
Anionic states of C6Cl6 probed in electron transfer experiments†
S.
Kumar
 a,
T.
Kilich
b,
M.
Łabuda
bc,
G.
García
d and
P.
Limão-Vieira
*a
a,
T.
Kilich
b,
M.
Łabuda
bc,
G.
García
d and
P.
Limão-Vieira
*a
aAtomic and Molecular Collisions Laboratory, CEFITEC, Department of Physics, Universidade NOVA de Lisboa, 2829-516 Caparica, Portugal. E-mail: plimaovieira@fct.unl.pt; Fax: +351-212948549; Tel: +351 212947859
bDepartment of Theoretical Physics and Quantum Information, Gdańsk University of Technology, Narutowicza 11/12, 80-233 Gdańsk, Poland
cBioTechMed, Gdańsk University of Technology, Gdańsk, Poland
dInstituto de Física Fundamental, Consejo Superior de Investigaciones Científicas (CSIC), Serrano 113-bis, 28006 Madrid, Spain
First published on 25th November 2021
Abstract
This is the first comprehensive investigation on the anionic species formed during collisions of fast neutral potassium (K) atoms with neutral hexachlorobenzene (C6Cl6) molecules in the laboratory frame range from 10 up to 100 eV. In such ion-pair formation experiments we also report a novel K+ energy loss spectrum obtained in the forward scattering giving evidence of the most accessible electronic states. The vertical electron affinity of (−3.76 ± 0.20) eV has been obtained and assigned to a purely repulsive transition from the C6Cl6 ground state to a 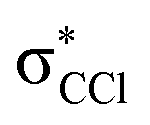 state of the temporary negative ion yielding Cl− formation. These experimental findings are also supported by state-of-the art theoretical calculations on the electronic structure of C6Cl6 in the presence of a potassium atom and are used for analysing the lowest unoccupied molecular orbitals participating in the collision process. From the time-of-flight mass spectra recorded in the wide collision energy range, more than 80% of the total anion yield is due to the undissociated parent anion C6Cl6−, C6Cl5− and Cl− formation. Other fragment anions such as C6Cl4−, C3Cl2−, C2Cl− and Cl2− that undergo complex internal reactions with the temporary negative ion formed after electron transfer account for less than 20% of the total yield. The joint experimental and theoretical methodologies employed in these electron transfer studies provide the most comprehensive and unique assignments of the hexachlorobenzene anionic species and the role of C6Cl6 electronic states in collision induced dissociation to date.
state of the temporary negative ion yielding Cl− formation. These experimental findings are also supported by state-of-the art theoretical calculations on the electronic structure of C6Cl6 in the presence of a potassium atom and are used for analysing the lowest unoccupied molecular orbitals participating in the collision process. From the time-of-flight mass spectra recorded in the wide collision energy range, more than 80% of the total anion yield is due to the undissociated parent anion C6Cl6−, C6Cl5− and Cl− formation. Other fragment anions such as C6Cl4−, C3Cl2−, C2Cl− and Cl2− that undergo complex internal reactions with the temporary negative ion formed after electron transfer account for less than 20% of the total yield. The joint experimental and theoretical methodologies employed in these electron transfer studies provide the most comprehensive and unique assignments of the hexachlorobenzene anionic species and the role of C6Cl6 electronic states in collision induced dissociation to date.
1 Introduction
Hexachlorobenzene (C6Cl6), as a class of volatile organochloride compounds, has been used as a pesticide across the globe1,2 and is found to be a prevailing environmental pollutant.3 Its global distribution reveals that it moves through the atmosphere from warmer source regions (where it is volatilized) and tends to condense at colder regions,4–6 undergoing a process known as global distillation.1,4 In the atmosphere, reactions with ˙OH radicals have been identified to follow a sink mechanism,1 yet the relatively low rate constant, 0.27 × 10−13 cm3 molecules−1 s−1 at 298 K, favours such a global distillation process.1 C6Cl6 can be eliminated by ozone coupled with hydrogen peroxide processes and these can be enhanced through combination with active carbon absorption methods.2,7 Additionally, the lack of comprehensive photolysis rates in the literature does not allow identifying ultra-violet photoabsorption as a route to remove and/or chemically change these molecular compounds in the Earth's atmosphere.Hexachlorobenzene has been investigated through experimental and theoretical methods, with the former involving ultraviolet photoabsorption,8,9 infrared photoabsorption,10,11 photoelectron spectroscopy,12 gas-phase reactions with molecular oxygen13 and reduction potentials with an electron capture detector (ECD),14 and the latter involving vibronic interactions and charge transfer15 and electron affinities of chlorinated benzene molecules.16–18 Photodegradation of C6Cl6 and theoretical prediction of its pathways using quantum chemical calculations have been reported by Yamada et al.19 As far as negative ion formation is concerned, a comprehensive literature survey reveals only the parent anion from resonance electron capture mass spectrometry, allowing the determination of an adiabatic electron affinity of 0.91 eV,13 whereas a value of 0.98 eV has been reported by Wiley and co-workers.17 Moreover, the generalized Kohn–Sham semicanonical projected random phase approximation method predicts a valence π* character for the C6Cl6− ground-state,16 while Robin9 has noted that the most relevant absorption features at 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 (1A1g → 1B1u) and 46000 cm−1 (1A1g → 1E1u) in neutral C6Cl6 have been assigned to intense halogen np → π* charge transfer transitions.
500 cm−1 (1A1g → 1B1u) and 46000 cm−1 (1A1g → 1E1u) in neutral C6Cl6 have been assigned to intense halogen np → π* charge transfer transitions.
The highest occupied molecular orbital configuration of C6Cl6 in D6h symmetry yields for the outer valence … (e1g)4 corresponding to the 1A1g sate, while the parent anion results from electron capture into the non-degenerate a1g lowest unoccupied molecular orbital with configuration … (e1g)4 (a1g)1 which corresponds to the 2A1g state, with no appreciable Jahn–Teller effect.15 Given the lack of any other relevant information on negative ion formation from hexachlorobenzene either by dissociative electron attachment or charge transfer processes, a comprehensive investigation of the underlying molecular mechanisms yielding C6Cl6− and its fragment anions is necessary for obtaining further knowledge on the electronic structure of such chemical compounds.
In this study, we present for the first time a comprehensive investigation of hexachlorobenzene negative ion formation in electron transfer processes, combining experimental and state-of-the art theoretical methods. In Section 2, we present a brief summary of the experimental setup and in Section 3 the computational details of the calculations that are used to interpret the experimental data. Section 4 is dedicated to results and discussion, which includes a complete description of the electronic state spectra of hexachlorobenzene probed by the experimental method and supported by quantum chemical calculations. The data have been used to assign the nature of the lowest-lying electronic states accessed in electron transfer processes in neutral potassium collisions with neutral hexachlorobenzene molecules. We finish with Section 5 by including a summary and conclusions.
2 Experimental methods
The crossed molecular beam setup used to investigate negative ion formation during collisions of neutral potassium (K) atoms with hexachlorobenzene (C6Cl6) molecules was previously described elsewhere,20 therefore only a general description is given here. Briefly, an effusive target molecular beam crosses a projectile beam of neutral hyperthermal K atoms and the product anions formed in the electron transfer process are analysed by using a dual-stage linear time-of-flight (TOF) mass spectrometer. The K beam is produced in a charge exchange chamber from the interaction of gas-phase neutral potassium atoms from an oven source with accelerated K+ ions emitted from a commercial ion source (HeatWave, USA) in the range of 10 to 100 eV in the lab frame. The intensity of the neutral potassium beam is monitored using a Langmuir–Taylor ionisation detector, before and after collecting the mass spectra. The TOF anion yield is normalized considering the primary beam current, pressure and acquisition time. The typical base pressure in the collision chamber was 4 × 10−5 Pa and the working pressure was 1 × 10−3 Pa. Mass calibration was performed on the basis of the well-known anionic species formed after potassium collisions with nitromethane20 and tetrachloromethane.21A hemispherical energy loss analyser was used to obtain the K+ signal post-collision in the forward scattering direction with the beam's optical path, where such experiments were not performed in coincidence with TOF mass spectrometry. The analyser was operated in constant transmission mode, hence keeping the resolution constant throughout the entire scans. The estimated energy resolution during the experiments was ∼1.2 ± 0.2 eV. The energy loss scale was calibrated using the K+ beam profile from the potassium ion source serving as the elastic peak. Hexachlorobenzene (C6Cl6) was supplied by Sigma Aldrich with a stated purity of ≥98%. The solid sample was used as obtained and gently heated up to 340 K through a temperature PID (proportional-integral-derivate controller) unit. In order to test for any thermal decomposition products within the hexachlorobenzene beam, mass spectra were recorded at different temperatures and no differences in the relative peak intensities as a function of temperature were observed.
3 Theoretical methods
Charge transfer processes are described with the aid of ab initio calculations, based on the evolution of the quasi-molecular system, formed by the potassium projectile and the molecular target along the reaction coordinate within the framework of the molecular representation. This methodology has been successfully used for diatomic systems and tested during ion-neutral molecule collisions,22–26 and later extended to atom-molecule interactions21,27,28 where the system evolves along the reaction coordinates corresponding to the distance R between the impact atom and the molecular target.29–32 In such approximations, we do not include the internal degrees of freedom of the molecular target explicitly, which is reasonable since collisional processes are very fast and thus nuclear vibrational and rotational motions are much slower than the collision time and can be considered frozen during the collision.The geometry of hexachlorobenzene at D2h symmetry was optimized at the MP2/def2-TVZP level of theory33 while in the presence of potassium atom the C2v symmetry was used. All calculations have been performed by means of the ORCA and MOLPRO packages of ab initio programmes.34,35 The potassium atom has been placed along the y axis as shown in Fig. 1 and the C6Cl6 target is kept frozen in its ground state (![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) :1A′) geometry during the collision process. A detailed analysis of the K–C6Cl6 interaction at R = [2.5, 5, 7.5, 10] Å, occurring between molecular states involved in this process, has been made in order to precisely determine the asymptotic molecular configurations of the calculated states. The calculation has been carried out in Cartesian coordinates, with no symmetries. All electrons of carbon and chlorine atoms have been included in the calculations and their 1s orbitals were treated as the frozen-core. For the potassium atom, the ECP18SDF core-electron pseudopotential36 with the associated basis set has been chosen. The natural molecular orbitals for K–C6Cl6 (see Fig. S1 and Table S1, ESI†) have been obtained at the state-averaged Complete Active Space Self Consistent Field (CASSCF)37–39 level of theory considering the static electron correlation for the reaction coordinate K–C6Cl6 at R = 5 Å distance, corresponding to the asymptotic region. The resultant highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) for K–C6Cl6 are shown in Fig. 2 and Fig. S1 (ESI†) together with the corresponding orbitals without the presence of the potassium atom (see Fig. S2 and Table S2, ESI†). Finally, Fig. S3 (ESI†) depicts the C6Cl6− highest doubly occupied, singly occupied (SOMO) and the lowest unoccupied molecular orbitals (RKS, B3LYP + D3).
:1A′) geometry during the collision process. A detailed analysis of the K–C6Cl6 interaction at R = [2.5, 5, 7.5, 10] Å, occurring between molecular states involved in this process, has been made in order to precisely determine the asymptotic molecular configurations of the calculated states. The calculation has been carried out in Cartesian coordinates, with no symmetries. All electrons of carbon and chlorine atoms have been included in the calculations and their 1s orbitals were treated as the frozen-core. For the potassium atom, the ECP18SDF core-electron pseudopotential36 with the associated basis set has been chosen. The natural molecular orbitals for K–C6Cl6 (see Fig. S1 and Table S1, ESI†) have been obtained at the state-averaged Complete Active Space Self Consistent Field (CASSCF)37–39 level of theory considering the static electron correlation for the reaction coordinate K–C6Cl6 at R = 5 Å distance, corresponding to the asymptotic region. The resultant highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) for K–C6Cl6 are shown in Fig. 2 and Fig. S1 (ESI†) together with the corresponding orbitals without the presence of the potassium atom (see Fig. S2 and Table S2, ESI†). Finally, Fig. S3 (ESI†) depicts the C6Cl6− highest doubly occupied, singly occupied (SOMO) and the lowest unoccupied molecular orbitals (RKS, B3LYP + D3).
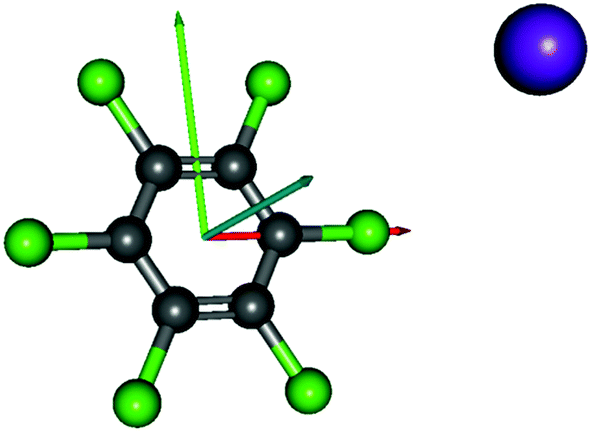 | ||
| Fig. 1 Molecular structure of C6Cl6 and orientation of the K–C6Cl6 collisional system; K, purple, x, red; y, green; z, blue. | ||
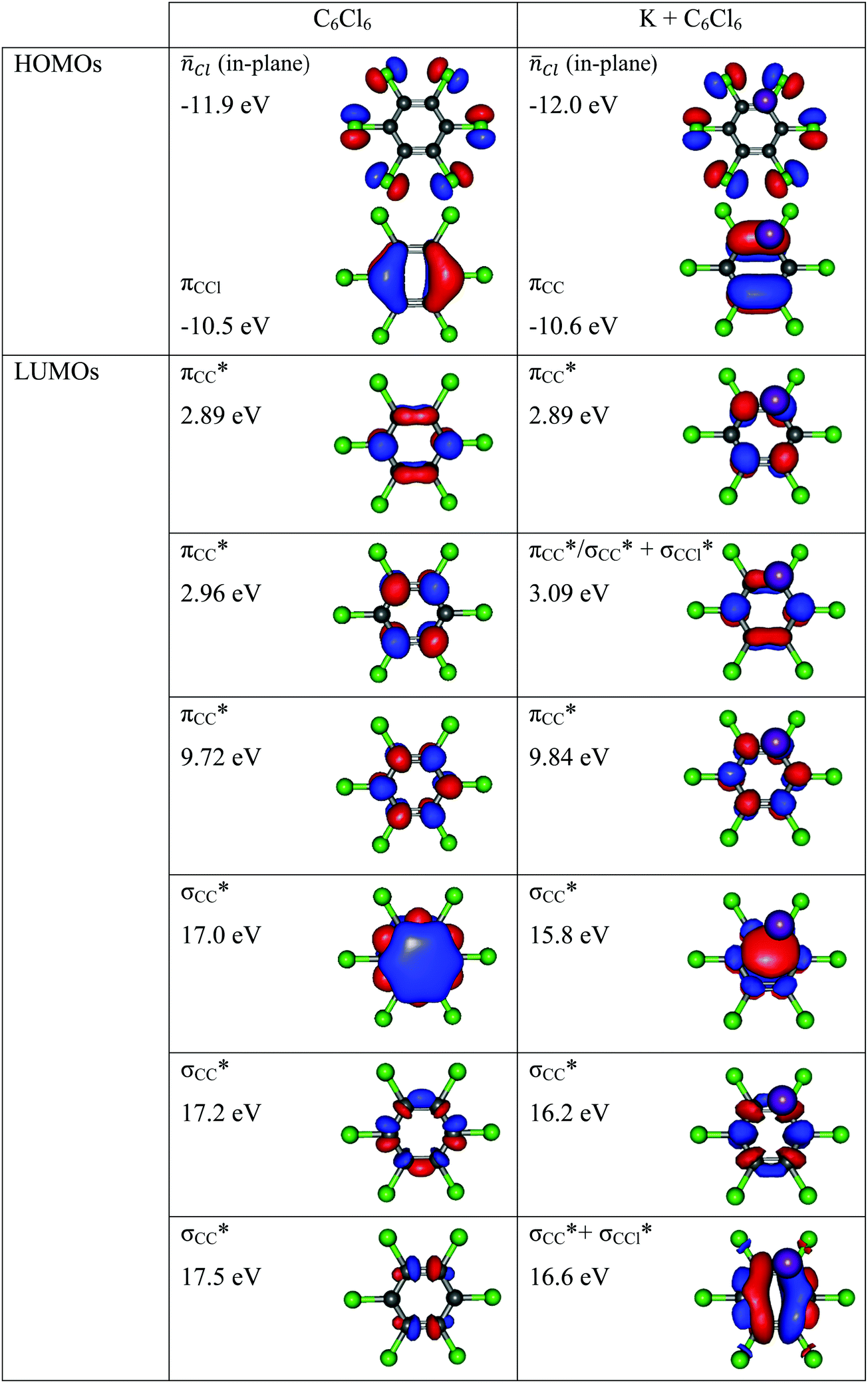 | ||
| Fig. 2 Calculated highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) for C6Cl6 and K + C6Cl6. Energies in eV. | ||
In order to determine the ionisation energy, electron affinity and vertical detachment energy (see Table S3, ESI†) and to look closely at the anion where the symmetry is broken, DFT calculations have been performed where Kohn–Sham orbitals were used rather than canonical HF orbitals, since the former improve the agreement with the experimental electron affinity of C6Cl6 by 0.08 eV. In order to describe an extra electron, diffuse functions with additional augmented basis functions were used at the restricted open shell Kohn–Sham, B3LYP + D3 level of theory.40 Energies of the neutral, the anion and the cation are −2989.57786 a.u., −2989.61393 a.u. and −2989.25288 a.u respectively. Detailed results are available in Table S4 in the ESI.†
4 Results and discussion
TOF mass spectra have been recorded in a wide range of lab-frame collision energies from 10 up to 100 eV (7.9 to 79.1 eV in the centre-of-mass frame), yielding negative ions assigned to Cl−, Cl2−, C2Cl−, C3Cl2−, C6Cl4−, C6Cl5− and C6Cl6− along with their respective isotopes. Fig. 3 shows a typical mass spectrum recorded at 100 eV (ECM = 79.1 eV), where the inset depicts the parent anion signal fitted with Gaussian functions to reproduce its isotope contributions at 282 u (51%), 284 u (100%), 286 u (81%) and 288 u (35%). Note that a TOF mass peak shows an apparent asymmetry relative to its maximum position, with typically the left-hand branch much steeper that the right-hand branch.41 Yet, for the sake of example, the Gaussian fitting used reproduces quite well in magnitude and in shape the TOF mass signal. Another relevant aspect pertains to the Cl2− TOF mass contribution, where the peak should show its isotope contributions at 70 u (100%), 72 u (∼65%), and 74 u (∼11%). A close inspection of this anion feature in Fig. 3 reveals that the peak contains three contributions but not in the expected intensity. Given that the limited TOF mass resolution (m/Δm ≈ 125) does not allow clearly resolving close fragment anions, yet a proper peak fitting, as that used for C6Cl6−, reproduces perfectly well the expected isotope distribution intensities (see Fig. S4 in the ESI†).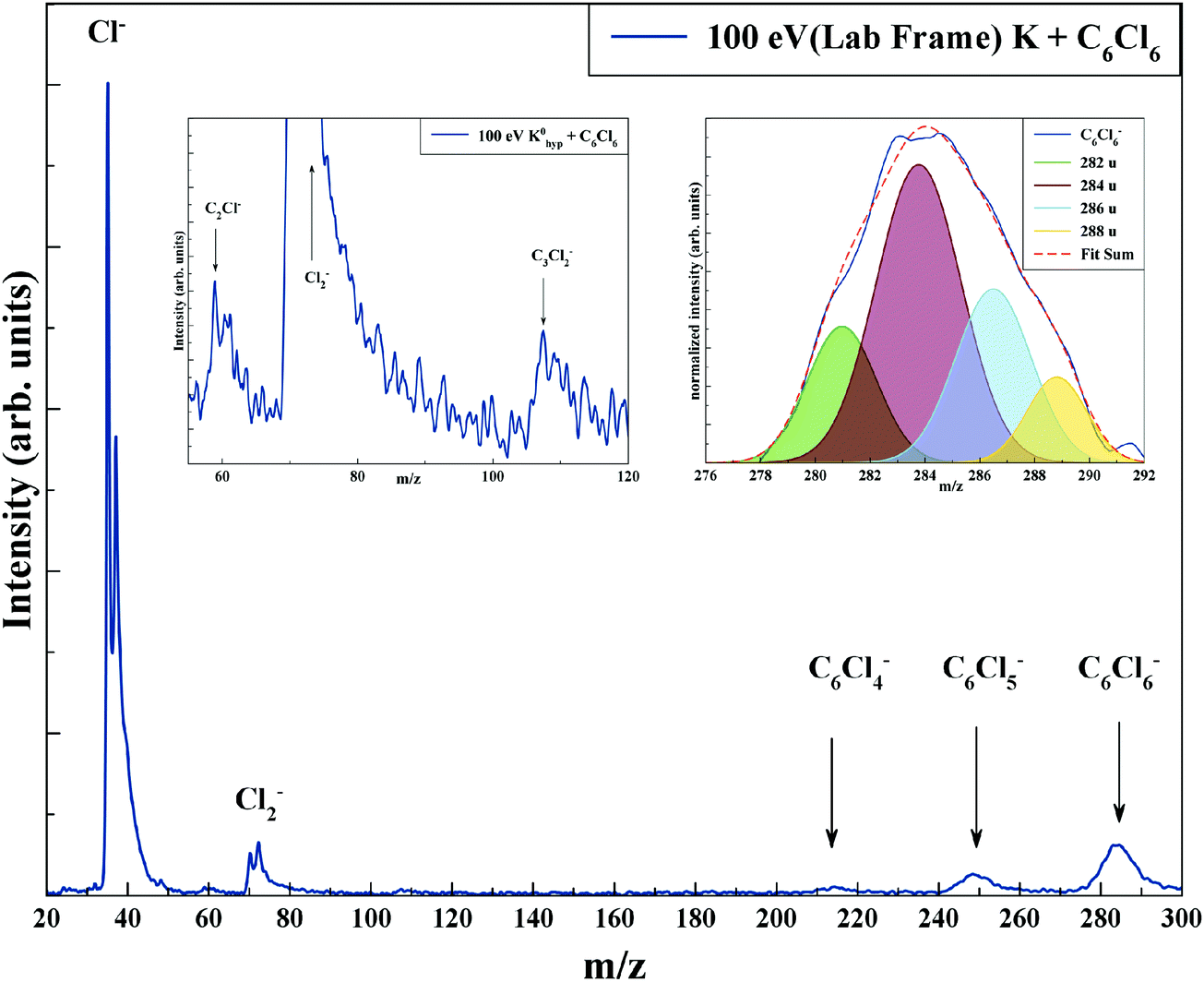 | ||
| Fig. 3 Time-of-flight negative ion mass spectra in potassium (K)–hexachlorobenzene (C6Cl6) collisions at 100 eV lab frame energy (79.1 eV in the centre-of-mass frame). See text for details. | ||
The energy loss spectrum of the potassium cations formed in the forward direction (θ ≅ 0°) of K atoms in collisions with C6Cl6 at 205 eV lab frame energy (ECM = 162 eV) is shown in Fig. 4. Hexachlorobenzene branching ratios (fragment anion yield/total anion yield) of the main negative ions formed as a function of collision energy in the centre-of-mass frame are depicted in Fig. 5.
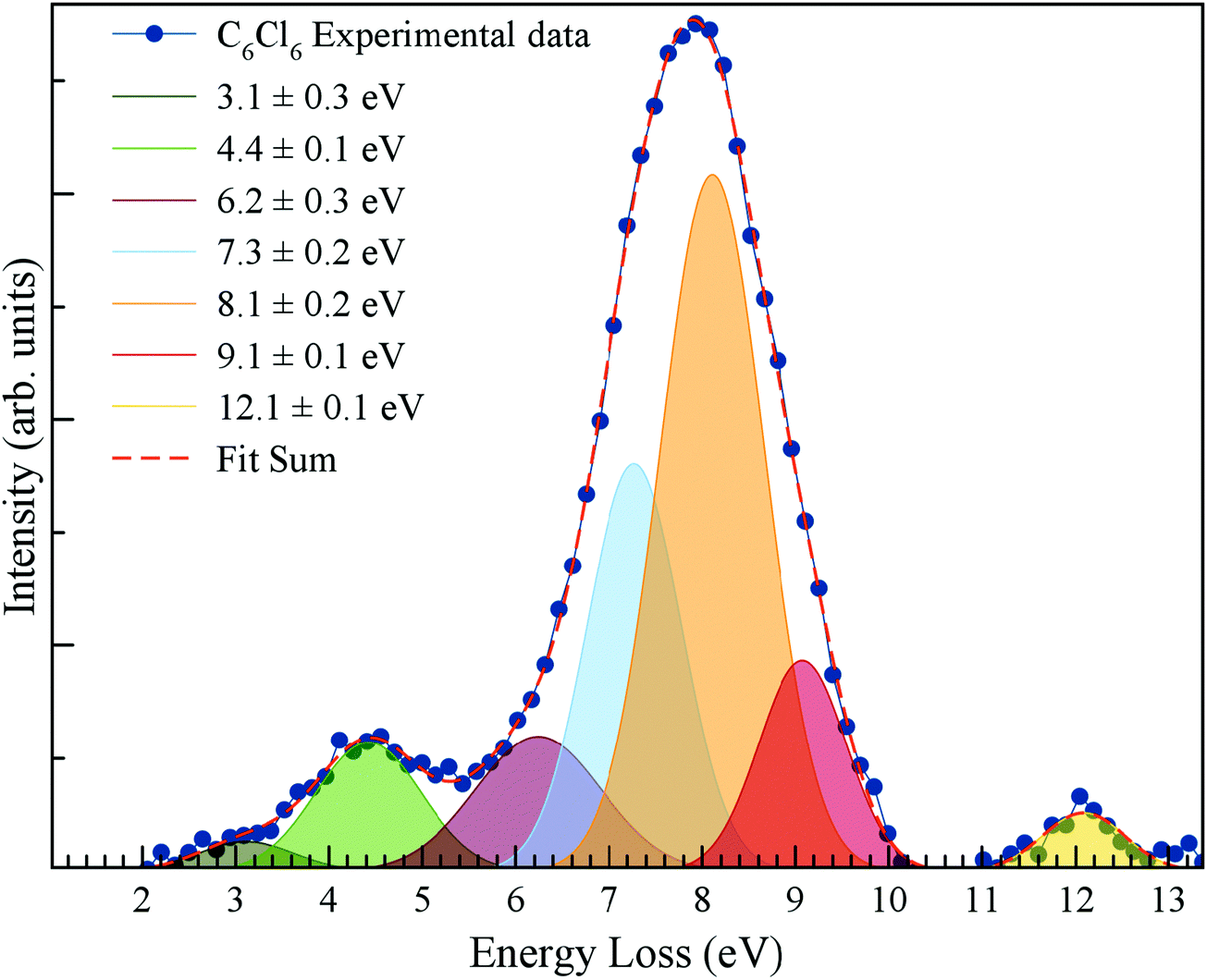 | ||
| Fig. 4 Energy loss spectrum of K+ ions formed in the forward direction of K atoms in collisions with C6Cl6 at 205 eV lab frame energy (162 eV in the centre-of-mass frame). See text for details. Error bars are within the experimental data points and within 10% associated with the fitting. | ||
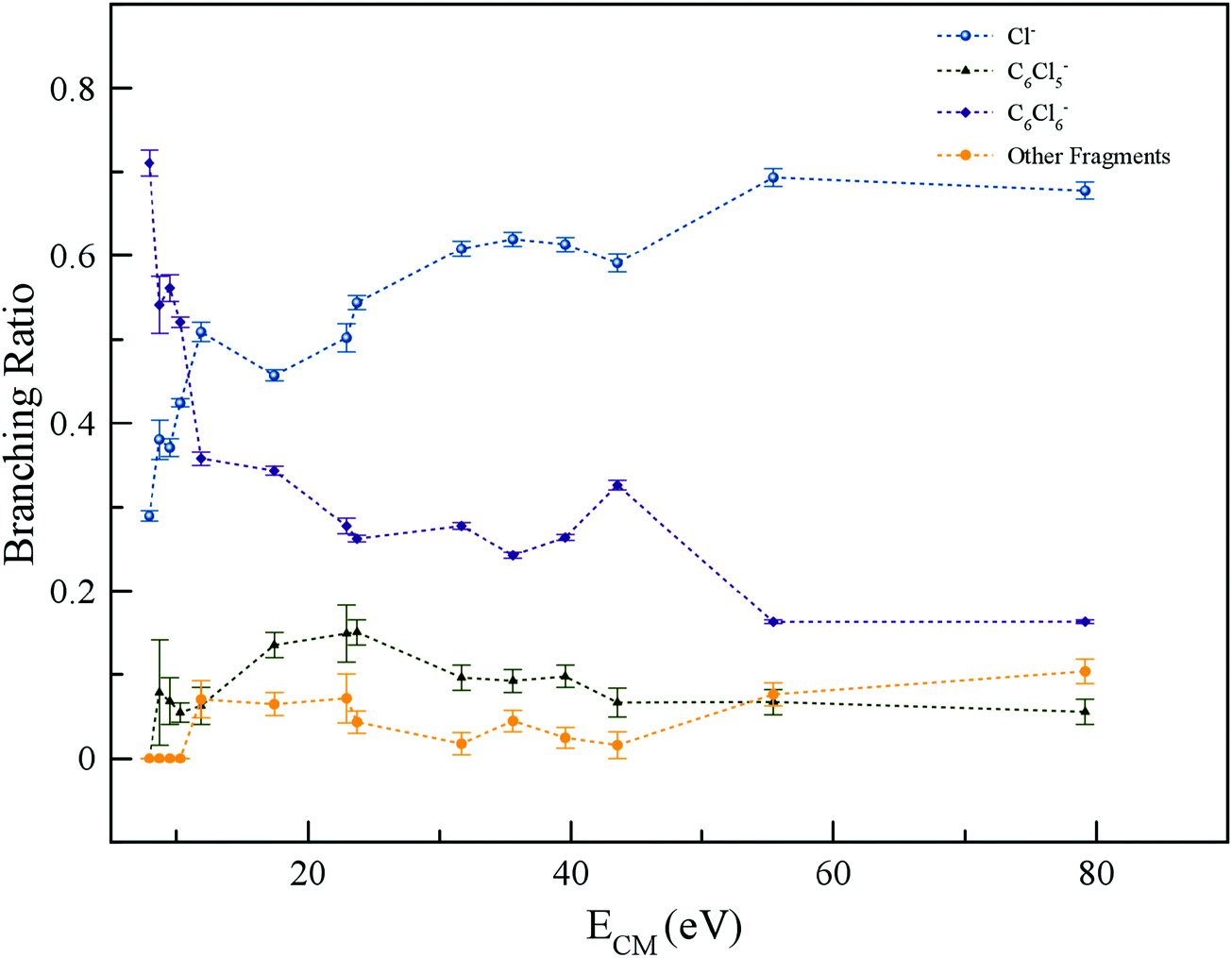 | ||
| Fig. 5 Hexachlorobenzene (C6Cl6) branching ratios (fragment anion yield/total anion yield) of the main negative ions formed as a function of the collision energy in the centre-of-mass frame. Error bars related to the experimental uncertainty associated with the ion yields have been added to a few data points in order to avoid congestion of the figure. The lines are just to guide the eye. See text for details. | ||
From a close inspection of TOF mass spectra in Fig. 3, the prevalent anion is assigned to Cl−, followed by the undissociated parent anion and Cl2−, while other fragment ions are considerably less intense, viz. those resulting from the loss of Cl units and ring breaking. We have performed additional calculations on C6Cl6− to obtain the energy values of the orbitals with the restricted open shell Kohn–Sham B3LYP + D3 level of theory (see Fig. S3, ESI†), where the electron spin densities show a preferential C–Cl bond excision yielding the chlorine anion (see the discussion below). The calculated lowest unoccupied molecular orbitals in Fig. 2 show that the LUMO+4 and LUMO+5 σ* states are slightly shifted to lower energies (0.8–1.0 eV) in comparison to the respective calculated MOs without the presence of the potassium atom.
4.1 K+ energy loss data
The energy loss spectrum of K+ ions formed during collisions of potassium atoms with hexachlorobenzene has been obtained at 162 eV in the centre-of-mass frame (205 eV lab frame) in the forward direction (θ ≅ 0°) and is depicted in Fig. 4. We observe for the first time some low-intensity features and a main peak with maximum intensity (Imax) at (8.1 ± 0.2) eV. Electron transfer processes from the collision of neutral potassium atoms (K) with neutral target molecules and yielding ion-pair formation, the energy loss required to access an electronic state, ΔE = IE(K) − EA(Imax), with the difference between the potassium atom ionisation energy and the target molecule's electron affinity for that state,42 which results in a vertical electron affinity of (−3.76 ± 0.20) eV. Gaussian fittings have been used to decompose the energy loss spectrum main peak, indicating the presence of adjacent contributions, as well as other low-intensity less resolved features with their vertical electron affinities listed in Table 1. Note that Voora16 has assigned the lowest-lying character of C6Cl6− to a valence-bound (vb) π* state with the electron affinity EA(C6Cl6) varying from 0.03 up to 0.30 eV, depending on the basis set used, and an associated error of 0.13 eV.| K+ energy loss features (eV) | VEA (eV) | Calculated VE of MO (eV) | Assignment | Ref. 16 | AEA13,17 (eV) |
|---|---|---|---|---|---|
| a Adiabatic electron affinity. b Valence-bound state, with calculated electron affinities of 0.03 eV (aug-cc-pVDZ + 7S7P), 0.30 eV (aug-cc-pVTZ + 7S7P) and 0.29 eV (aug-cc-pVTZ). See Voora16 for details. | |||||
| 3.1 ± 0.3 | 1.24 ± 0.30 | 0.98a | 0.91; 0.98 | ||
| 4.4 ± 0.1 | −0.06 ± 0.10 | π* | vbb-π* | ||
| 6.2 ± 0.3 | −1.86 ± 0.30 | −1.92 | 71, 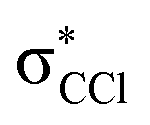 |
||
| 7.3 ± 0.2 | −2.96 ± 0.20 | −3.10 | LUMO+1 | ||
| 8.1 ± 0.2 | −3.76 ± 0.20 |
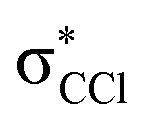
|
|||
| 9.1 ± 0.1 | −4.76 ± 0.10 | π* | |||
| 12.1 ± 0.1 | −7.76 ± 0.10 | n Cl → (n + 1)s | |||
| n Cl → (n + 2)s | |||||
4.2 C6Cl6− formation
The TOF mass spectra recorded in the wide collision energy range show that the undissociated parent anion accounts for more than 70% of the total ion yield for ECM < 10 eV, whereas above 50 eV its BR is ∼20% (Fig. 5). These results indicate that electron transfer is very efficiently captured by hexachlorobenzene and the process may proceed according to the reaction:| K + C6Cl6 → (K+ C6Cl6−) → K+ + (C6Cl6−)# → K+ + C6Cl6− | (1) |
The attachment of an electron is accompanied by significant energy release comprising the kinetic energy of the incoming electron and the molecule's electron affinity, which was calculated to be −0.14 eV (vertical) and 0.98 eV (adiabatic) at the RKS/B3LYP + D3/aug-cc-pVTZ level of theory (Table S3, ESI†), with the latter in good agreement with 0.91 eV and 0.98 eV from studies by Knighton et al.13 and Wiley et al.,17 respectively. Since the collision energy is always above the threshold of ion-pair formation, the excess energy of the TNI (metastable) can lead to statistical and/or direct dissociation.41 Formation of metastable parent anions and their detection within the TOF mass window (<80 μs) have been observed in electron transfer experiments with cyclic and non-cyclic molecular compounds. The former may enable an efficient redistribution of the excess energy through the different internal degrees of freedom, e.g. nitroimidazoles,43,44 and the latter can be rationalised in terms of an efficient door-way for enhanced bond breaking such as nitromethane.20 Hexachlorobenzene is a highly symmetric molecule with 30 vibrational degrees of freedom, thus providing an effective mean for energy redistribution, enhancing the C6Cl6− yield in the low-energy collision range. As the collision energy is further increased, the parent anion yield decreases, meaning that in such an energy range, more energy is transferred to the target molecule enhancing fragmentation.
In the energy loss data of Fig. 4, the weak feature peaking at (3.1 ± 0.3) eV yields a positive electron affinity of (1.24 ± 0.3) eV. Note that the asymptotic limit of C6Cl5 + Cl− is 0.56 eV, i.e. 0.35 eV below the ground state of the neutral15,19 (C6Cl6) meaning that the feature at 1.24 eV does not lead to bond breaking which results in C6Cl6− formation. There is no available information in the literature on the precise energy value of C–Cl stretching mode of the parent anion (372 cm−1 for the neutral ground state12); thus the lower limit of the electron affinity (0.94 eV) is in very good agreement with the adiabatic values reported by Knighton et al.13 and Wiley et al.,17 0.91 and 0.98 eV, respectively, as well as the calculated value at the RKS/B3LYP + D3/aug-cc-pVTZ level of theory (see Table 1 and Table S3, ESI†).
4.3 C6Cl5− and Cl− formation
The TOF mass spectra in Fig. 3 are dominated by Cl− formation in the collision energy range above 10 eV, amounting to ∼70% of the total anion yield above 40 eV (see BRs in Fig. 5). The dominance of such anion formation does not seem unexpected given the considerable high electron affinity of Cl (3.6131 eV45). Moreover, in the energy range above 40 eV, the Cl− branching ratio shows a rather constant behaviour which is reminiscent of the fast collision dynamics (collision time < 37 fs) dictated by direct vertical access within the Franck–Condon region to the ionic states. Hence, those prominent strong antibonding character potential energy surfaces above the ground state are attainable. The collision is therefore mostly dictated by electron promotion into a σ*(C–Cl) molecular orbital as depicted in Fig. 2 (LUMO+5). Formation of C6Cl5− and/or Cl− occurs after cleavage of a C–Cl bond in the TNI and is a result from a complementary reaction with respect to the negative charge:| K + C6Cl6 → (K+ C6Cl6−) → K+ + (C6Cl6−)# → K+ + C6Cl5− + Cl | (2) |
| K + C6Cl6 → (K+ C6Cl6−) → K+ + (C6Cl6−)# → K+ + Cl− + C6Cl5 | (3) |
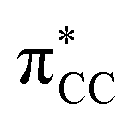 character. In such a delocalized system, all six identical chlorine atoms compete for the extra charge, with the excess energy being channelled into the available degrees of freedom, resulting in a stable parent anion rather than prevalent bond breaking. The shape of a selection of C6Cl6− molecular orbitals calculated at the RKS/B3LYP + D3 level of theory (see Fig. S3, ESI†) reveals that the SOMO has a πCCl character while the next MO shows a strong
character. In such a delocalized system, all six identical chlorine atoms compete for the extra charge, with the excess energy being channelled into the available degrees of freedom, resulting in a stable parent anion rather than prevalent bond breaking. The shape of a selection of C6Cl6− molecular orbitals calculated at the RKS/B3LYP + D3 level of theory (see Fig. S3, ESI†) reveals that the SOMO has a πCCl character while the next MO shows a strong 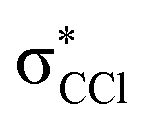 antibonding nature. Effective bond breaking yielding Cl− can only be achieved by efficient non-adiabatic curve crossing between
antibonding nature. Effective bond breaking yielding Cl− can only be achieved by efficient non-adiabatic curve crossing between 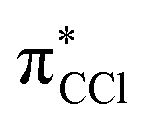 and
and 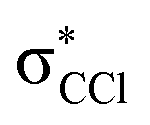 (see Fig. S1 (ESI†) and 72, π* and 73,
(see Fig. S1 (ESI†) and 72, π* and 73, 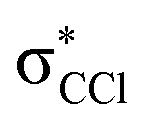 in Fig. S3 (ESI†)), indicating that the nuclear wave packet in the C–Cl coordinate survives long enough for the system to change its character, resulting in the formation of a chlorine anion. However, as the collision energy is increased, the number of electronic excited states being accessed also increases, the MOs are mostly
in Fig. S3 (ESI†)), indicating that the nuclear wave packet in the C–Cl coordinate survives long enough for the system to change its character, resulting in the formation of a chlorine anion. However, as the collision energy is increased, the number of electronic excited states being accessed also increases, the MOs are mostly 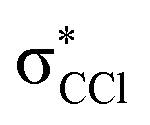 in character (Fig. S3, ESI†) and so direct bond cleavage resulting in Cl− formation is expected to be more favourable. The Cl− BR in Fig. 5 shows clearly that trending behaviour as the collision energy is increased above ECM = 20 eV. Moreover, in the high energy collision region, one should not discard that such anion formation can also proceed through shape and/or core-excited resonances, and an example for the latter is relaxing into a dissociative state by internal conversion.
in character (Fig. S3, ESI†) and so direct bond cleavage resulting in Cl− formation is expected to be more favourable. The Cl− BR in Fig. 5 shows clearly that trending behaviour as the collision energy is increased above ECM = 20 eV. Moreover, in the high energy collision region, one should not discard that such anion formation can also proceed through shape and/or core-excited resonances, and an example for the latter is relaxing into a dissociative state by internal conversion.
The K+ energy loss spectrum obtained in the forward direction shows the main feature at (8.1 ± 0.2) eV (Fig. 4) with a vertical electron affinity of (−3.76 ± 0.20) eV. This can be assigned to a purely repulsive transition from the C6Cl6 ground state to a 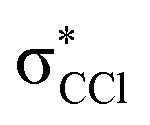 state of the temporary negative ion yielding Cl− formation, given that this is the most intense fragment anion formed in potassium–hexachlorobenzene collisions above 10 eV.
state of the temporary negative ion yielding Cl− formation, given that this is the most intense fragment anion formed in potassium–hexachlorobenzene collisions above 10 eV.
The energy loss feature peaking at 6.2 ± 0.3 eV (Fig. 4) results from accessing an electronic state at 1.86 ± 0.30 eV above the neutral molecule. The calculated molecular orbital for hexachlorobenzene anion at 1.92 eV (Fig. S3, ESI†) shows a remarkable 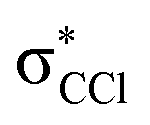 antibonding character with the extra charge sitting on the C6Cl5 radical (Fig. S3, ESI†). If we now take the ionisation energy of the potassium atom as 4.34 eV45 and the C6Cl5 electron affinity (see above) from the appearance energy (AE) in the energy loss spectrum (∼4.5 eV), we can estimate the C6Cl5–Cl bond dissociation energy. Thus, D(C6Cl5–Cl) = AE(C6Cl5−) − IE(K) + EA(C6Cl5) = (3.26 ± 0.30) eV, which is in excellent accordance with the calculated value of 3.297 eV (318.1 kJ mol−1) by Yamada and co-workers19.
antibonding character with the extra charge sitting on the C6Cl5 radical (Fig. S3, ESI†). If we now take the ionisation energy of the potassium atom as 4.34 eV45 and the C6Cl5 electron affinity (see above) from the appearance energy (AE) in the energy loss spectrum (∼4.5 eV), we can estimate the C6Cl5–Cl bond dissociation energy. Thus, D(C6Cl5–Cl) = AE(C6Cl5−) − IE(K) + EA(C6Cl5) = (3.26 ± 0.30) eV, which is in excellent accordance with the calculated value of 3.297 eV (318.1 kJ mol−1) by Yamada and co-workers19.
4.4 Other fragments
The branching ratios in Fig. 5 show that other fragment anions account for ≤20% of the total anion yield and these have been assigned to C6Cl4−, C3Cl2−, C2Cl− and Cl2−. The possible reaction mechanisms that can be involved in such anions’ formation are:| K + C6Cl6 → (K+ C6Cl6−) → K+ + (C6Cl6−)# → K+ + C6Cl4− + Cl2 | (4a) |
| K + C6Cl6 → (K+ C6Cl6−) → K+ + (C6Cl6−)# → K+ + C6Cl4− + Cl + Cl | (4b) |
| K + C6Cl6 → (K+ C6Cl6−) → K+ + (C6Cl6−)# → K+ + C3Cl2− + C3Cl4 | (5) |
| K + C6Cl6 → (K+ C6Cl6−) → K+ + (C6Cl6−)# → K+ + C2Cl− + C4Cl4 + Cl | (6) |
| K + C6Cl6 → (K+ C6Cl6−) → K+ + (C6Cl6−)# → K+ + Cl2− + C6Cl4 | (7) |
The TOF mass spectra in the inset in Fig. 3 show two weak signals that have been assigned to C3Cl2− and C2Cl− with proposed mechanisms in reactions (5) and (6). We note that these anions are only formed at 100 eV collision energy lab frame, while the latter has been reported by MacNeil and Thynne41 in ion-pair formation from ionisation of tetrachloroethylene. Due to the lack of any other relevant information in the literature regarding either resonances from dissociative electron attachment or any gas-phase thermochemistry data, it is impossible to estimate any thermodynamic thresholds for reactions (5) and (6) producing C3Cl2− and C2Cl− and their associated neutral radicals.
Finally, we detain ourselves with Cl2− formation from reaction (7) where two C–Cl bonds have to be broken and a molecular chlorine anion has to be formed. From the electron affinity value of Cl2 to be (2.5 ± 0.2) eV,45 the above bond dissociation energy D(C6Cl5–Cl) = (3.26 ± 0.30) eV, taking the available data on D(C–Cl) = (3.3 ± 0.3) eV46 and D(Cl–Cl) = 2.52 eV,47 and after adding the potassium ionisation energy, the appearance energy of reaction (7) is given by:
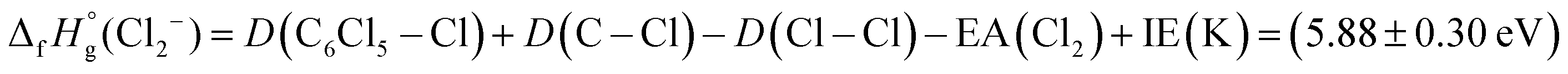 | (7.1) |
The K+ energy loss features at (9.1 ± 0.1) and (12.1 ± 0.1) eV (Fig. 4) with vertical electron affinities of (−4.76 ± 0.10) and (−7.76 ± 0.10) eV are tentatively assigned to core-excited resonances of π* character and Rydberg excitation, respectively (Table 1). The former is closely related to electronic excitation of the neutral molecule at 4.96 eV to the 1B1u state,8 while Robin reports a value of 5.27 eV;9 the latter can be assigned to the series nCl → ns converging to (e1g−1) at 9.19 eV.12 Due to the large number of states which occur in this high energy region, Rydberg assignment is rather difficult to perform, so the series are labelled either as (n + 1) or (n + 2).
5 Conclusions
We have presented a novel comprehensive study on negative ion formation during the collision of neutral potassium atoms with neutral hexachlorobenzene molecules in the lab frame energy range from 10 up to 100 eV using TOF mass spectrometry. The TOF mass spectra are dominated by the undissociated parent anion C6Cl6−, and Cl− and together with C6Cl5−, these anions contribute to more than 80% of the ions recorded, while other fragments assigned to C6Cl4−, C3Cl2−, C2Cl− and Cl2−, contributed less than 20% of the total anion yield. Theoretical calculations on the vertical excitation energies were performed at different levels of theory to help us in assigning the electronic transitions.Potassium cation post-collision energy loss data have been obtained in the forward direction (θ ≅ 0°) at 162 eV in the centre-of-mass frame (205 eV lab frame), with a dominant feature assigned to an electronic transition with a vertical electron affinity of (−3.76 ± 0.20) eV. This has been assigned to a purely repulsive transition from the C6Cl6 ground state to a 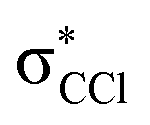 state of the temporary negative ion yielding Cl− formation. The energy loss data have revealed a weak feature at (3.1 ± 0.3) eV, yielding a positive electron affinity of (1.24 ± 0.3) eV, where its lower limit value (0.94 eV) is assigned to C6Cl6 adiabatic electron affinity and is in very good agreement with the values reported in the literature. Moreover, the C6Cl5− and Cl2− thresholds of formation have been obtained from the experimental energy loss data, with an estimated bond dissociation energy D(C6Cl5–Cl) = (3.26 ± 0.30) eV for the former.
state of the temporary negative ion yielding Cl− formation. The energy loss data have revealed a weak feature at (3.1 ± 0.3) eV, yielding a positive electron affinity of (1.24 ± 0.3) eV, where its lower limit value (0.94 eV) is assigned to C6Cl6 adiabatic electron affinity and is in very good agreement with the values reported in the literature. Moreover, the C6Cl5− and Cl2− thresholds of formation have been obtained from the experimental energy loss data, with an estimated bond dissociation energy D(C6Cl5–Cl) = (3.26 ± 0.30) eV for the former.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK acknowledges the Portuguese National Funding Agency (FCT) through PD/BD/142831/2018, and together with PLV the research grants CEFITEC (UIDB/00068/2020) and PTDC/FIS-AQM/31281/2017. This work was also supported by Radiation Biology and Biophysics Doctoral Training Programme (RaBBiT, PD/00193/2012); UCIBIO (UIDB/04378/2020). All the calculations were performed at the CI TASK Computer Centre in Gdańsk. The work of MŁ is based upon support from COST Action CA18212 – Molecular Dynamics in the GAS phase (MD-GAS). GG acknowledges partial financial support from the Spanish Ministerio de Ciencia e Innovación (Project No. PID2019-104727RB-C21) and CSIC (Project LINKA20085).References
- W. W. Brubaker and R. A. Hites, Experimental section, Environ. Sci. Technol., 1998, 32, 766–769 CrossRef CAS
.
- M. P. Ormad, N. Miguel, A. Claver, J. M. Matesanz and J. L. Ovelleiro, Pesticides removal in the process of drinking water production, Chemosphere, 2008, 71, 97–106 CrossRef CAS PubMed
-
Toxicologic profile of hexachlorobenzene, U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry, Division of Toxicology and Human Health Sciences, Environmental Toxicology Branch, Atlanta, Georgia, 2015 Search PubMed
- S. L. Simonich and R. A. Hites, Global distribution of persistent organochlorine compounds, Science, 1995, 269, 1851–1854 CrossRef CAS PubMed
- G. W. Patton, M. D. Walla, T. F. Bidleman and L. A. Barrie, Polycyclic aromatic and organochlorine compounds in the atmosphere of northern Ellesmere Island, Canada, J. Geophys. Res., 1991, 96, 10867–10877 CrossRef
- M. Oehme, Further evidence for long-range air transport of polychlorinated aromates and pesticides: North America and Eurasia to the Arctic, Ambio, 1991, 20, 293–297 Search PubMed
- P. Roche and M. Prados, Removal of pesticides by use of ozone or hydrogen peroxide/ozone, Ozone: Sci. Eng., 1995, 17, 657–672 CrossRef CAS
- H. Scharping, C. Zetzsch and H. A. Dessouki, The UV absorption spectra of the trichlorobenzenes and the higher chlorinated benzenes in the gas phase and in n-hexane solution, J. Mol. Spectrosc., 1987, 123, 382–391 CrossRef CAS
-
M. B. Robin, Higher Excited States of Polyatomic Molecules, Volume II, Academic Press, 1975 Search PubMed
- S. Saëki, The assignment of the molecular vibrations of hexachlorobenzene, Bull. Chem. Soc. Jpn., 1962, 35, 322–326 CrossRef
- J. R. Scherer and J. C. Evans, Vibrational spectra and assignments for sixteen chlorobenzenes, Spectrochim. Acta, 1963, 19, 1739–1775 CrossRef CAS
- B. Ruščić, L. Klasinc, A. Wolf and J. V. Knop, Photoelectron spectra of and ab initio calculations on chlorobenzenes. 3. Hexachlorobenzene, J. Phys. Chem., 1981, 85, 1495–1497 CrossRef
- W. B. Knighton, J. A. Bognar and E. P. Grimsrud, Reactions of selected molecular anions with oxygen, J. Mass Spectrom., 1995, 30, 557–562 CrossRef CAS
- P. P. Romańczyk, G. Rotko and S. S. Kurek, Dissociative electron transfer in polychlorinated aromatics. Reduction potentials from convolution analysis and quantum chemical calculations, Phys. Chem. Chem. Phys., 2016, 18, 22573–22582 RSC
- T. Kato and T. Yamabe, Vibronic interactions and charge transfer in negatively charged chloroacenes, Chem. Phys., 2006, 321, 149–158 CrossRef CAS
- V. K. Voora, Molecular electron affinities using the generalized Kohn–Sham semicanonical projected random phase approximation, J. Phys. Chem. Lett., 2021, 12, 433–439 CrossRef CAS PubMed
- J. R. Wiley, E. C. M. Chen, E. S. D. Chen, P. Richardson, W. R. Reed and W. E. Wentworth, The determination of absolute electron affinities of chlorobenzenes, chloronaphthalenes and chlorinated biphenyls from reduction potentials, J. Electroanal. Chem., 1991, 307, 169–182 CrossRef CAS
- W. Xu and A. Gao, DFT study on the electron affinities of the chlorinated benzenes, THEOCHEM, 2005, 732, 63–70 CrossRef CAS
- S. Yamada, Y. Naito, M. Takada, S. Nakai and M. Hosomi, Photodegradation of hexachlorobenzene and theoretical prediction of its degradation pathways using quantum chemical calculation, Chemosphere, 2008, 70, 731–736 CrossRef CAS PubMed
- R. Antunes, D. Almeida, G. Martins, N. J. Mason, G. Garcia, M. J. P. Maneira, Y. Nunes and P. Limão-Vieira, Negative ion formation in potassium–nitromethane collisions, Phys. Chem. Chem. Phys., 2010, 12, 12513–12519 RSC
- K. Regeta, S. Kumar, T. Cunha, M. Mendes, A. I. Lozano, P. J. S. Pereira, G. García, A. M. C. Moutinho, M. C. Bacchus-Montabonel and P. Limão-Vieira, Combined experimental and theoretical studies on electron transfer in potassium collisions with CCl4, J. Phys. Chem. A, 2020, 124, 3220–3227 CrossRef CAS PubMed
- E. Bene, Á. Vibók, G. J. Halász and M. C. Bacchus-Montabonel,
Ab initio molecular treatment of charge transfer processes induced by collision of C2+ ions with the OH radical: A linear approach, Chem. Phys. Lett., 2008, 455, 159–163 CrossRef CAS
- M. C. Bacchus-Montabonel and Y. S. Tergiman, Radiation damage on biomolecular systems: Dynamics of ion induced collision processes, Comput. Theor. Chem., 2012, 990, 177–184 CrossRef CAS
- M. C. Bacchus-Montabonel and Y. S. Tergiman, Charge transfer dynamics of carbon ions with uracil and halouracil targets at low collision energies, Chem. Phys. Lett., 2011, 503, 45–48 CrossRef CAS
- M. C. Bacchus-Montabonel, M. Łabuda, Y. S. Tergiman and J. E. Sienkiewicz, Theoretical treatment of charge-transfer processes induced by collision of Cq+ ions with uracil, Phys. Rev. A, 2005, 72, 052706 CrossRef
- E. Erdmann, M.-C. Bacchus-Montabonel and M. Łabuda, Modelling charge transfer processes in C2+-tetrahydrofuran collision for ion-induced radiation damage in DNA building blocks, Phys. Chem. Chem. Phys., 2017, 19, 19722–19732 RSC
- M. Mendes, B. Pamplona, S. Kumar, F. F. da Silva, A. Aguilar, G. García, M. C. Bacchus-Montabonel and P. Limão-Vieira, Ion-pair formation in neutral potassium-neutral pyrimidine collisions: Electron transfer experiments, Front. Chem., 2019, 7, 1–10 CrossRef
- D. Almeida, M.-C. Bacchus-Montabonel, F. Ferreira da Silva, G. García and P. Limão-Vieira, Potassium-uracil/thymine ring cleavage enhancement as studied in electron transfer experiments and theoretical calculations, J. Phys. Chem. A, 2014, 118, 6547–6552 CrossRef CAS PubMed
-
L. Salem, Electrons in Chemical Reactions: First Principles, Wiley Interscience, New York, 1982 Search PubMed
- M. C. Bacchus-Montabonel and Y. S. Tergiman, An ab initio study of ion induced charge transfer dynamics in collision of carbon ions with thymine, Phys. Chem. Chem. Phys., 2011, 13, 9761–9767 RSC
- M. C. Bacchus-Montabonel,
Ab initio treatment of ion-induced charge transfer dynamics of isolated 2-deoxy-D-ribose, J. Phys. Chem. A, 2014, 118, 6326–6332 CrossRef CAS PubMed
- M. C. Bacchus-Montabonel and F. Calvo, Nanohydration of uracil: Emergence of three-dimensional structures and proton-induced charge transfer, Phys. Chem. Chem. Phys., 2015, 17, 9629–9633 RSC
- A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: A general-purpose quantum chemistry program package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS
- A. Nicklass, M. Dolg, H. Stoll and H. Preuss, Ab initio energy-adjusted pseudopotentials for the noble gases Ne through Xe: Calculation of atomic dipole and quadrupole polarizabilities, J. Chem. Phys., 1995, 102, 8942–8952 CrossRef CAS
-
B. O. Roos, Ab initio methods in Quantum Chemistry II, Wiley-VCH, Chichester, 1987 Search PubMed
- H. J. Werner and P. J. Knowles, A second order multiconfiguration SCF procedure with optimum convergence, J. Chem. Phys., 1985, 82, 5053–5063 CrossRef CAS
- P. J. Knowles and H. J. Werner, An efficient second-order MC SCF method for long configuration expansions, Chem. Phys. Lett., 1985, 115, 259–267 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- P. Limão-Vieira, A. M. C. Moutinho and J. Los, Dissociative ion-pair formation in collisions of fast potassium atoms with benzene and fluorobenzene, J. Chem. Phys., 2006, 124, 054306 CrossRef PubMed
- A. W. Kleyn and A. M. C. Moutinho, Negative ion formation in alkali-atom – molecule, J. Phys. B: At., Mol. Opt. Phys., 2001, 4075, R1–R44 CrossRef
- M. Mendes, M. Probst, T. Maihom, G. García and P. Limão-Vieira, Selective bond excision in nitroimidazoles by electron transfer experiments, J. Phys. Chem. A, 2019, 123, 4068–4073 CrossRef CAS PubMed
- M. Mendes, G. García, M. C. Bacchus-Montabonel and P. Limão-Vieira, Electron transfer induced decomposition in potassium–nitroimidazoles collisions: An experimental and theoretical work, Int. J. Mol. Sci., 2019, 20, 6170 CrossRef CAS PubMed
- http://webbook.nist.gov/chemistry/, http://webbook.nist.gov/chemistry/.
- E. Illenberger, Energetics of negative ion formation in dissociative electron attachment to CCl4, CFCl3, CF2Cl2, and CF3Cl, Ber. Bunsenges. Phys. Chem., 1982, 86, 252–261 CrossRef CAS
- H.-U. Scheunemann, E. Illenberger and H. Baumgartel, Dissociative electron attachment to CCl4, CHCl3, CH2C12 and CH3Cl, Ber. Bunsenges. Phys. Chem., 1980, 84, 580–585 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 to S3 and Tables S1 to S3 with the results of theoretical calculations for the selection of K + C6Cl6, C6Cl6 highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs), as well as ionisation energies, electron affinities and vertical detachment energies. See DOI: 10.1039/d1cp04500h |
| This journal is © the Owner Societies 2022 |