunter
 *a,
J. D.
Sellars
bc and
M. R.
Probert
*a
*a,
J. D.
Sellars
bc and
M. R.
Probert
*a
aChemistry, School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: l.hunter6@newcastle.ac.uk; michael.probert@newcastle.ac.uk
bBiosciences Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne NE2 4HH, UK
cSchool of Pharmacy, Faculty of Medical Sciences, King George VI Building, Newcastle upon Tyne NE1 7RU, UK
First published on 10th November 2022
Abstract
Flexible coordination polymer networks reveal a plethora of structural transformations when exposed to high pressure as an external stimulus. Transformations are driven through organic linkers in a variety of ways, such as bond rotations, rearrangements and conformational shifts. We have successfully synthesised two sets of isostructural lanthanide coordination polymer networks that differ as a consequence of the lanthanide contraction. We have demonstrated that a pressure-induced reversible phase transition is possible between the two structurally distinct sets of networks. The system demonstrates a bond rearrangement due to an alteration of carboxylic acid binding modes from the 2,5-bis(allyloxy)terephthalate linker in addition to bond rotations within the flexible carbon backbone of the linker between 30–35 kbar.
1. Introduction
Coordination polymer networks (CPs) and metal–organic frameworks (MOFs) were originally sought for their structural stability and rigidity. However, more recently the focus has been to engineer dynamic ‘sponge-like’, porous, frameworks that offer greater opportunity for unique structural transformations. These dynamic frameworks allow transformations that lead to interesting and unique applications, such as enhanced proton conductivity,1 plastically flexible crystals,2 controlled drug delivery,3 increased gas storage capabilities4 and uses as mechanical shock absorbers.5,6Several external stimuli, such as light,7 heat8 and pressure9–12 can be utilised to create conditions to manipulate the dynamic nature of these frameworks. The study that first brought attention to flexible MOFs centred on MIL-53, a chromium-based MOF, termed the ‘breathing framework’. Initially, it was recognised that MIL-53's diamond shaped pores could reversibly shrink by 5 Å in length in response to guest absorption and removal of H2O.13,14 Nevertheless, it was later realised that the use of pressure up to 10 kbar is another route to probe this transition.15,16
To explore potential structural transformations further, the incorporation of flexible organic linkers in CPs and MOFs allows a greater variety of physical responses to be investigated under extreme conditions. Networks can respond with familiar movements under high pressure conditions, including unit cell contractions,17 bond orientation transformations, and negative linear compressibility.18–20 More notable changes include solid state amorphisation,21 phase transitions22 and bond connectivity changes.23 Nevertheless, a transformation involving bond connectivity transitions/rearrangements under extreme pressure conditions are rare, with reversibility of this even more scarce.24 Herein, we report the synthesis of seven new lanthanide-based coordination polymer networks, that exhibit a distinct structural change in line with the lanthanide contraction (Fig. 1). The variation in global structure between sets is a repercussion of the decrease in coordination environment from 9 coordinate to 8 coordinate across the lanthanide series. Moreover, early lanthanide polymer networks undergo an interesting bond connectivity change at 30–35 kbar, whereby the carboxylic acid binding mode switches to that seen in lanthanide polymer networks later in the series, whilst retaining crystallinity, i.e. the pressure increase mimics the same structural change as the lanthanide contraction effect.
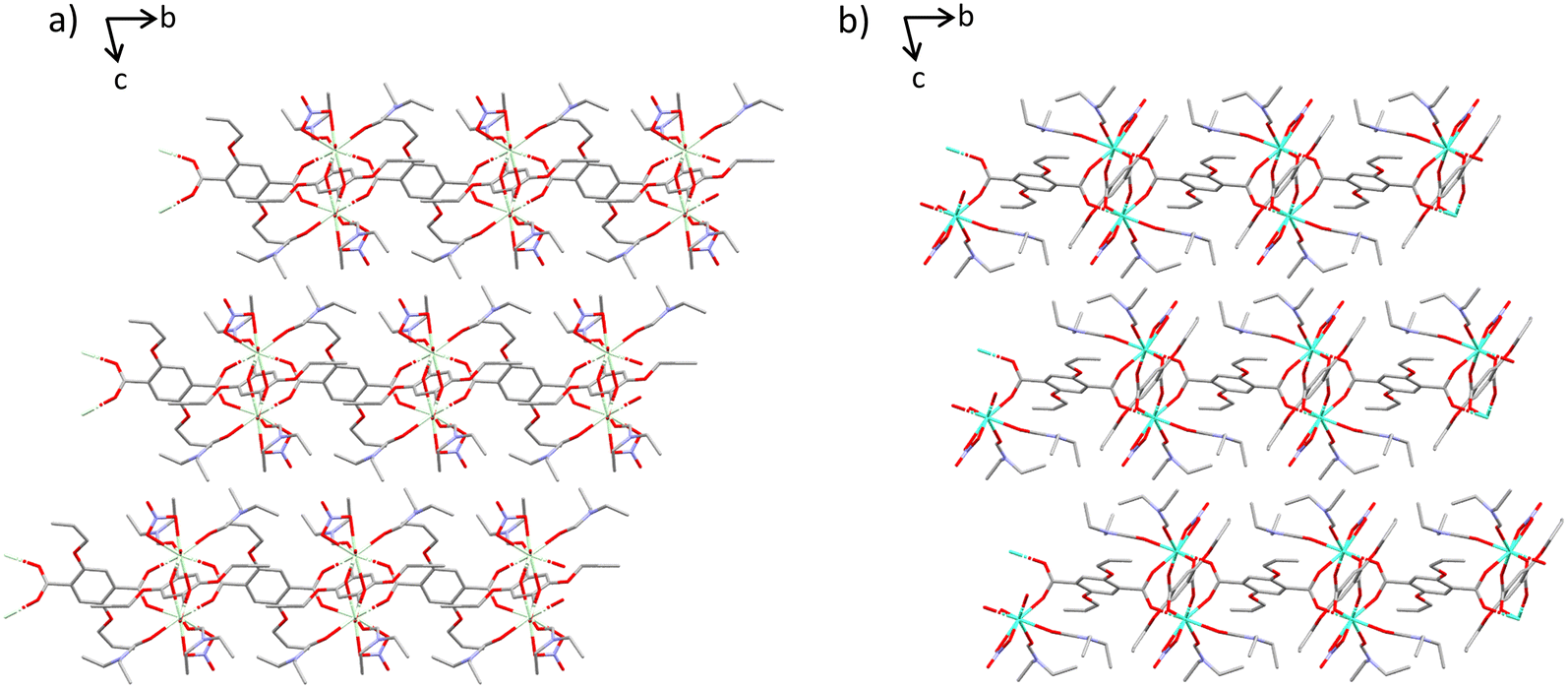 | ||
| Fig. 1 a.) Complexes 1–3 viewed down the a-axis showing the 2D layered nature of the coordination polymers, linked by dimeric subunits; b.) Complexes 4–7 viewed down the a-axis. These representations show the similarities between the two sets of polymers in terms of both the local and global structure. Hydrogen atoms are omitted for clarity. Ln metal – cream/green, C – grey, O – red, N – blue. | ||
2. Crystal structure analysis
The lanthanide coordination polymer networks synthesised are all 2D non interpenetrating dense frameworks and all frameworks crystallise in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. There are two subsets of network crystallised via the same conditions, this is hypothesised as a direct consequence of the lanthanide contraction (Fig. 1). Early metals in the series Ce–Nd, complexes 1–3, possess a coordination number of nine, whereas metals later in the lanthanide series Eu–Dy, complexes 4–7, have a coordination number of eight. This trend follows the decreasing ionic radius across the lanthanide series. Complexes 1–3 are isostructural, as are complexes 4–7. All seven complexes consist of a dimeric subunit containing four linkers, two nitrate ions and four diethyl formamide solvent molecules. The difference in unit cell parameters, coordination number and consequently the secondary building unit structure between the two sets of networks arises from the bridging mode of the carboxylate-based linkers. Complexes 1–3 contain dimeric subunits with two distinct carboxylate binding modes. Two linkers bridge the metal centres via the commonly seen bidentate bridge μ2–η2, whilst the other two linkers bridge the metal centres via an μ2–η1, η2 mode. In complexes 4–7 all four linkers bridge the two lanthanide metals only in an μ2–η2 mode. This gives rise to a distorted paddle wheel like geometry (Fig. 2c and d), commonly seen in Cu(II) MOF chemistry.25
space group. There are two subsets of network crystallised via the same conditions, this is hypothesised as a direct consequence of the lanthanide contraction (Fig. 1). Early metals in the series Ce–Nd, complexes 1–3, possess a coordination number of nine, whereas metals later in the lanthanide series Eu–Dy, complexes 4–7, have a coordination number of eight. This trend follows the decreasing ionic radius across the lanthanide series. Complexes 1–3 are isostructural, as are complexes 4–7. All seven complexes consist of a dimeric subunit containing four linkers, two nitrate ions and four diethyl formamide solvent molecules. The difference in unit cell parameters, coordination number and consequently the secondary building unit structure between the two sets of networks arises from the bridging mode of the carboxylate-based linkers. Complexes 1–3 contain dimeric subunits with two distinct carboxylate binding modes. Two linkers bridge the metal centres via the commonly seen bidentate bridge μ2–η2, whilst the other two linkers bridge the metal centres via an μ2–η1, η2 mode. In complexes 4–7 all four linkers bridge the two lanthanide metals only in an μ2–η2 mode. This gives rise to a distorted paddle wheel like geometry (Fig. 2c and d), commonly seen in Cu(II) MOF chemistry.25
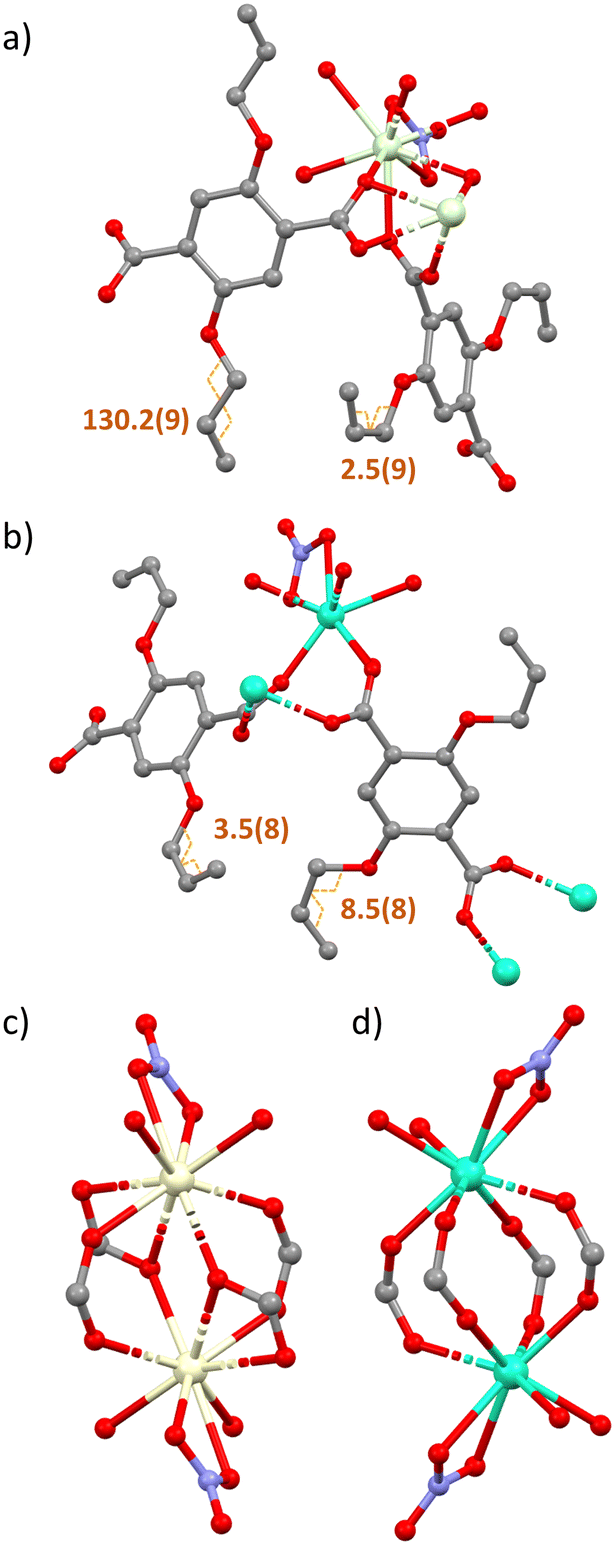 | ||
| Fig. 2 A closer inspection of the molecular subunits and secondary building units of complexes 1–3 (a and c) and 4–7 (b and d). Complexes 1–3 incorporate eclipsed and staggered allyloxy groups, whereas complexes 4–7 contain only the eclipsed orientation. Orange labels in figures a) and b) denote the allyloxy torsion angles in degrees. Images c and d take a closer look at the secondary building units, clearly showing the different binding modes in each isostructural set. Hydrogen atoms are omitted for clarity. Ln metal – cream/green, C – grey, O – red, N – blue. | ||
Once grown, both sets of structures create a dense sheet that lies in the ab plane. There are no solvent accessible voids dispersed throughout the structures with all networks occupying approximately 65% of the unit cell volume. The largest spherical void radius within networks 1–3 is 1.2 Å, whereas for networks 4–7 it is 1.0 Å (Fig. S2†). One more notable difference between complexes 1–3 and 4–7 is the rotameric form of the extended allyloxy chains on the linkers. Complexes 1–3 have two separate orientations of carbon chain within the same polymer network. When viewing down the C–O bond that extends from the aromatic centroid, you can see the torsion angle of the chain sits in both an eclipsed and staggered positioning within the same polymer (Fig. 2b). The eclipsed torsion angle sits at 2.5°(9) and the staggered torsion angle sits at 130.2°(9). In complexes 4–7 only the eclipsed orientation of the linker exists.
3. High pressure experiments
To study the dynamic nature of the complexes 1–7, high pressure studies were conducted, with a general procedure outlined below. A crystal was placed in a diamond anvil cell (DAC) containing a pentane/isopentane 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with diamond culets of 0.8 mm. The crystal occupies a sample chamber created by a stainless-steel gasket of 0.25 mm thickness, pre-indented to 0.15 mm with a precision drilled hole of 300 μm. A spherical ruby contained within the sample chamber facilitates pressure determination utilising fluorescence measurements.26 To begin, the pressure was increased in 5 kbar increments until there was a notable change in unit cell parameters. The DAC was mounted directly onto the goniometer of XIPHOS II, a four circle Huber diffractometer with Ag–Kα IμS generator. High pressure data were collected and processed using the Bruker APEX software suites, which incorporates SAINT and SADABS for integration, cell refinement, and scaling.27 The SHELX program suite was used for structure solution and refinement of all structures within the OLEX2 interface.28 The data completeness of structures measured at high pressure is severely reduced due to the constraints placed on data collection when using a diamond anvil cell, for information see relevant CIF files.
:1 mixture with diamond culets of 0.8 mm. The crystal occupies a sample chamber created by a stainless-steel gasket of 0.25 mm thickness, pre-indented to 0.15 mm with a precision drilled hole of 300 μm. A spherical ruby contained within the sample chamber facilitates pressure determination utilising fluorescence measurements.26 To begin, the pressure was increased in 5 kbar increments until there was a notable change in unit cell parameters. The DAC was mounted directly onto the goniometer of XIPHOS II, a four circle Huber diffractometer with Ag–Kα IμS generator. High pressure data were collected and processed using the Bruker APEX software suites, which incorporates SAINT and SADABS for integration, cell refinement, and scaling.27 The SHELX program suite was used for structure solution and refinement of all structures within the OLEX2 interface.28 The data completeness of structures measured at high pressure is severely reduced due to the constraints placed on data collection when using a diamond anvil cell, for information see relevant CIF files.
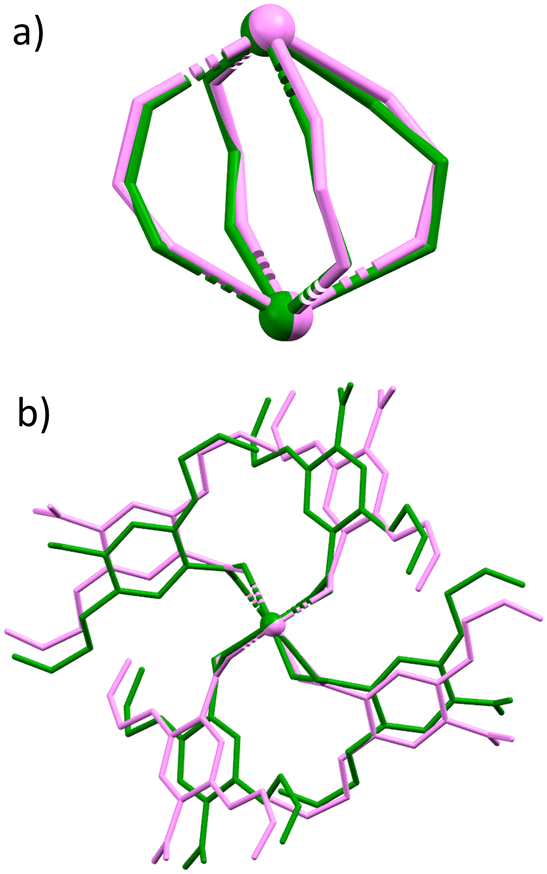
A crystal of complex 2 was placed in the diamond anvil cell and was subjected to pressure increases of 5 kbar increments up to 35 kbar. Between 30 and 35 kbar a notable, first order, phase transition was observed (Fig. S1†). At this pressure, the unit cell axis lengths stay relatively constant whilst the angles show a significant shift compared to the ambient pressure structure. Parameters alter from α = 109.416(3)°, β = 95.138(3)°, γ = 95.947(3)°, to α = 98.203(4)°, β = 111.376(4)°, γ = 91.2570(2)° in addition to a considerable decrease in unit cell volume from 1420.2(2) Å3 to 1208.43(11) Å3. This displacement in cell parameters is accommodated in several ways. Most interestingly, the coordination number of the Praseodymium metal centre decreases by one as a Pr–O bond is broken. This is facilitated by the binding mode of two carboxylate ligands changing from μ2–η1, η2 to μ2–η2 (Fig. 3a and b). Consequently, the secondary building unit (SBU) opens and the metal–metal distance increases from 4.112(4) Å to 4.329(2) Å. Alongside these significant structural changes, when the complex is viewed down the axis eclipsing the two metal centres a clockwise rotation is observed with the four linkers, that when combined with the increase in metal–metal centre distance imitates a corkscrew-like motion (Fig. 3c). Notwithstanding this, further adaptations can be observed, with the alkene ‘arms’ at atmospheric pressure occupying both an eclipsed and staggered geometry whilst at higher pressures they transform to an exclusively eclipsed orientation. All these structural transformations, once coupled together, convert the unit cell parameters of complex 2, to mirror those observed in complexes 4–7.
 | ||
| Fig. 3 Overlaid views of complex 2 at atmospheric pressure (orange) and at 35 kbar (purple). a) and b) Show two different orientations of the overlaid secondary building units, whereby you can visualise the change in carboxylic acid binding mode. c.) Shows a view along eclipsing metal centres, where you can see the anti-clockwise rotation of linkers under high pressure. | ||
Whilst these changes are notable, we wanted to understand the potential reversibility of this process. To achieve this, the pressure within the diamond anvil cell was reduced in increments of 5 kbar. At 25 kbar the unit cell parameters were seen to rebound to near their former values of α = 109.464(3)°, β = 94.951(3)°, γ = 96.1010(3)° whilst the volume returned to 1458.24(11) Å3. Upon return to atmospheric pressure and performing a full data collection, we see clearly the reversion of the SBU, with reinstatement of the coordination number of Pr back to nine and the binding mode of the two carboxylic acid linkers alters to μ2–η1, η2 and the extra M–O bond is reformed, all whilst retaining crystallinity throughout these measurements. As a result, this system represents a fully reversible single crystal to single crystal transition when exposed to external pressure manipulation. Further images of this transformation can be visualised in Fig. S4 of the ESI.†
To understand how complexes 4–7 behaved under high pressure conditions, a crystal of complex 6 was placed in a diamond anvil cell using the same experimental methods. Pressure was applied to the DAC in 5 kbar increments up to 25 kbar. When applying a pressure of 5 kbar we see an initial compression of the unit cell, resulting in a volume decrease of ∼185 Å3 (Fig. S3†). The volume of the unit cell is now comparable to complex 2 at 35 kbar. When applying further pressure up to 25 kbar, unit cell parameters stay relatively constant within the margin of error reported, no significant transition was observed. By 25 kbar axis lengths decrease by less than 5.5% in all directions with compression at its largest along the a-axis from 10.4991(13) Å to 9.8113(6) Å. When overlaying the SBU's measured at atmospheric pressure and 25 kbar it is apparent that the changes observed are negligible with a minor displacement in the orientation of linkers in each of the dimeric subunits when viewing down the eclipsed metal centres (Fig. 4b).
 | ||
| Fig. 4 Overlaid views of complex 6 at atmospheric pressure (green) and at 25 kbar (pink). a.) Shows the overlaid secondary building units. b.) Shows a view along eclipsing metal centres. The image shows the distortion of the network units, whereby you can observe the tilt angle changes for all four linkers. | ||
Conclusion
We have successfully synthesized seven new coordination polymers using the 2,5-bis(allyloxy)terephthalate linker and lanthanide nitrate salts. These CPs can be separated into two sets of isostructural networks which are dependent upon the size of the metal ion incorporated. When placing the CP's containing larger lanthanide ions under pressures up to 35 kbar, the unit cell parameters alter to mimic the structures already synthesised using the smaller lanthanide ions. This change is facilitated in two ways. Firstly, a bond rearrangement from a μ2–η1, η2 to a μ2–η2 carboxylic acid binding mode, and also bond rotations of the flexible ‘arm’ from a staggered to eclipsed torsion. The noted first order phase transition is reversible and networks can return to their former unit cell parameters at 25 kbar.Associated content
Supporting information contains the synthetic route to 2,5-bis(allyloxy)terephthalate, IR, crystallographic information file data, void space plots, and high pressure data.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors wish to thank Newcastle University's School of Natural and Environmental Sciences for funding this body of research.Notes and references
- D. Umeyama, S. Horike, C. Tassel, H. Kageyama, Y. Higo, K. Hagi, N. Ogiwara and S. Kitagawa, APL Mater., 2014, 2, 124401–124405 CrossRef.
- X. Liu, A. A. L. Michalchuk, B. Bhattacharya, N. Yasuda, F. Emmerling and C. R. Pulham, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- K. Jiang, L. Zhang, Q. Hu, D. Zhao, T. Xia, W. Lin, Y. Yang, Y. Cui, Y. Yang and G. Qian, J. Mater. Chem. B, 2016, 4, 6398–6401 RSC.
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006–100045 CrossRef.
- P. G. Yot, Z. Boudene, J. Macia, D. Granier, L. Vanduyfhuys, T. Verstraelen, V. Van Speybroeck, T. Devic, C. Serre, G. Férey, N. Stock and G. Maurin, Chem. Commun., 2014, 50, 9462–9464 RSC.
- X. Zhou, Y. Miao, K. S. Suslick and D. D. Dlott, Acc. Chem. Res., 2020, 53, 2806–2815 CrossRef CAS PubMed.
- I. M. Walton, J. M. Cox, C. A. Benson, D. G. Patel, Y. S. Chen and J. B. Benedict, New J. Chem., 2016, 40, 101–106 RSC.
- A. Karmakar, P. G. M. Mileo, I. Bok, S. B. Peh, J. Zhang, H. Yuan, G. Maurin and D. Zhao, Angew. Chem., Int. Ed., 2020, 132, 11096–11102 CrossRef.
- L. Bolinois, T. Kundu, X. Wang, Y. Wang, Z. Hu, K. Koh and D. Zhao, Chem. Commun., 2017, 53, 8118–8121 RSC.
- I. E. Collings and A. L. Goodwin, J. Appl. Phys., 2019, 126, 181101–181113 CrossRef.
- P. Zhao, T. D. Bennett, N. P. M. Casati, G. I. Lampronti, S. A. Moggach and S. A. T. Redfern, Dalton Trans., 2015, 44, 4498–4503 RSC.
- W. Cai, A. Gładysiak, M. Anioła, V. J. Smith, L. J. Barbour and A. Katrusiak, J. Am. Chem. Soc., 2015, 137, 9296–9301 CrossRef CAS PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- A. V. Neimark, F. X. Coudert, C. Triguero, A. Boutin, A. H. Fuchs, I. Beurroies and R. Denoyel, Langmuir, 2011, 27, 4734–4741 CrossRef CAS PubMed.
- (a) L. B. I. Beurroies, M. Boulhout, P. L. Llewellyn, B. Kuchta, G. Férey, C. Serre and R. Denoyel, Angew. Chem., Int. Ed., 2010, 49, 7526–7529 CrossRef PubMed; (b) P. Serra-Crespo, A. Dikhtiarenko, E. Stavitski, J. Juan-Alcañiz, F. Kapteijn, F. X. Coudert and J. Gascon, CrystEngComm, 2015, 17, 276–280 RSC.
- M. Wahiduzzaman, N. Reimer, J. P. Itié, N. Stock, G. Maurin and P. G. Yot, Polyhedron, 2018, 155, 144–148 CrossRef CAS.
- P. G. Yot, Q. Ma, J. Haines, Q. Yang, A. Ghoufi, T. Devic, C. Serre, V. Dmitriev, G. Férey, C. Zhong and G. Maurin, Chem. Sci., 2012, 3, 1100 RSC.
- A. Sussardi, C. L. Hobday, R. J. Marshall, R. S. Forgan, A. C. Jones and S. A. Moggach, Angew. Chem., Int. Ed., 2020, 59, 8118–8122 CrossRef CAS PubMed.
- W. Li, M. R. Probert, M. Kosa, T. D. Bennett, A. Thirumurugan, R. P. Burwood, M. Parinello, J. A. K. Howard and A. K. Cheetham, J. Am. Chem. Soc., 2012, 134, 11940–11943 CrossRef CAS PubMed.
- A. B. Cairns, J. Catafesta, C. Levelut, J. Rouquette, A. Van Der Lee, L. Peters, A. L. Thompson, V. Dmitriev, J. Haines and A. L. Goodwin, Nat. Mater., 2013, 12, 212–216 CrossRef CAS PubMed.
- K. W. Chapman, D. F. Sava, G. J. Halder, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 18583–18585 CrossRef CAS PubMed.
- M. Zhou, K. Wang, Z. Men, C. Sun, Z. Li, B. Liu, G. Zou and B. Zou, CrystEngComm, 2014, 16, 4084–4087 RSC.
- J. K. Clegg, A. J. Brock, K. A. Jolliffe, L. F. Lindoy, S. Parsons, P. A. Tasker and F. J. White, Chem. – Eur. J., 2017, 23, 12480–12483 CrossRef CAS PubMed.
- E. C. Spencer, M. S. R. N. Kiran, W. Li, U. Ramamurty, N. L. Ross and A. K. Cheetham, Angew. Chem., Int. Ed., 2014, 53, 5583–5586 CrossRef CAS PubMed.
- Y. Chen, B. Wang, X. Wang, L. H. Xie, J. Li, Y. Xie and J. R. Li, ACS Appl. Mater. Interfaces, 2017, 9, 27027–27035 CrossRef CAS PubMed.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 CrossRef CAS.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2165369, 2165370, 2165373, 2165377, 2165388, 2165389, 2165394, 2165395, 2165397, 2165431, 2214554 and 2214557. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00499b |
| This journal is © The Royal Society of Chemistry 2022 |