
The crystal structure, thermal expansion and far-IR spectrum of propanal (CH3CH2CHO) determined using powder X-ray diffraction, neutron scattering, periodic DFT and synchrotron techniques†
Helen E.
Maynard-Casely
 *a,
Nikita S.
Yevstigneyev
b,
Samuel G.
Duyker
c and
Courtney
Ennis
b
*a,
Nikita S.
Yevstigneyev
b,
Samuel G.
Duyker
c and
Courtney
Ennis
b
aAustralian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia. E-mail: helen.maynard-casely@ansto.gov.au
bDepartment of Chemistry, University of Otago, Dunedin, 9054, New Zealand
cSydney Analytical, Core Research Facilities, The University of Sydney, Sydney NSW, 2006, Australia
First published on 6th December 2021
Abstract
The crystal structure of propanal has been determined using powder X-ray diffraction (PXRD), where this common laboratory aldehyde is measured to crystallise in spacegroup P21/a, Z = 4 with a unit cell a = 8.9833(6) Å, b = 4.2237(2) Å, c = 9.4733(6) Å and β = 97.508(6)°, resulting in a volume of 356.37(4) Å3 at 100 K and atmospheric pressure. The thermal expansion observed from 100 K until the sample melted (∼164 K) was found to be anisotropic. An additional neutron diffraction study was carried out, reaching a temperature of 3 K and found no further phase transformations from the determined structure at lower temperatures. The investigated temperature regime correlates to astronomical surfaces, including outer Solar System bodies and interstellar dust mantles, where propanal is thought to be generated by energetic processing of composite molecular ices. Results from the structure determination were applied to model propanal ice using periodic density functional theory for the calculation of intermolecular frequencies, where the simulated far-infrared spectrum of solid propanal can now be used for future molecular astronomy.
Introduction
Propanal (propionaldehyde, CH3CH2CHO) is a molecule of astronomical interest having been located in interstellar sources such as the star-forming region Sagittarius B2(N)1 and low-mass protostar IRAS 16293–2422,2 as confirmed by ground-based telescopes through rotational transitions. A saturated complex organic molecule (COM), propanal is expected to be formed on interstellar grain surfaces3 before its eventual release to the gas-phase under conditions approaching its vacuum sublimation temperature of 120–150 K.4 In regards to Solar System sources, the Rosetta mission revealed propanal emanating from the comet 67P/Churyumov-Gerasimenko, as detected by on-board mass spectrometers.5 Here, the combined abundance of propanal and its structural isomer acetone was derived as 4.7 ± 2.4 × 10−5 relative to water.6To account for the observed abundance of aldehydes in such sources, the chemical evolution of icy grain mantles and planetary surfaces has been explored in laboratory experiments simulating a range of astrophysical environments. Mechanisms of propanal formation previously investigated include (i) the reduction of carbon monoxide dominant ices by hydrogenation processes,7,8 and (ii) the oxidation of hydrocarbon9 and alcohol10 ices. Here, both pathways depend on the initial molecular composition and a supply of hydrogen and oxygen atoms to facilitate non-equilibrium chemistry in the solid-state. Such substituted aldehydes (R-CHO) are essential precursors towards α-amino acids via Strecker synthesis,11 with the aldehyde R-group retained in the synthesis pathway. Therefore, the presence of propanal, ammonia, and hydrogen cyanide in a composite ice environment could lead to the formation of 2-aminobutyric acid (2ABA) – a non-proteinogenic amino acid – upon hydrolysis of an aminonitrile intermediate. Organic acids like 2ABA are central to origin-of-life chemistry, participating in reactions of biological importance. Indeed, both propanal12 and its 2ABA Strecker product have been detected in samples of the Murchison meteorite13,14 to support the theory of exogenous delivery of biomolecular building-blocks to the early-Earth.
To accompany rotational (Earth-based telescopes)15 and mass spectrometric (spacecraft) detection of astromolecules in the gas-phase, vibrational spectroscopy is commonly utilised for the exploration of outer Solar System surfaces using infrared (IR) wavelengths. However, to date propanal has evaded detection by IR techniques. The non-detection is likely due (i) to its trace abundance expected within H2O dominant ice and (ii) that its mid-IR spectrum (and related optical properties) have only recently been compiled under planetary surface conditions.4 The identification of aldehydes could also be pursued in the lower frequency far-IR region, as successfully deployed by the Cassini-CIRS instrument during its search for hydrocarbons16 and nitriles17 in the Titan atmosphere. However, assignment of the solid propanal far-IR spectrum has not yet been performed, meaning the character of intermolecular lattice bands remain unknown for wavelengths below 200 cm−1.18,19 Assignment of these features has been hampered by the lack of fundamental data for this common laboratory reagent; including the CH3CH2CHO crystal structure being absent in the literature.
Determining the structure for a molecular crystal allows for the calculation of external lattice (translation and libration) frequencies using periodic density functional theory (p-DFT) approaches, which can be subsequently used to simulate a far-IR spectrum. By elucidating intermolecular bonding preferences, such as the formation of dimer moieties, their influence on solid-state reactions can be rationalised. Although the accumulation of pure CH3CH2CHO crystals in planetary and interstellar environments is not expected due to its low abundance, a more complete description of propanal's intermolecular bonding and thermal behaviour will build understanding of its interactions within a water-dominant ice. These interactions could lead to the formation of hydrates,20 or co-crystal materials with CO, CO2, and hydrocarbons in mixed-phase ices.21,22
To plug this gap in fundamental physical data available for the propanal molecule, in this paper the crystal structure of pure CH3CH2CHO ice has been determined. Thermal expansion of this phase was then examined over a wide range of temperatures: from 3 K until melting. The resolved structure was then used to calculate and compile vibrational frequencies, which are then applied to assign the far-IR spectrum of propanal ice films collected at the Australian Synchrotron. As an additional test for the predictive abilities of our DFT protocol, the laboratory propanal diffraction patterns and the IR spectra are compared to several theoretical structures of solid propanal; constructed from the known (modified) molecular crystals of (i) propionitrile (CH3CH2CN), (ii) propanoic acid (CH3CH2COOH), (iii) N-methyl formamide (CH3NHCHO) and (iv) acetone (CH3COCH3). With the physical conditions of our experiments overlapping with those associated with planetary and interstellar environments, it is anticipated these data will contribute to the detection of propanal deposits residing on icy surfaces in planetary and interstellar environments, during future spacecraft and telescope surveys.
Experimental
Diffraction measurements
Powder diffraction analysis was undertaken both with laboratory X-ray diffraction and neutron diffraction facilities under atmospheric pressure conditions. Powder X-ray diffraction (PXRD) patterns were measured on a STOE STADI P diffractometer operating in Debye–Scherrer geometry with Mo Kα radiation and three Mythen detectors in a stationary mode, covering a 2θ range of 0–55°. The sample was contained in a thin-walled glass (WJM-Glas) capillaries that were rotated during the measurements. The sample was syringed with a smaller diameter capillary into the glass capillaries, along with some strands of silica wool to promote crystallisation. The capillary was then flame-sealed prior to being placed onto the instrument. Temperature control was achieved using an Oxford Cryosystems nitrogen cryostream. The sample was cooled from room temperature at a rate of 6 K min−1 with diffraction measurements recorded simultaneously for 2 minutes per pattern to monitor for crystallisation. Once the sample had crystallised, cooling continued to 100 K. Once at 100 K, a longer pattern was collected for structure solution attempts (10 minutes) before the sample was warmed at a rate of 1 K min−1 with data being collected every 2 minutes to give the thermodiffractogram presented in Fig. 1.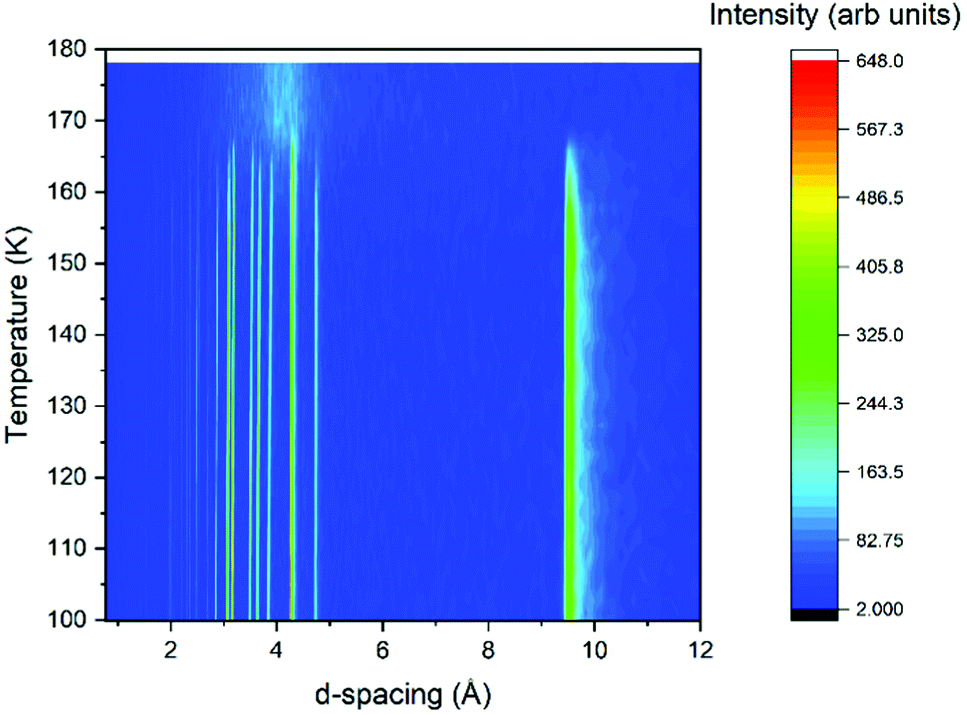 | ||
| Fig. 1 Thermodiffractogram of variable temperature X-ray powder diffraction data collected on the propanal sample. | ||
Neutron powder diffraction experiments were performed on the Wombat23 high intensity diffractometer with a neutron wavelength of 2.41 Å. Liquid propanal (the hydrogenous form was used despite the additional background from incoherent scattering from the hydrogen atoms) was loaded into a 9 mm diameter vanadium can and affixed within a C-bracket of copper on a steel sample stick, with the whole assembly placed within an Orange Cryostats cryofurnace.24
Calculations
Quantum calculations for crystalline propanal were performed by p-DFT using the CRYSTAL17 (C17) program.25 C17 input files require an initial geometry to describe the repeating unit cell, where lattice parameters, space group symmetry, and fractional atomic coordinates were obtained from the structure refinement. C17 code has been successfully applied to optimise the molecular structure and compile vibrational frequencies at an accuracy sufficient to assign far-IR intermolecular lattice modes associated with molecular crystals of astrophysical interest.26,27 Consistent with these studies, the B3LYP hybrid functional with user-defined triple-ζ 6-311G(d) basis sets28 were applied for the description of the electronic structure of the repeating propanal unit cell, where long-range interactions were corrected by implementing D3 dispersion terms.29Prior to p-DFT modelling of crystalline propanal from the confirmed experimental structures, theoretical propanal crystal structures were derived from the unit cells published for solid propionitrile (CH3CH2CN),30 propanoic acid (CH3CH2COOH),31N-methyl formamide (CH3NHCHO),32 and acetone (CH3COCH3).33 Here, hydrogen positions and atom labels were modified about the respective heavy atom (C3) backbones to generate Z = 4 propanal cells. Iterative structure optimisation procedures were completed to threshold energy (RMS: 8 × 10−4 eV) and gradient convergence utilising relaxed lattice parameters and removing symmetry constraints (space group: P1). Here, the stationary point on the potential energy surface was confirmed as a local minimum by the absence of imaginary frequencies resultant from harmonic frequency calculations. Simulated IR spectra and powder diffraction patterns were generated for comparison with the experimental data recorded for propanal ice.
Synchrotron spectroscopy
Thin films of propanal ice were produced at the Far-IR and THz Beamline at the Australian Synchrotron using the transmission infrared spectroscopy setup described previously.26 Within the high-vacuum chamber (2 × 10−8 Torr), the vapour from de-gassed CH3CH2CHO (Sigma Anhydrous ≥99.5%) was deposited on a far-IR transparent silicon monocrystal substrate (Edmund Optics) attached to the cold finger of a closed-cycle helium cryocooler (Sumitomo CH202) at 20 K. Vapour deposition was performed over 2–5 minutes at a chamber background pressure of ca. 5 × 10−5 Torr. Although the film thickness could not be measured with our setup (likely 2–5 μm regime via comparison to previous O-bearing ice samples26), the far-IR spectrum was monitored to ensure amorphous lattice features avoided saturation upon transformation to the crystal-phase during warming, where IR features of the crystal have intrinsically higher intensity.Far-IR spectra were collected using the beamline spectrometer (Bruker IFS125) fitted with polyethylene outer chamber windows, a 5 μm Mylar beamsplitter, and a liquid helium cooled bolometer for the detection of the synchrotron source edge radiation; thereby accessing the 50–600 cm−1 region. All spectra were recorded at 4 cm−1 resolution averaged from 50 individual scans. Temperature dependent far-IR studies were performed using a temperature control unit (Lakeshore 330) for a 50 Ω heater producing a linear 0.5 K min−1 temperature ramp for the silicon substrate. Spectra were collected at 2 K temperature steps measured by a silicon diode sensor (Lakeshore).
Results and discussion
Determining the crystal structure of solid propanal
Structure solution for propanal was completed using the PXRD pattern collected at 100 K for 10 minutes. From these data the unit cell was indexed within the TOPAS34 program suite; refined as a primitive monoclinic cell of a = 12.1690(14) Å, b = 4.2226(3) Å, c = 8.977(1) Å and β = 129.521(6)°, resulting in a volume of 355.84(7) Å3. Examination of systematic absences and intensity distributions for this indexing suggest a P21/c spacegroup, with the structure solution progressed in the P21/a setting (β angle between 90 and 120°). The program Free Objects for Crystallography35 (FOX version 1.9.7.1) was used to perform the structure solution using the Parallel Tempering alogorithm. The 100 K pattern was again used for this procedure, along with a rigid model of the propanal molecule obtained from a crystal structure in the Cambridge Structure Database36 (refcode HUHDAS) that contains propanal as an adsorbed solvent.37 This propanal model applied was the CH3-SYN configuration, which has been previously shown to be the dominant conformer at 300 K.38 Optimisation resulted in a viable crystal structure (verified by the crystal packing and resultant density). This structure was then further refined via Rietveld refinement39 in the TOPAS suite, where the fit to these data, and the resultant crystal structure, are presented in Fig. 2. The resultant density of the 1.082 g cm−3 at 100 K compares well with the value of 1.105 g cm−3 determined by Yarnall et al. measured by quartz crystal microbalance.4 The Crystallographic Information File (CIF) for this structure is provided as part of the ESI.†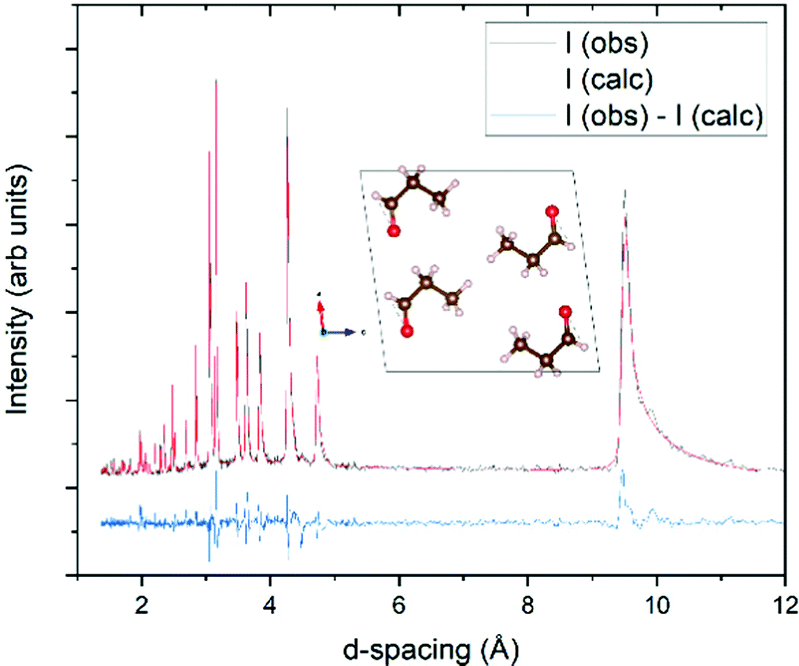 | ||
| Fig. 2 Rietveld fit of the structure determined for propanal at 100 K, with a Rwp of 11.4%. Background, peak shape parameters, lattice parameters, and scale factor were refined at all stages of the procedure. Lattice parameters are fit to the P21/a spacegroup, with a unit cell: a = 8.9833(6) Å, b = 4.2237(2) Å, c = 9.4733(6) Å and β = 97.508(6)°. After the atomic positions had been constrained, only isotropic displacement parameters on the oxygen and carbon atoms were allowed to refine further. For the last cycle of refinement, a 4th order spherical harmonic model was applied to account for minor preferred orientation in the sample. Inset: View of the propanal crystal structure down the c axis, with red spheres representing oxygen atoms, brown carbon atoms, and pink hydrogen atoms. Structure drawn with the VESTA42 program. | ||
As the structure of propanal determined here has been derived from PXRD data, it is not appropriate to discuss the conformation of the molecule itself (especially as this was modelled as a rigid body and derived from a different structure). However, discussion can be directed at the intermolecular interactions and crystal packing of solid propanal, particularly in the context of other small molecular aldehydes. As has been reported previously, small aldehyde molecular structures provide fundamental insight toward weak hydrogen bonding motifs established between highly polar carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups and C–H bonds.40 The solution of the propanal crystal structure provides a higher understanding of intermolecular bonding types in the series of solid aldehydes progressing from the methanal (formaldehyde), ethanal (acetaldehyde) and, now, propanal crystal structures. First, in the methanal structure (refcode KEMZIL in the CSD) CO dipole-type interactions are the strongest in forming (Z = 4) tetramer clusters within the unit cell. These are then weakly bound to neighbouring tetramers via CO⋯H–C interactions,40 defined as a weak hydrogen-bonding motif. Secondly, the analysis of close contacts in the ethanal crystal structure reveals that CO⋯H–C interactions instead govern the crystal structure.41
O groups and C–H bonds.40 The solution of the propanal crystal structure provides a higher understanding of intermolecular bonding types in the series of solid aldehydes progressing from the methanal (formaldehyde), ethanal (acetaldehyde) and, now, propanal crystal structures. First, in the methanal structure (refcode KEMZIL in the CSD) CO dipole-type interactions are the strongest in forming (Z = 4) tetramer clusters within the unit cell. These are then weakly bound to neighbouring tetramers via CO⋯H–C interactions,40 defined as a weak hydrogen-bonding motif. Secondly, the analysis of close contacts in the ethanal crystal structure reveals that CO⋯H–C interactions instead govern the crystal structure.41
Enabled by the current results, examination of the solid propanal structure suggests that crystal packing is the result of a combination of CO dipole and weak hydrogen bonds; as observed in methanal. As there exists ambiguity in the hydrogen atom positions due to the procedure used for crystal structure determination, short contacts are quantified by carbon to oxygen distances, as shown in Fig. 3.
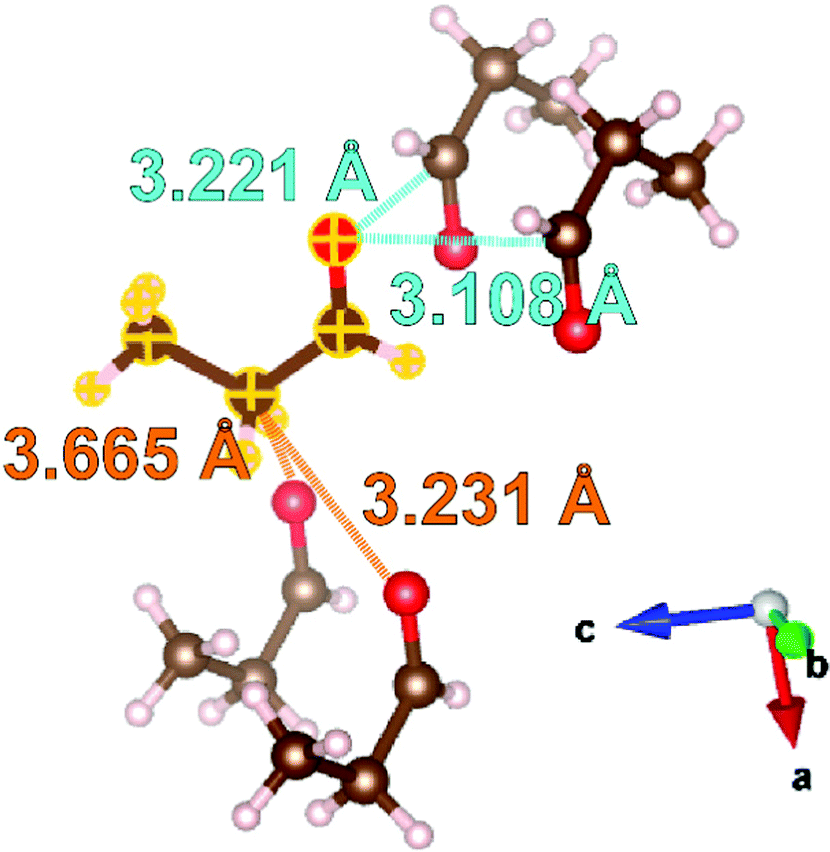 | ||
| Fig. 3 Examination of the intermolecular interactions within the structure of propanal determined at 100 K. Distances from the molecule highlighted with yellow outlines are expressed as carbon-to-oxygen distances. The intermolecular interactions similar to the CO dipole interactions in methanal are labelled in light blue, and the interactions similar to CO⋯C–H contacts in ethanal are labelled in orange. Oxygen atoms within the selected molecules shown are represented by red spheres, carbon by brown spheres, and hydrogen by pink spheres. Structures drawn with the VESTA42 program. | ||
Fig. 3 illustrates four intermolecular short contacts found in the propanal structure at 100 K. Comparison can be drawn to the methanal structure with both displaying short contacts between the formyl groups of nearest neighbour molecules. In methanal, the corresponding CO⋯CO distances are 2.866 and 3.245 Å,40 which are comparable to 3.221 and 3.108 Å found in the propanal structure. However, there are also two short contacts resulting from CO⋯H–C interactions; labelled in orange in Fig. 3. These are comparable to the CO⋯H–C interactions that typify the ethanal structure measured to be 3.560, 3.657, and 3.693 Å at 5 K41 (note CO⋯C–H distance used here for consistency), compared with 3.231 and 3.665 Å in propanal. Given this is a snapshot at only one (100 K) temperature, it is challenging to confirm which of the intermolecular bonding types is dominant for propanal, but further insight may be gleamed from the following thermal expansion studies.
Thermal behaviour of solid propanal
The response of solid propanal structure to temperature was investigated, with lattice parameters extracted by Rietveld refinements for each temperature step compiled in Fig. 1. The resultant (relative) change in each unit cell dimension is charted in Fig. 4, which shows that the crystal structure expands anisotropically.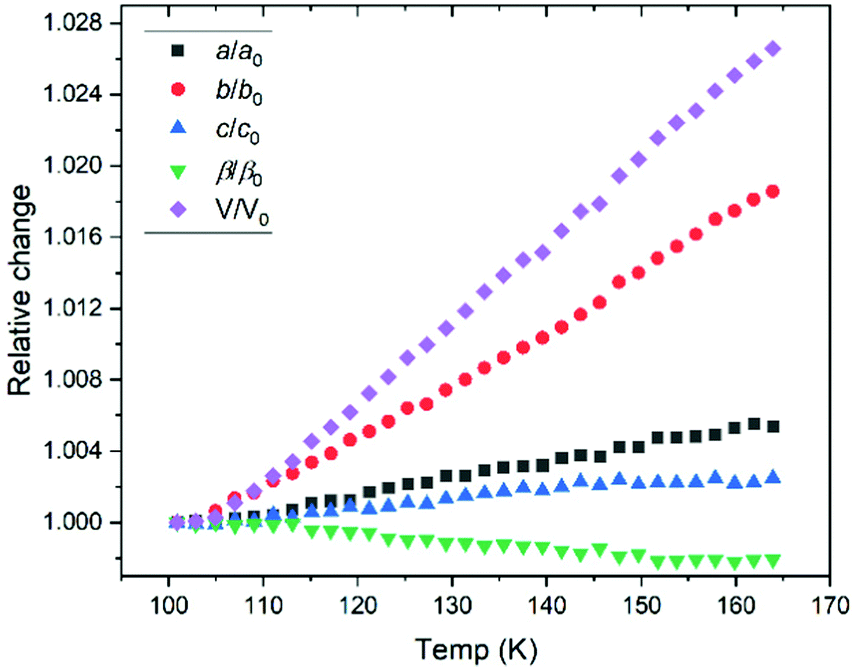 | ||
| Fig. 4 Relative change in lattice parameter over the range of temperatures studied by X-ray powder diffraction. The tabulated values of lattice parameters are presented in the ESI.† | ||
The largest thermal expansion takes place along the b axis, giving an insight toward the relative importance of the intermolecular bonding, suggesting that the CO⋯H–C interactions are indeed pivotal in stabilising the crystal structure. This is due to CO⋯H–C interactions aligned primarily with the a–c plane (Fig. 3), where both of these axes show smaller expansion. Aligning with the b–c plane, the CO dipole-type interactions possess a stronger dipole interaction along the c direction, resulting in a larger expansion along the b axis. Hence, while the propanal structure exhibits both CO dipole-type interactions between the formyl groups, and CO⋯H–C contacts with the side chain, insight from variable temperature studies indicate that hydrogen-bonding type interactions dominate.
Neutron powder diffraction was performed to locate low temperature transitions for the propanal crystal structure below 100 K, as low temperature polymorphs are relatively common for small organic molecules (e.g. for acetonitrile43).
The sample for the neutron diffraction study crystallised with strong preferred orientation, and as such full Rietveld refinement was not possible (Fig. 5). However a Pawley refinement44 was undertaken where it was found that the pattern at 3 K matched the same P21/a unit cell determined at 100 K by PXRD. As a result, it is concluded that no phase transformation had occurred at temperatures as low as 3 K and, hence, the experimental crystal structure of propanal is expected to be consistent with the form of planetary and interstellar ices.
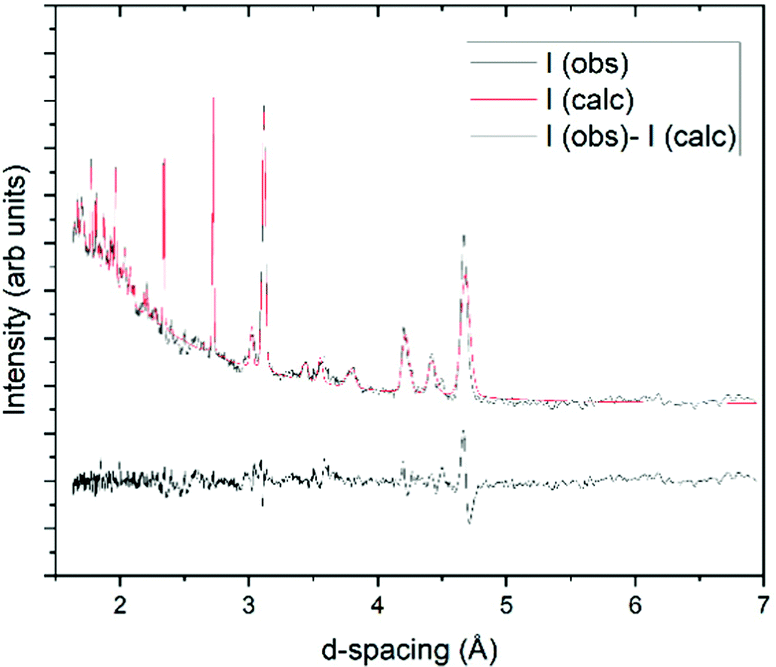 | ||
| Fig. 5 Pawley refinement fit for neutron powder diffraction data collected at 3 K. This was refined to a P21/a cell with a = 8.9394(17) Å, b = 4.1520(6) Å, c = 9.4645(6) Å, β = 97.518(9)° and V = 348.27(11) Å3. | ||
Far-infrared spectroscopy
Synchrotron FTIR spectroscopy of CH3CH2CHO ice deposited at 20 K shows a broad absorption feature with a peak maximum at 117 cm−1, and two narrower, higher intensity features at 172 and 269 cm−1 (Table 1). Warming to 90 K prompts a rapid transition to a crystalline phase, as evidenced by the 117 cm−1 band converting to a series of narrow peaks (FWHM: 20–30 cm−1) in the far-IR spectrum (Fig. 6). This transition temperature is consistent with that observed in the mid-IR.4 Between 90 and 100 K, the 172 cm−1 peak blue-shifts to 182 cm−1 while the 269 cm−1 band shifts only slightly to higher wavenumber but narrows in profile.| Far-IR/cm−1 | p-DFT/cm−1 | Assignment | |
|---|---|---|---|
| 20 K | 120 K | ||
| 63 | 66 | Translation (a/c-plane) | |
| 72 | Translation (b/c-plane) | ||
| 87 | 86 | Lattice libration | |
| 102 | 103 | Lattice libration | |
| 117 | 119 | 121 | Lattice libration |
| 172 | 182 | 183 | Methyl torsion |
| 269 | 270 | 264 | ν 15 CCC deformation |
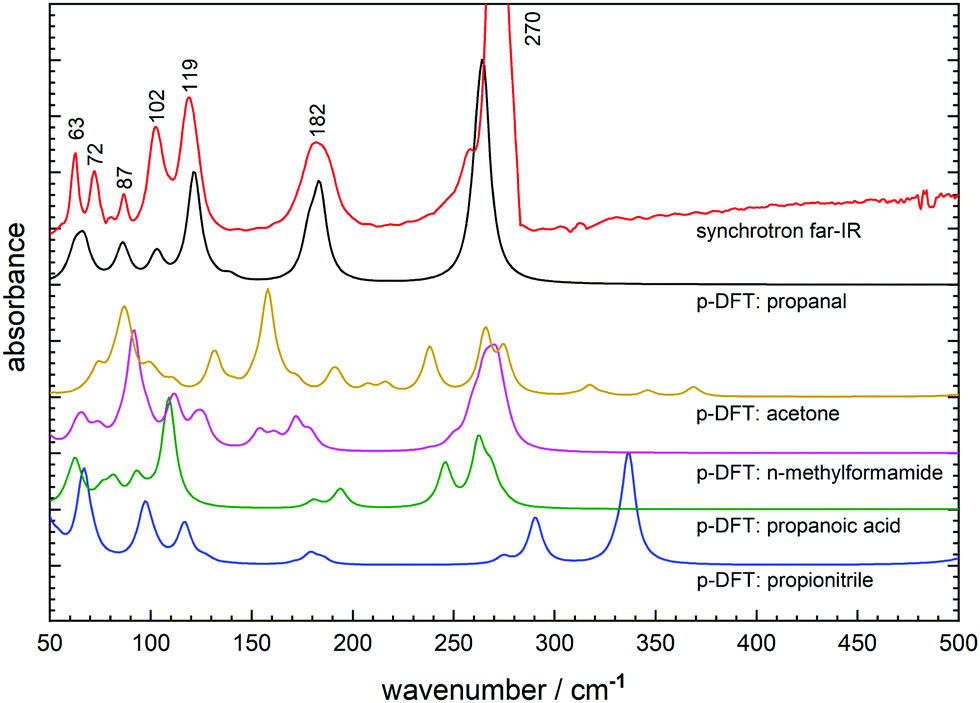 | ||
| Fig. 6 Synchrotron far-IR spectrum of solid propanal at 100 K (red) compared to the p-DFT simulated far-IR spectra for the experimentally-derived propanal crystal structure (black) and propanal crystal cells derived from propionitrile (blue), propanoic acid (green), N-methylformamide (magenta) and acetone (yellow). | ||
The series of five new peaks clustered below 120 cm−1 are of moderate intensity and have not been assigned in previous literature. Therefore p-DFT frequency calculations have been performed to determine the vibrational modes associated with each far-IR signature, with the simulated far-IR spectrum (10 cm−1 Lorentzian fit) depicted in Fig. 6. Visualisation of the calculated molecular (atomic) displacements associated with intermolecular (intramolecular) modes show that the two lowest frequency peaks at 63 and 72 cm−1 correspond to propanal translational modes within the crystal structure (Fig. S2a, ESI†) while the remaining three lattice vibrations at 87, 102, and 119 cm−1 correspond to hindered rotation (libration) modes (Fig. S2b–d, ESI†). Finally, the two lowest lying fundamental vibrations are confirmed as the methyl torsion mode at 182 cm−1 and the bright C–C–C backbone deformation mode at 270 cm−1. Note that these crystalline vibrational frequencies are significantly shifted from the gas-phase propanal positions as described in the literature,18,45 with the methyl rotation particularly perturbed when observed in the condensed-phase (Δν = ∼70 cm−1).
Also shown in Fig. 6 are the simulated far-IR spectra from the propanal crystal structures generated from modified (then DFT optimised) unit cells of other small molecules. Here, many of the far-IR simulations provide unsatisfactory comparison to the solved propanal structure, where even intramolecular modes display significant shifts or peak-splitting from their measured positions. In all cases, the far-IR lattice features are poorly reproduced; confirming that the prediction of molecular crystal geometries is not a simple undertaking and that biased starting geometries can generate incorrect final structures after energy optimisation. It can be considered that the closest match is provided by the propanoic acid starting geometry featuring an arrangement of CH3CH2COOH dimers displaying R22(8) hydrogen bonding synthon. Here, a dimer arrangement was maintained in constructing a unit cell for its reduced propanal counterpart; where the unit cell contains CH3CH2CHO units arranged by R22(6) motifs. The resultant p-DFT predicted lattice features (Fig. 6: green) best mimics the spectrum derived from experiment (Fig. 6: black) in both peak position and relative intensity. However, the intramolecular-type bands are poorly reproduced by p-DFT where both peak splitting and a reduced relative intensity can be seen in the propanoic acid based prediction. This is most likely a result of poor simulation of these large amplitude torsional modes that can be highly sensitive to perturbation in the solid-state, and also noting that the anharmonic character of these modes is not treated by the theory.
Conclusions
The behaviour of solid propanal has been characterised using diffraction and spectroscopic techniques, allowing for its crystal structure to be resolved between 3 K and melting, thermal expansion from 100–170 K to be mapped, and its low frequency vibrational modes to be assigned by p-DFT simulations. In undertaking this study, we have highlighted the following observations as of particular interest:(a) In determining the crystal structure of solid propanal, an important sequence relating to the intermolecular bonding of small aldehyde structures can now be observed. Crystalline aldehydes show a balance between (i) dipole-type bonding of the formyl groups and (ii) hydrogen bonding moieties formed by side chain C-H groups to neighbouring carboxyl groups. Single crystal investigations, potentially with Hartree–Fock refinements, would be enormously informative in further investigating these intermolecular interactions with key applications to the crystal engineering community.
(b) This work also demonstrates the importance of examining variable temperature behaviour, particular when a structure is revealed from PXRD. This allowed for ambiguity in the hydrogen atom positions to be circumvented, and the relative strength of the intermolecular interactions probed.
(c) Propanal ice has a distinctive far-IR spectrum comprising of the signatures of numerous lattice and torsion vibrations. This low frequency fingerprint is qualitatively more complex than molecular ices produced from organics of comparative size, including species of interest to planetary and interstellar astrochemistry. Therefore, these new assignments may be of benefit to future spacecraft and astronomical surveys where far-IR frequencies can be employed to locate this important aldehyde within composite ice surfaces.
(d) Crystal structure prediction of molecular crystals comprising of small organic molecules using p-DFT geometry optimisation methods has, in this case, not generated a match to our experimentally-determined propanal unit cell. Here, far-IR simulations have shown that starting geometries which most closely describe real intermolecular interactions, such as hydrogen-bonding dimer motifs, produce the best simulations; although not at a level sufficient to accurately assign vibrational features at low frequency.
Although unlikely to exist as pristine, crystal deposits in planetary and interstellar environments, this new information on the fundamental structure, intermolecular bonding, and spectroscopy of the key C3 aldehyde propanal should benefit its study in condensed-phase experiments. For example, its role in Strecker synthesis pathways toward amino acids can now be further rationalised by taking into account its solid-state geometry within composite molecular ices.
Author contributions
H. E. M.-C. and C. E. conceived, procured funding, and administered the project. All authors contributed to data curation, formal analysis (including software and visualisation), and writing of the original draft. Corresponding author H. E. M.-C. completed review of the final draft and submitted the manuscript.Conflicts of interest
The authors confirm that there are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the Australian Centre of Neutron Scattering for provision of instrument time through program proposal 7728. This research was facilitated by access to Sydney Analytical, a core research facility at the University of Sydney. The authors also wish to thank ANSTO Australian Synchrotron and Dr Dominique Appadoo at the THz and Far-IR beamline for assistance during merit beamtime proposal M15686. N. S. Y. wishes to thank Prof. Lyall Hanton at the University of Otago for assistance throughout his Department of Chemistry Student Scholarship and the New Zealand Synchrotron Group for travel funding allowing him to perform facility experiments.References
- J. M. Hollis, P. R. Jewell, F. J. Lovas, A. Remijan and H. Møllendal, Astrophys. J., Lett., 2004, 610, L21 CrossRef CAS.
- J. Lykke, A. Coutens, J. Jørgensen, M. Van der Wiel, R. Garrod, H. Müller, P. Bjerkeli, T. Bourke, H. Calcutt and M. Drozdovskaya, Astron. Astrophys., 2017, 597, A53 CrossRef.
- D. Quan and E. Herbst, Astron. Astrophys., 2007, 474, 521–527 CrossRef CAS.
- Y. Y. Yarnall, P. A. Gerakines and R. L. Hudson, Mon. Not. R. Astron. Soc., 2020, 494, 4606–4615 CrossRef CAS.
- F. Goesmann, H. Rosenbauer, J. H. Bredehöft, M. Cabane, P. Ehrenfreund, T. Gautier, C. Giri, H. Krüger, L. Le Roy and A. J. MacDermott, Science, 2015, 349 Search PubMed.
- M. Schuhmann, K. Altwegg, H. Balsiger, J.-J. Berthelier, J. De Keyser, S. A. Fuselier, S. Gasc, T. I. Gombosi, N. Hänni and M. Rubin, ACS Earth Space Chem., 2019, 3, 1854–1861 CrossRef CAS.
- M. Jonusas, J.-C. Guillemin and L. Krim, Mon. Not. R. Astron. Soc., 2017, 468, 4592–4600 CrossRef CAS.
- D. Qasim, G. Fedoseev, K.-J. Chuang, V. Taquet, T. Lamberts, J. He, S. Ioppolo, E. van Dishoeck and H. Linnartz, Astron. Astrophys., 2019, 627, A1 CrossRef CAS.
- R. Hudson, M. Loeffler and K. Yocum, Astrophys. J., 2017, 835, 225 CrossRef.
- R. L. Hudson and M. Moore, Astrophys. J., 2018, 857, 89 CrossRef.
- K. Harada, Nature, 1963, 200, 1201 CrossRef CAS PubMed.
- G. Jungclaus, G. Yuen, C. Moore and J. Lawless, Meteoritics, 1976, 11, 231–237 CrossRef CAS.
- J. R. Cronin and C. B. Moore, Science, 1971, 172, 1327–1329 CrossRef CAS PubMed.
- M. H. Engel and B. Nagy, Nature, 1982, 296, 837–840 CrossRef CAS.
- O. Zingsheim, H. S. Müller, F. Lewen, J. K. Jørgensen and S. Schlemmer, J. Mol. Spectrosc., 2017, 342, 125–131 CrossRef CAS.
- C. Nixon, D. Jennings, J.-M. Flaud, B. Bézard, N. Teanby, P. Irwin, T. Ansty, A. Coustenis, S. Vinatier and F. Flasar, Planet. Space Sci., 2009, 57, 1573–1585 CrossRef CAS.
- S. Vinatier, B. Bézard, C. A. Nixon, A. Mamoutkine, R. C. Carlson, D. E. Jennings, E. A. Guandique, N. A. Teanby, G. L. Bjoraker and F. M. Flasar, Icarus, 2010, 205, 559–570 CrossRef CAS.
- G. Sbrana and V. Schettino, J. Mol. Spectrosc., 1970, 33, 100–108 CrossRef CAS.
- S. Frankiss and W. Kynaston, Spectrochim. Acta, Part A, 1972, 28, 2149–2161 CrossRef CAS.
- J. Ghosh, G. Vishwakarma, S. Das and T. Pradeep, J. Phys. Chem. C, 2021, 125, 4532–4539 CrossRef CAS.
- H. E. Maynard-Casely, M. L. Cable, M. J. Malaska, T. H. Vu, M. Choukroun and R. Hodyss, Am. Mineral., 2018, 103, 343–349 CrossRef.
- C. Ennis, M. L. Cable, R. Hodyss and H. E. Maynard-Casely, ACS Earth Space Chem., 2020, 4, 1195–1200 CrossRef CAS.
- A. J. Studer, M. E. Hagen and T. J. Noakes, Phys. B: Condens. Matter, 2006, 385–386(Part 2), 1013–1015 CrossRef CAS.
- D. Brochier, Institut Laue-Langevin technical report, 1977 Search PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1360 Search PubMed.
- C. Ennis, R. Auchettl, D. Appadoo and E. Robertson, Mon. Not. R. Astron. Soc., 2017, 471, 4265–4274 CrossRef CAS.
- C. Ennis, R. Auchettl, D. Appadoo and E. Robertson, Phys. Chem. Chem. Phys., 2018, 20, 23593–23605 RSC.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, J. Chem. Phys., 2005, 123, 174101 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. E. Brand, Q. Gu, J. A. Kimpton, R. Auchettl and C. Ennis, J. Synchrotron Radiat., 2020, 27, 212–216 CrossRef CAS PubMed.
- F. J. Strieter, D. H. Templeton, R. Scheuerman and R. Sass, Acta Crystallogr., 1962, 15, 1233–1239 CrossRef.
- J. Neuefeind, PhD, RWTH Aachen, 1994 Search PubMed.
- D. R. Allan, S. J. Clark, R. M. Ibberson, S. Parsons, C. R. Pulham and L. Sawyer, Chem. Commun., 1999, 751–752 RSC.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- S. Ohta, Y. Iwabuchi, R. Mukai, M. Ishizaki, T. Toda, M. Kurihara and M. Okazaki, Cryst. Growth Des., 2020, 20, 4046–4053 CrossRef CAS.
- P. Van Nuffel, L. Van den Enden, C. Van Alsenoy and H. Geise, J. Mol. Struct., 1984, 116, 99–118 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- T. S. Thakur, M. T. Kirchner, D. Bläser, R. Boese and G. R. Desiraju, Phys. Chem. Chem. Phys., 2011, 13, 14076–14091 RSC.
- R. Ibberson, O. Yamamuro and T. Matsuo, J. Mol. Struct., 2000, 520, 265–272 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- O. K. Antson, K. Tilli and N. Andersen, Acta Crystallogr., Sect. B: Struct. Sci., 1987, 43, 296–301 CrossRef.
- G. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- G. Metha, M. Buntine, D. McGilvery and R. Morrison, J. Mol. Spectrosc., 1994, 165, 32–56 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2112476. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cp04477j |
| This journal is © the Owner Societies 2022 |