Recognition of quinolone antibiotics by the multidrug efflux transporter MexB of Pseudomonas aeruginosa†
Received
25th February 2022
, Accepted 13th May 2022
First published on 29th June 2022
Abstract
The drug/proton antiporter MexB is the engine of the major efflux pump MexAB-OprM in Pseudomonas aeruginosa. This protein is known to transport a large variety of compounds, including antibiotics, thus conferring a multi-drug resistance phenotype. Due to the difficulty of producing co-crystals, only two X-ray structures of MexB in a complex with ligands are available to date, and mechanistic aspects are largely hypothesized based on the body of data collected for the homologous protein AcrB of Escherichia coli. In particular, a recent study (Ornik-Cha, Wilhelm, Kobylka et al., Nat. Commun., 2021, 12, 6919) reported a co-crystal structure of AcrB in a complex with levofloxacin, an antibiotic belonging to the important class of (fluoro)-quinolones. In this work, we performed a systematic ensemble docking campaign coupled to the cluster analysis and molecular-mechanics optimization of docking poses to study the interaction between 36 quinolone antibiotics and MexB. We additionally investigated surface complementarity between each molecule and the transporter and thoroughly assessed the computational protocol adopted against the known experimental data. Our study reveals different binding preferences of the investigated compounds towards the sub-sites of the large deep binding pocket of MexB, supporting the hypothesis that MexB substrates oscillate between different binding modes with similar affinity. Interestingly, small changes in the molecular structure translate into significant differences in MexB–quinolone interactions. All the predicted binding modes are available for download and visualization at the following link: https://www.dsf.unica.it/dock/mexb/quinolones.
1 Introduction
Molecular recognition between protein and ligand complexes is driven by inter-molecular forces and shape complementarity.1 In the case of non-covalent bonds, binding can have either a polar (electrostatic interactions and hydrogen bonds) or an apolar (van der Waals and hydrophobic interactions) characteristic.2 In general, the higher the complementarity between polar and apolar surfaces, the higher the affinity of a compound to a protein target.3–5 For this reason, a detailed molecular-level knowledge of binding pockets of target proteins and binding modes of ligands is crucial.6 Molecular docking is a computational technique that, starting from a protein and a ligand alone, predicts putative structures of their complexes.7 In this work, we applied such a technique to the urgent issue of antimicrobial resistance.8 The massive use of antimicrobials in clinics and farms has made them increasingly ineffective, leading to the compromised treatment of bacterial infections.9–12 The World Health Organization pointed nosocomial ESKAPE pathogens11,13 with growing multidrug resistance (MDR) as high-priority.14 Gram-negative ESKAPE pathogens such as Pseudomonas aeruginosa are of particular concern and new antibiotics are urgently needed.15 Among the different mechanisms of resistance developed by bacteria, the ability to expel antimicrobial compounds out of the cell represents one of the major causes of the MDR.16 Efflux pumps belonging to the resistance-nodulation-division (RND) superfamily of proteins play a key role in the MDR phenotype of Gram-negative bacteria.17,18 The major RND efflux pump of P. aeruginosa is MexAB-OprM,19,20 which extends from the inner membrane to the outer membrane, and has been found to capture and expel various ligand classes with different physico-chemical properties.21 Among them, (fluoro)-quinolones (FQs) represent one of the most used drugs in the treatment of Gram-negative infection22 (Fig. 1); however, the synergistic action of the outer membrane (i.e., permeation barrier) and efflux pumps, in combination with an increasing frequency of target mutations, limits the therapeutic effects of these compounds.23–25
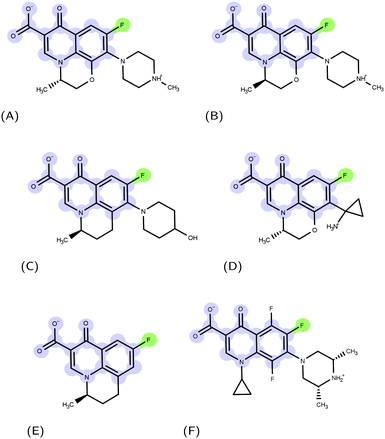 |
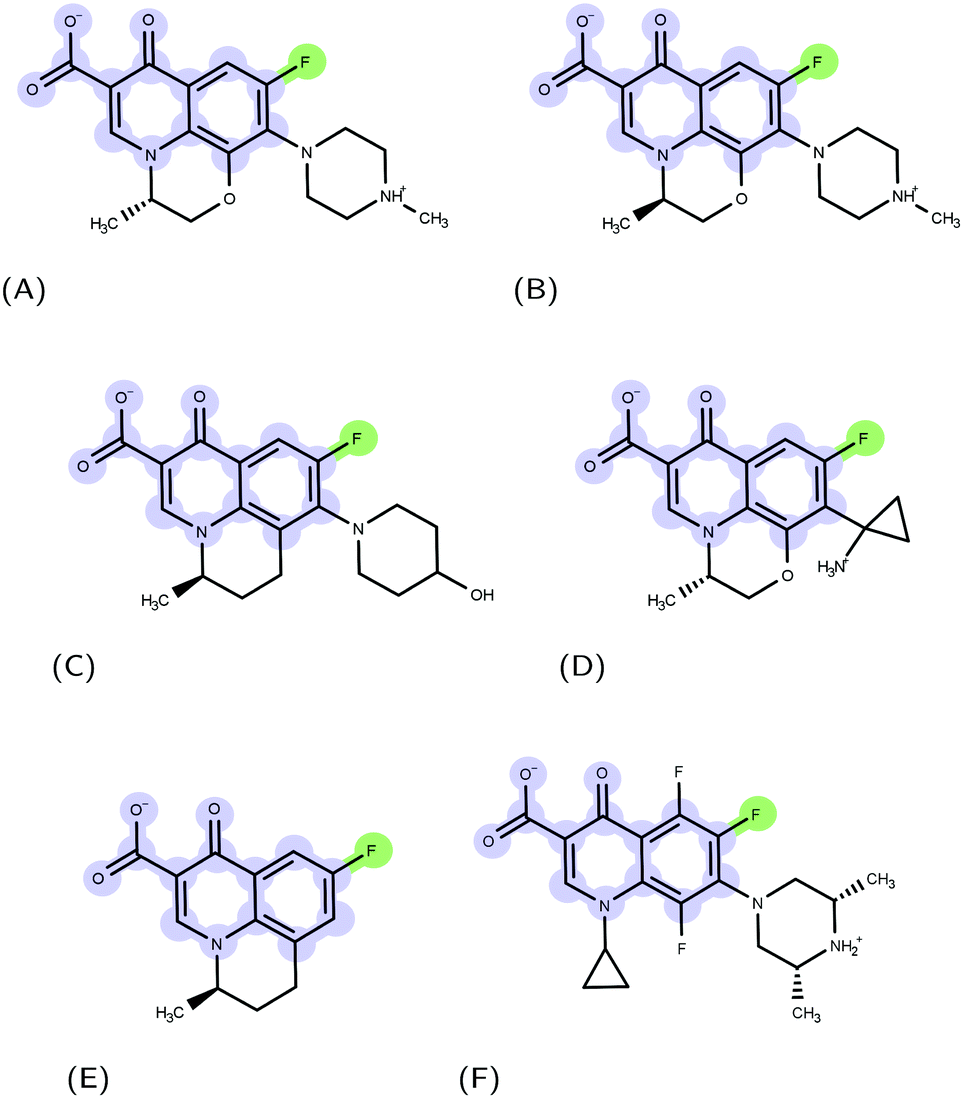
| | Fig. 1 Examples of FQ structures: (A) levofloxacin (LFX), (B) ofloxacin (OFX), (C) nadifloxacin (NFX), (D) pazufloxacin (PFX), (E) flumequine (FMQ), and (F) orbifloxacin. The molecular scaffold common to all quinolones and the fluorine atom characterizing fluoroquinolones are highlighted in blue and green, respectively. | |
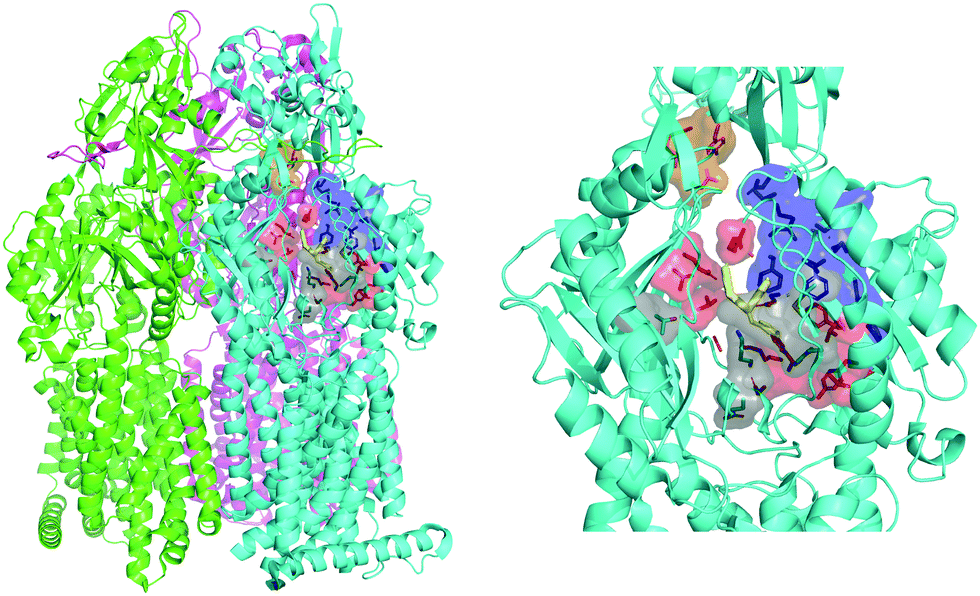
The engine of the MexAB-OprM complex is represented by the homo-trimeric inner membrane transporter MexB, homologous of the prototypical bacterial RND transporter AcrB from Escherichia coli.26 The two proteins share the same tertiary and quaternary structures with an overall sequence identity and similarity of 69.8 and 83.2%, respectively.27 The available crystal and cryo-EM studies of AcrB and MexB provided insights into the complex structure and dynamics of these systems,16,26,28–30 including the information about putative entry channels (see, e.g.,31,32) and the knowledge of ligand binding sites. According to the available crystal structures, the three monomers, although identical in their sequence, may adopt three different states, the so-called loose (L), tight (T), and open (O) (Fig. 2, see, e.g.,16,26,30). The two main binding sites were identified, a proximal pocket in the L monomer and a distal cavity in the T monomer33 (called the deep binding pocket, in the following DPT). It is thought that all compounds extruded by the transporter pass through the DPT, which is characterized by a cluster of phenylalanine and other hydrophobic residues, called hydrophobic trap (HT).16,29 Similar to AcrB,16 the pocket can be divided into three different sub-regions: an interface separating the DPT from the proximal pocket and containing the switch loop,34 a wide cave region, and a narrow groove located in the deeper portion (Fig. 2).
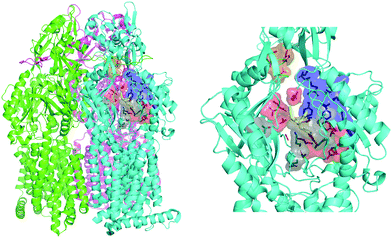 |
| | Fig. 2 MexB structure: the left panel highlights L, T, and O monomers of MexB colored in green, cyan, and magenta, respectively (PDB Id: 3W9J29). The detailed visualization on the right shows the different DPT sub-regions of MexB considered in this work: the interface is colored in gray (residues S79, T91, K134, F573, F617, M662, and E673), the cave is colored in red (residues Q46, T89, T130, N135, F136, V139, Q176, K292, Y327, V571, R620, and F628) and the groove is colored in blue (residues K151, F178, G179, R180, D274, S276, I277, A279, S287, P326, F610, V612, F615, and V47; S48, Q125, G126, R128, Q163, D174, F175, and Q273 that are located near the exit gate, colored in orange). The switch loop is represented as a yellow cartoon. | |
For AcrB, several crystal structures in a complex with antibiotics of different classes are available. In particular, the first crystal structure of a FQ molecule (levofloxacin, LFX) bound to the DPT of AcrB has been recently published.32 In contrast, only two crystal structures of MexB in a complex with a ligand (the inhibitor ABI-PP29 and the substrate LMNG35) are available. Computational studies can provide the detailed atomistic information on the binding of compounds to MexB.36,37 In the absence of structural data, in this work, we performed a systematic investigation on 36 FQs targeting MexB (the list of compounds is reported in Table S1, ESI†). Given the mentioned experimental evidence of the prominent role of DPT in the binding of MexB and AcrB substrates, here we focused only on this binding pocket. Ensemble docking followed by cluster analysis and molecular-mechanics optimization of docking poses were conducted to gain insights into the details of molecular recognition and found characteristic binding modes of FQs to MexB. Special emphasis has been placed to LFX and very similar compounds, differing only for small substitutions. We analysed the distribution of docking poses in the three sub-regions and the overall complementarity between the different FQs and the transporter. We found that all compounds can assume vastly diverse binding modes within the pocket, a result consistent with the highly promiscuous nature of MexB21 and the diffusive binding (or oscillation) hypothesis.27,38 According to this hypothesis, AcrB/MexB substrates oscillate between different binding modes with similar affinity within the DPT. The PDB files of the predicted binding modes of each FQ can be downloaded and visualized online at the web address https://www.dsf.unica.it/dock/mexb/quinolones.
2 Materials and methods
We combined ensemble docking with the cluster analysis of docking poses followed by energy minimization at the molecular-mechanics (MM) level and hydrophobic/hydrophilic surface matching calculations.
2.1 Molecular docking
Molecular docking calculations were performed using two different packages: the latest version of AutoDock VINA 1.2.3,39,40 implementing a stochastic global optimization approach, and the recently developed GNINA software,41 whose scoring function is based on convolution neural networks. For consistency, the same input files, structures, and settings were employed in both programs. Since AutoDock VINA 1.2.3 was applied using two scoring functions, VINA and AutoDock4 (AD4), three sets of independent runs were performed for each compound. The default settings were used in all cases, except for the exhaustiveness parameter (giving a measure of the exhaustiveness of the local search), for which we tested different values (ranging from 64 to 1024, default 8, see below). The protein and ligand input .pdbqt files were prepared using AutoDock tools.42 We used a rectangular box of size 30 × 30 × 30 Å3, centered at the center of mass of the DPT of the RND protein. While AutoDock VINA has proven to yield reliable results in previous studies of bacterial RND transporters,43,44 to assess the overall quality of the three docking scoring functions adopted in this study, we first performed re-docking calculations on the only available co-crystal structure of MexB (PDB Id: 3W9J in a complex with ABI-PP29) and on two AcrB crystal structures: that with the highest resolution (PDB Id: 4DX5, in a complex with minocycline45) and the one in a complex with the FQ molecule, LFX (PDB Id: 7B8T32). The receptor and ligand were kept frozen at the crystallographic conformation and hydrogen atoms were added according to physiological pH using the pdb2pqr46 and Marvin ChemAxon47 programs, respectively. Given the presence of the basic piperazine group, LFX was considered in two different protonation states: with a total net charge of −1 and in the zwitterionic form.48 For minocycline, we considered the zwitterionic form depicted in Fig. S1 (ESI†). We tested four different exhaustiveness values (i.e., 64, 128, 512, and 1064) and the performance of the protocol was evaluated by computing the ligand root-mean-square-displacement (RMSD) between the docking poses and the corresponding crystal structure. As shown in Table S2, (ESI†), highlighting docking poses with RMSD ≤ 2.5 Å, the poses reproducing the crystal structures were mainly ranked in the top positions in all cases. For the different protonation states of LFX, we found similar results, with a slightly better RMSD values for the zwitterionic form. The increase of the exhaustiveness parameters seems to be significant when increasing from 64 to 128, while values greater than 128 do not yield better results. Therefore, an exhaustiveness of 128 was chosen as the reference value for all the following runs.
Docking calculations using the ensemble of conformations of both the receptor and ligand make it possible to account indirectly for docking partner flexibility, generate multiple binding modes, and pinpoint the most frequently contacted protein residues.44,49,50 Before applying systematically the ensemble docking protocol to the full set of FQs, we tested its ability to reproduce the co-crystal conformations used for re-docking calculations. Specifically, for LFX and minocycline, we considered the AcrB crystal structures with PDB Ids 2GIF,512J8S52 (AcrB apo), 4DX545 (AcrB – minocycline), 4DX745 (AcrB – doxorubicin), 5JMN53 (AcrB – fusidic acid) and 3W9H29 (AcrB–ABI-PP). For ABI-PP, we used a combination of three crystal structures of MexB (PDB Ids: 2V50,543W9I, and 3W9J29) and three conformations extracted from molecular dynamics (MD) simulations.27 Given the importance of the membrane environment for the correct folding of transmembrane proteins,55–57 in these simulations, MexB was embedded in a double layer membrane made of phosphatidylethanolamine (POPE) and phosphatidylglycerol (POPG). This latter set of MexB structures combining X-ray conformations and MD snapshots was employed for the systematic investigation of FQs. For all considered ligands, FQs, minocycline and ABI-PP, we used ten conformations obtained from the cluster analysis of 1 μs-long all-atom MD simulations in water.58 For 11 FQs containing the piperazine group, we considered the two protonation states. In each docking run, we generated 10 poses obtaining a total of 1800 conformations per compound (3 docking protocols × 6 MexB structures × 10 FQ configurations × 10 poses). A summary of protein and ligand ensembles and docking settings is reported in Table S3 (ESI†).
2.2 Clustering, MM and surface-matching calculations
Following the same protocol used in a previous study,32 all 1800 docking poses generated by ensemble docking were clustered using a hierarchical algorithm,59 yielding 10 structural clusters. Using the Amber18 package,60 ten cluster representatives were energy optimized to refine protein–ligand interactions. The force field of ligands was retrieved from the freely available database AB-DB,58 while the protein was parameterized using the ff14SB force field.61 The resulting complexes were re-scored by the VINA affinity score39 and each representative was assigned to a DPT sub-region (i.e. interface, cave, and groove) according to the frequency of contacts to protein residues lining each sub-pocket (cut-off distance 3.5 Å). For compounds in which the number of contacts of two regions were the same, we splitted the contribution into the two regions. The visual inspection of all cluster representatives enabled distinct binding modes to be identified.
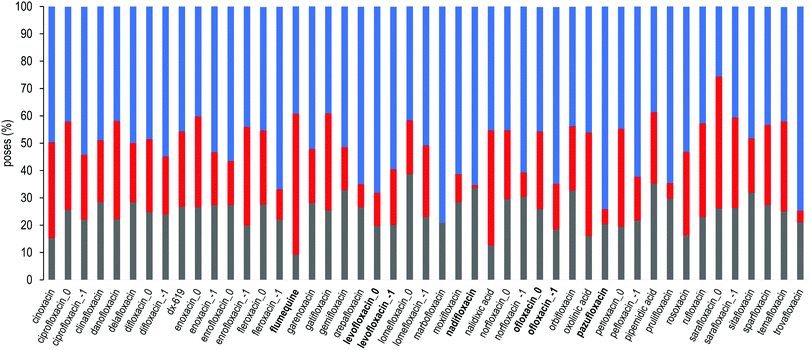
Finally, to further gain insights into the affinity of each compounds to the DPT of MexB, we used the PLATINUM web-server62 to obtain a quantitative characterization of the hydrophobic/hydrophilic surface match (SM). This method is based on empirical molecular hydrophobicity potential (MHP), which is used to calculate molecular hydrophobic/hydrophilic properties. Namely, the MHP at any point j, due to N atoms, is defined as follows:63,64
| | 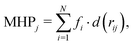 | (1) |
where
i and
fi are the atom number and the corresponding hydrophobicity constant and
d(
rij) is the distance function between the atom
i and the point
j. In this work, we adopted the same settings used in previous studies
44,65 to compute the fraction of the total (lipophilic and hydrophylic) SM
TOT and the lipophilic SM
L surface matches. Further technical details can be found in the ESI.
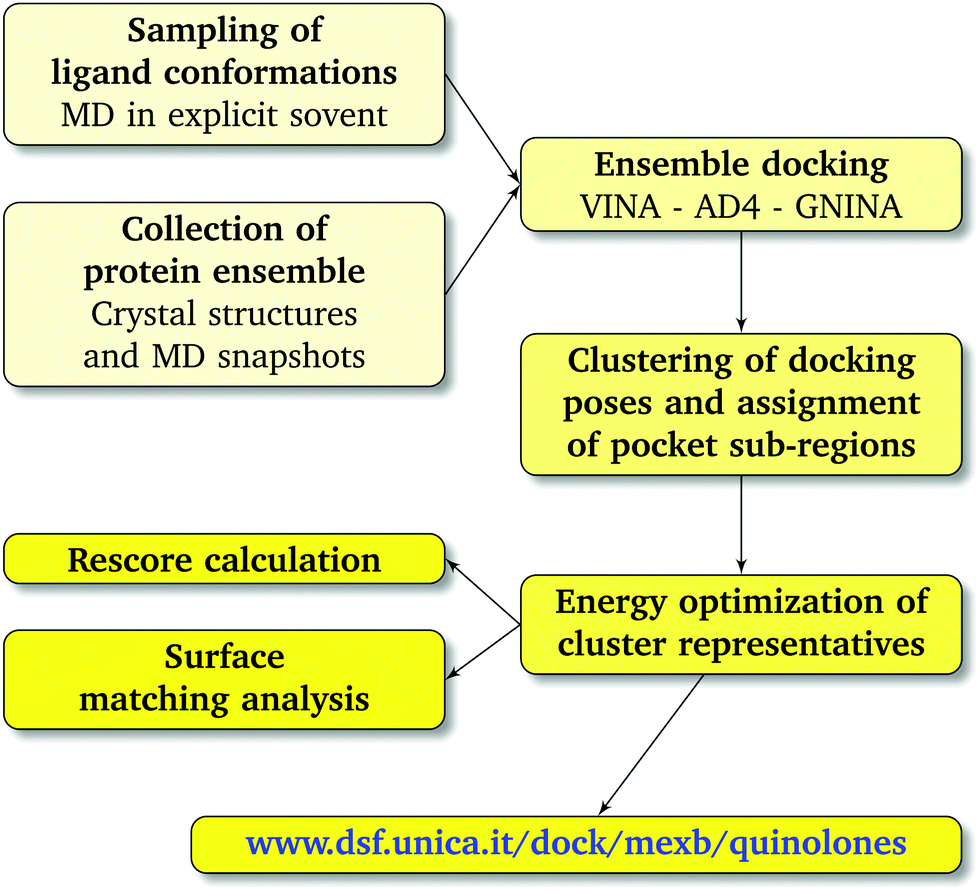
† The weighted average affinity and surface match values were associated with each cluster representative using its population as weight. The workflow used in this study is schematically represented in
Fig. 3. Energy-minimized cluster representatives for each compound can be downloaded and visualized online through the NGL-viewer.
66 All graphics are rendered using PyMOL.
67
 |
| | Fig. 3 Computational workflow used to investigate the binding of FQ antibiotics to the multidrug efflux transporter MexB of P. aeruginosa. | |
3 Results and discussion
We first applied our protocol to a reduced set of systems for which co-crystal structures are available, including the AcrB–LFX complex.32 The same methodology was then systematically extended to all FQs.
3.1 Available co-crystal structures
Table 1 presents the outcomes of our ensemble docking / cluster analysis / energy minimization and re-scoring for the ABI-PP–MexB, minocycline–AcrB, and LFX–AcrB complexes. For each conformational cluster, we report the corresponding population and the RMSD with respect to the co-crystallized ligand.
Table 1 Validation of the computational protocol – RMSD values with respect to the corresponding crystal structure are expressed in Å. Cluster population (%) is reported in parenthesis. Only the results of the top five most populated clusters are reported. LFX (0) and LFX (−1) refer to levofloxacin in the zwitterionic and non-zwitterionic (net charge −1) states, respectively
| Cluster |
AcrB |
MexB |
| LFX (0) |
LFX (−1) |
Minocycline |
ABI-PP |
| 0 |
1.3 (43) |
1.8 (51) |
14.8 (21) |
13.9 (19) |
| 1 |
3.3 (26) |
3.4 (22) |
3.3 (21) |
12.7 (15) |
| 2 |
4.8 (22) |
6.5 (14) |
9.0 (18) |
6.1 (14) |
| 3 |
9.1 (4) |
8.3 (9) |
6.4 (16) |
9.2 (13) |
| 4 |
16.5 (2) |
10.6 (1) |
15.4 (11) |
11.8 (13) |
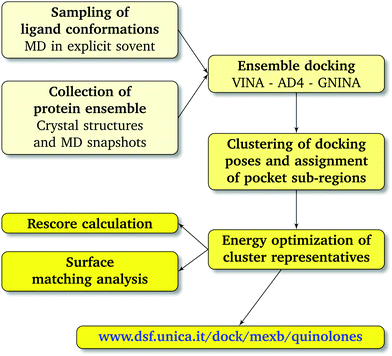
As shown in Table 1 and Fig. 4, in all considered benchmark cases, our protocol was able to find the binding mode of the co-crystal structure with reasonable accuracy. More specifically, the second and third populated clusters of minocycline and ABI-PP correspond to the experimental structures (RMSD 3.3 and 6.1 Å, respectively). The relatively high value found for ABI-PP is partly due to the high flexibility of this compound (13 rotatable bonds vs. 2 for minocycline) and the high RMSD of the ten fixed input conformations used for docking with respect to the crystal structure (Table S4, ESI†). A better agreement was found in the case of the less flexible LFX (2 rotatable bonds), although the co-structure of AcrB with this compound (PDB Id: 7B8T) was not included in the AcrB ensemble. The most populated clusters of the zwitterionic and −1 charged forms (43 and 51%, respectively) were found to reproduce the experimental conformation (RMSD 1.3 Å and 1.8 Å). Similar to re-docking (see the Methods section), even though the difference between the two protonation states is small, slightly better performance was found by using the zwitterionic form. This suggest that the zwitterionic form is the most likely protonation state of LFX under the crystallization conditions.
 |
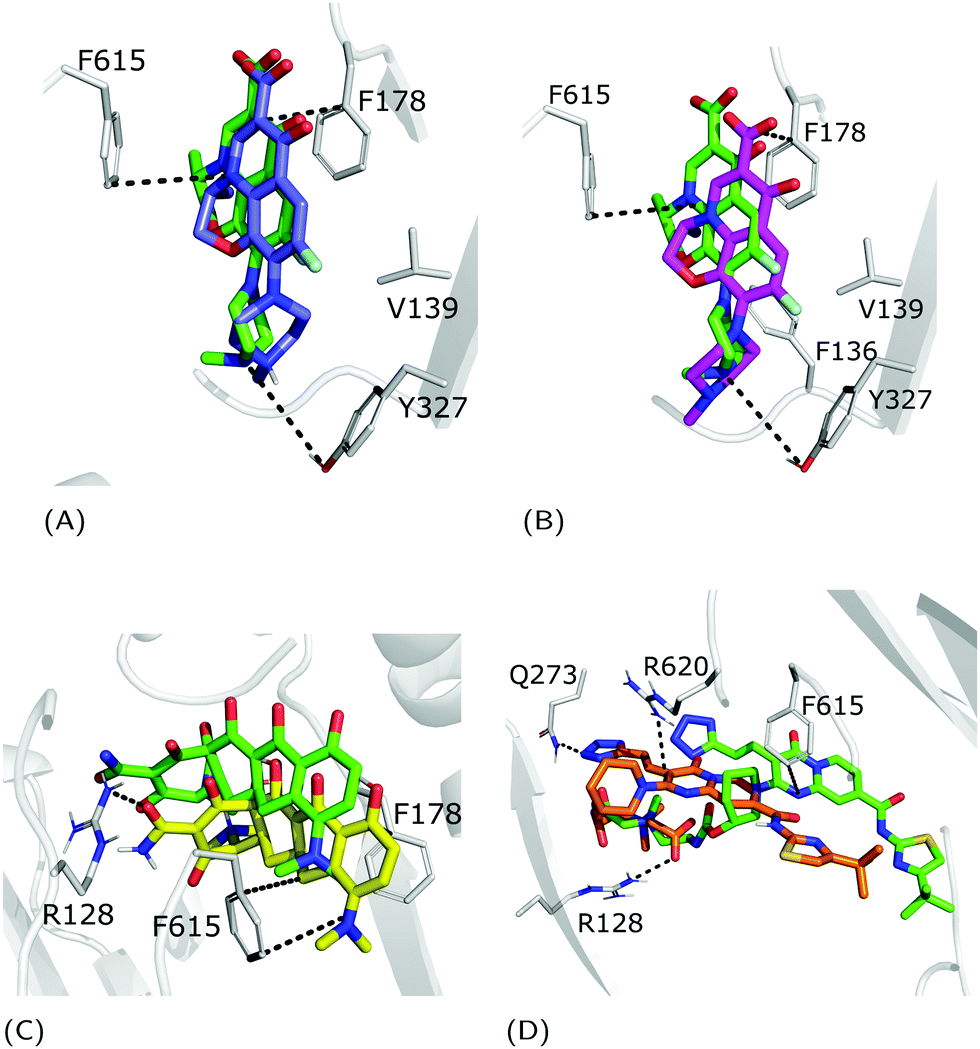
| | Fig. 4 Superimposition between the crystal structure (colored in green) and the docking poses: (A) zwitterionic LFX in violet, (B) non-zwitterionic LFX (net charge −1) in magenta, (C) minocycline in yellow, and (D) ABI-PP in orange. LFX and minocycline are in a complex with AcrB (PDB Ids: 7B8T32 and 4DX5,45 respectively), while ABI-PP is in a complex with MexB (PDB Id 3W9J29). Protein–ligand interactions are represented as dotted lines. | |
The weighted averages of AutoDock VINA re-scores and surface complementary indexes reported in Table 2 are consistent with the above findings and the localization of the three molecules in the corresponding crystal structures.32 In detail, we found that LFX and ABI-PP bind preferentially in the cave, while minocycline is a groove-binder. Surface-matching data follow the overall trend of the docking scores suggesting that the binding affinity is primarily driven by shape and surface complementarities.
Table 2 Autodock VINA affinity score obtained by rescoring and PLATINUM surface matching, both associated to each DPT sub-region and weighted on the cluster population – SMTOT refers to the total surface match (lipophilic and hydrophilic), and SML to the lipophilic match
|
|
Weighted score (arbitrary units) |
Weighted SMTOT |
Weighted SML |
| Interface |
Cave |
Groove |
Interface |
Cave |
Groove |
Interface |
Cave |
Groove |
| AcrB |
| LFX (0) |
−0.9 |
−4.4 |
−2.5 |
0.1 |
0.3 |
0.1 |
0.1 |
0.4 |
0.2 |
| LFX (−1) |
−0.1 |
−5.4 |
−2.6 |
0.0 |
0.5 |
0.2 |
0.0 |
0.6 |
0.2 |
| Minocycline |
−1.9 |
−2.7 |
−3.6 |
0.1 |
0.2 |
0.2 |
0.1 |
0.2 |
0.2 |
| MexB |
| ABI-PP |
−0.9 |
−3.7 |
−2.7 |
0.0 |
0.2 |
0.2 |
0.1 |
0.3 |
0.2 |
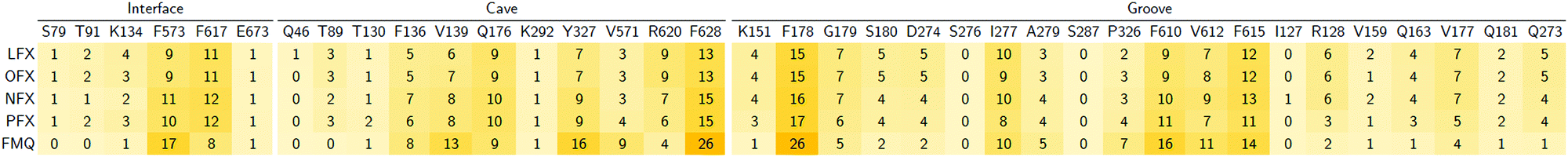
In particular, focusing on the multiple binding modes found for LFX in AcrB, while most populated clusters (with a fraction of > 10%) are located in the same region of the crystal structure, the least populated clusters (i.e., population < 10%) spread from the groove to the exit gate. These findings are quantitatively supported by the heat map generated from the statistical contact analysis of all docking poses (Table 3). Interestingly, the most contacted residues are amongst those found in the crystal structure and whose prominent role was confirmed by site-directed mutagenesis experiments: F178, Y327, F610 and F628.32 In addition, other residues appear to be relevant in the map, such as R620 and V571 of the cave, and G179, N274 and P326 of the groove. We speculate that additional binding modes involving these residues represent further steps in the efflux pathway.
Table 3 Heat map of contacts (%) between all poses of LFX with the DPT of AcrB – Contacts within 3.5 Å found in the experimental structure (PDB Id: 7B8T32) are reported in red boldface
3.2 Systematic application to FQs
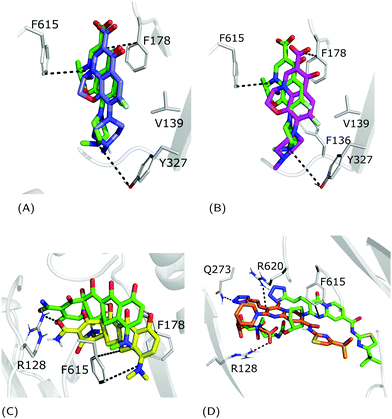
For each FQ, we first looked at the fractional population of docking poses in the three sub-regions of the DPT (Fig. 5). Overall, almost all compounds were found to bind MexB preferentially in the groove, with the exception of flumequine and sarafloxacin that were found mostly in the cave. Conversely, marbofloxacin was not found in the cave region, and pazufloxacin, nadifloxacin, grepafloxacin, trovafloxacin and prulifloxacin were found for less than 1% in this region. The distribution of all other compounds varied in these three regions. Similarly, Fig. S2 (ESI†) reports the weighted average docking affinity, normalized to the molecular weight, associated with each sub-region. As expected from the poses distribution, the groove sub-region is the one with the higher affinity, while the scores of the interface and cave are comparable. Sarafloxacin presents both the lowest and the highest scores in the groove and the cave, respectively. Conversely, marbofloxacin and trovafloxacin have the highest scores in the groove and the lowest in the cave. The distribution of the scores at the interface is more homogeneous as compared to that observed at the cave and the groove (standard deviation: 0.5, 0.9 and 1.1, respectively). As mentioned above, 11 FQs were simulated in two different protonation states: one with a net charge of −1, and one in the neutral zwitterionic form. Fig. 5 highlights the differences in the distribution of the protonation states. In some cases, we found the same trend (i.e., ciprofloxacin and norfloxacin), while in the majority of FQs we found little differences (e.g., difloxacin, enrofloxacin, and fleroxacin). Generally, the groove is the most populated sub-region and the differences between the interface and the cave are minimal. This could suggest that, inside the distal pocket, different protonation states can find multiple binding modes. Taken together, the above results highlight that there is no common pattern through the compounds, and even small changes in the structure lead to differences in the binding modes. Therefore, from the general overview of docking poses spatial distribution and binding affinity, it is interesting to identify prevalent, distinct binding modes. In the following, we focused first on LFX, for which the co-structure with AcrB is available,32 moving then to four FQs structurally similar to LFX, namely ofloxacin (OFX), nadifloxacin (NFX), pazufloxacin (PFX) and flumequine (FMQ).
 |
| | Fig. 5 Sub-region distribution of the docking poses (%): gray bars refer to the interface, red to the cave, and blue to the groove. FQs are in ascending order according to the molecular weight. FQs containing the piperazine group are reported in two net charges (i.e., 0 and −1). LFX and related compounds (Fig. 1) are highlighted in boldface. | |
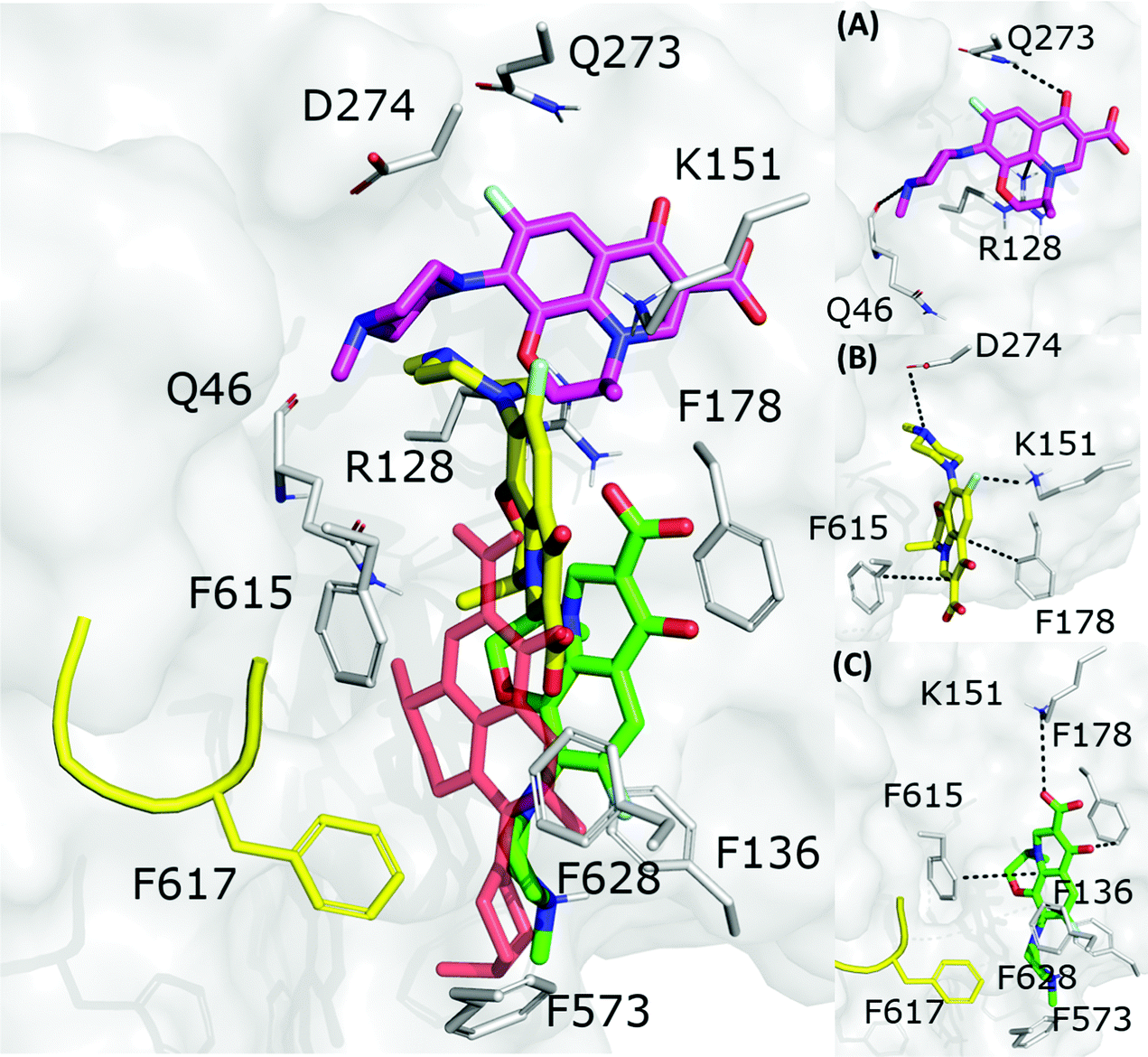
3.2.1 Levofloxacin.
The fractional population of the LFX docking poses in MexB reflects the general trend discussed above: 68, 12, and 20% for the groove, cave, and interface, respectively. Looking at all cluster representatives, we identified three main binding modes. The first binding mode (BM1) is located in the groove, where the central aromatic core is involved in a π–π stacking interaction with F178, F615 and F628, the carboxyl group faces K151 and the 7-piperazyl ring points towards the HT (Fig. 6C). In the second binding mode (BM2), the ligand lies in the groove interacting by π–π stacking interactions with both F178 and F615, and by an electrostatic interaction with D274 through the protonated amine of the 7-piperazyl ring (Fig. 6B). Finally, the third binding mode (BM3) is in the deeper portion of the groove, near the exit gate (Fig. 6A). Here, LFX contacts R128 by a cation–π interaction and is involved in H-bonds with Q46 and Q273. Noteworthily, although we found analogous binding modes of LFX in both AcrB and MexB, in the former LFX was mainly docked according to BM1, and only a small percentage explored the rest of the pocket, in particular the deep portion of the groove (BM3). In contrast, in MexB, 19% of the poses were found interacting as BM3. The distal pockets of AcrB and MexB are well conserved; however, even minor changes can modify the electrostatic properties, thus affecting protein–ligand interactions.27 Most of the key residues of BM3 are conserved, with the exception of R128, which in AcrB is S128.
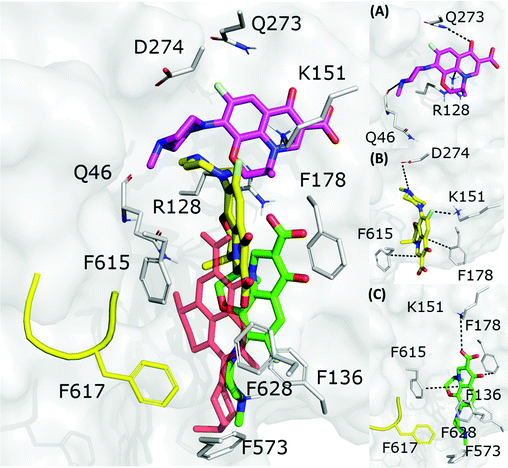 |
| | Fig. 6 LFX binding modes in MexB: left panel: BM1 is colored in green, BM2 in yellow and BM3 in magenta. The switch loop is represented as a yellow cartoon, and the superimposition of LFX co-crystallized in AcrB (PDB Id: 7B8T32) is red and semi-transparent. Right panel: Detailed visualization of interactions (reported as dotted lines) between LFX and MexB in (A) BM3, (B) BM2 and (C) BM1. | |
3.2.2 FQs structurally similar to levofloxacin.
We investigated how small modifications of LFX affect the prevalent interactions within the DPT of MexB by analysing FQs structurally similar to LFX (Fig. 1). Like Table 3 for AcrB, Table 4 presents the percentage of contacts of LFX and the similar FQs with the DPT of MexB. For LFX, amongst the residues with the highest percentage of contacts (>10%), F178 and F628 were found to be relevant in the binding of AcrB according to X-ray and mutagenesis,32 suggesting their role in MexB as well. In addition, the heat map reveals the importance of other residues for LFX and related compounds, such as Q176, I277, F573, F610, F615, and F617. To the best of our knowledge, point mutation studies on the impact of the DPT residues of MexB on resistance to LFX and similar compounds are not available to date. In silico simulations coupled with experimental mutational analyses could help elucidate the role of these residues.
Table 4 Heat map of contacts (%) between the DPT residues in MexB and all poses of LFX, OFX, NFX, PFX and FMQ
Focusing on the differences in Table 4, we found that OFX is the FQ most similar to LFX, followed by NFX, PFX and FMQ (overall differences in contacts: 0.14%, 0.76%, 0.94%, and 3.25%, respectively). Afterward, we carefully compared the main binding modes of OFX, NFX, PFX and FMQ with BM1, BM2, and BM3 of LFX. OFX results are largely comparable to those of LFX, in terms of both binding modes (BM1, BM2 and BM3) and contacts, suggesting that the distal pocket of MexB is large enough to overcome the specificity towards enantiomers. NFX differs from LFX mainly for the lacking of a basic protonated group, which is replaced by hydroxyl piperidine. Despite this, we found NFX poses in BM1, BM2 and BM3 (Fig. 7A). Differently, PFX possesses a basic amine group, but it lacks the piperazine ring which is replaced by cyclopropane, leading to a smaller steric hindrance. As a result, BM1 for PFX is rotated by 90°, with the cyclopropane moiety inserted into the HT, the aromatic rings involved in a stacking interaction with F178 and F628, and the carboxyl group pointing in the opposite direction of the HT. In BM2, we found the same interactions with F178 and F615, but the molecule is rotated by 180° forming a salt bridge between the carboxyl moiety and K151. BM3 was present only in small percentages, while the interface localization, in proximity to the switch loop, is more populated (20%) (Fig. 7B). FMQ contains the central core of NFX and lacks both the piperazine ring and the basic group. This yielded to the accumulation of docking poses at the HT, with a similar orientation of PFX not only in BM1, but also in BM2. Meanwhile, the poses placed in BM3 were not found (Fig. 7C).
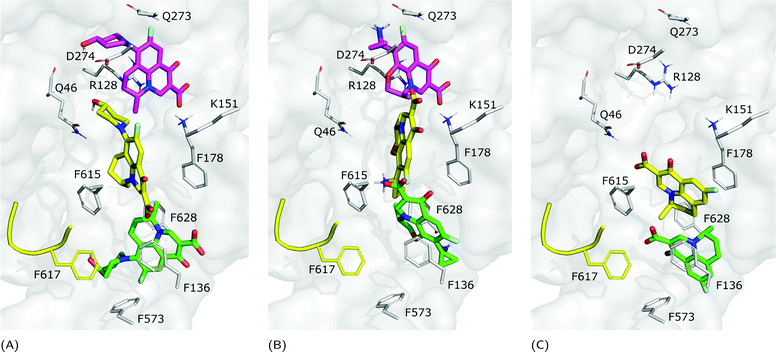 |
| | Fig. 7 Binding modes of (A) NFX, (B) PFX and (C) FMQ. BM1 is colored in green, BM2 is colored in yellow, and BM3 is colored in magenta. BM3 was not found in FMQ. The switch loop is represented as a yellow cartoon. | |
The representatives of each binding mode, after the energy refinement, underwent a surface matching calculation by PLATINUM (see Materials and methods). The results are listed in Table 5. Overall, the higher lipophilic matches (SML) were reached in BM1, followed by BM2, reflecting the hydrophobic characteristic of the two sites, which are rich in aromatic residues (e.g., HT). In contrast, BM3 presents several hydrophilic residues (e.g., Q46, R128, and Q273); therefore, the matches are guided by polar interactions. Given the small and rigid shape of FMQ, the docking protocol was able to maximize the surface complementarity, especially with aromatic rings. Therefore, FMQ obtained the highest matching score.
Table 5 Surface matches between MexB DPT and FQs, for different binding modes – SMTOT refers to the total surface match (lipophilic and hydrophilic), and SML to the lipophilic match. The population (%) of the corresponding structural cluster is also reported
|
|
BM1 |
BM2 |
BM3 |
| % |
SMTOT |
SML |
% |
SMTOT |
SML |
% |
SMTOT |
SML |
| LFX |
31 |
0.3 |
0.5 |
13 |
0.4 |
0.5 |
18 |
0.3 |
0.0 |
| OFX |
22 |
0.6 |
0.8 |
16 |
0.4 |
0.5 |
17 |
0.3 |
0.2 |
| NFX |
13 |
0.7 |
0.8 |
36 |
0.6 |
0.7 |
12 |
0.3 |
0.2 |
| PFX |
43 |
0.4 |
0.6 |
13 |
0.4 |
0.5 |
9 |
0.5 |
0.2 |
| FMQ |
50 |
0.8 |
0.9 |
25 |
0.9 |
0.9 |
5 |
0.5 |
0.5 |
Furthermore, we compared LFX with orbifloxacin (Fig. 1F), which was reported as a competitive inhibitor of MexB.50 The structure of orbifloxacin differs from LFX also in the central core, and we found indeed some differences in the binding modes. Specifically, some of the most populated clusters are located at the interface (33%, score: −2.1 kcal mol−1) with different orientations, near the switch loop and the residues S79 and T91. BM1 and BM2 are also present, while BM3 is poorly explored.
4 Conclusions
Computational methods allow the molecular recognition process in biological systems, such as protein–ligand complexes, to be investigated. The binding is guided by weak interactions and surface complementarity between the two molecules, and molecular docking can be exploited to study these phenomena. In this work, we applied this method to systems strongly involved in the appearance of the MDR, which is an urgent issue to world public health. Among these systems, efflux pumps are transmembrane protein complexes that are able to extrude a wide range of antibiotics out of the bacterial cell. Here, we focused on MexB, the major RND transporter of P. aeruginosa, for which only one co-crystal structure with an inhibitor is available to date. Given the high sequence identity with the homologous protein AcrB of E. coli, we used the AcrB–LFX crystal structure as a reference. First, we validated the computational protocol by reproducing the experimental conformations of AcrB and MexB complexes (Fig. 4). Then, we performed a systematic ensemble docking campaign of 36 different FQs within the DPT of MexB, followed by cluster analysis, re-scoring and surface-matching calculations. Our findings suggest that multiple binding modes in different sub-pockets of the DPT are possible, supporting the diffusive binding hypothesis.27,38 In particular, focusing on LFX, we identified three main binding modes (i.e., BM1, BM2 and BM3, Fig. 6), which were found also in other FQs with similar structures (namely OFX, NFX, PFX and FMQ, Fig. 7). However, compounds with different structures compared to LFX were found to assume diverse modes of binding (e.g., orbifloxacin). Given the lack of MexB experimental structures co-crystallized with FQs, we made available the binding modes of each compound at https://www.dsf.unica.it/dock/mexb/quinolones, where users can both directly visualize the complexes and download the corresponding PDB files. Our results constitute a solid starting point for further studies making use of more advanced techniques68,69 aimed at investigating the dynamics of compounds inside the transporter and rationalize the available microbiology data.24
Author contributions
Conceptualization: G. M., A. V. V., and P. R.; methodology: G. M., A. B., A. V. V., and P. R.; software: G. M., A. V. V., and A. B.; validation: S. G. and G. M.; formal analysis: S. G. and G. M.; investigation: S. G. and G. M.; writing original draft: S. G. and G. M.; writing – review and editing: S. G., G. M., A. B., A. V. V., H. I. Z., and P. R.; visualization: S. G. and G. M.; project administration: H. I. Z and P. R.; funding acquisition: H. I. Z. and P. R. All the authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases/NIH Grant no. R01AI136799 and “Progetti biennali d'Ateneo Finanziati dalla Fondazione di Sardegna – annualità 2020.” We thank Giovanni Serra (University of Cagliari) for technical assistance with computational resources. One of the authors (P.R.) thanks Giorgio Benedek for having introduced him to the realm of surfaces. Without Giorgio's guidance the passage from silicon and metal surfaces to biosurfaces would have been much harder.
References
- S. Kaushik and Chia-en A. Chang, Front. Mol. Biosci., 2021, 8, 659687 CrossRef CAS PubMed.
- R. Baron and J. A. McCammon, Annu. Rev. Phys. Chem., 2013, 64, 151–175 CrossRef CAS PubMed.
- A.-L. Noresson, O. Aurelius, C. T. Öberg, O. Engström, A. P. Sundin, M. Håkansson, O. Stenström, M. Akke, D. T. Logan, H. Leffler and U. J. Nilsson, Chem. Sci., 2018, 9, 1014–1021 RSC.
- E. P. Barros, J. M. Schiffer, A. Vorobieva, J. Dou, D. Baker and R. E. Amaro, J. Chem. Theory Comput., 2019, 15, 5703–5715 CrossRef CAS PubMed.
- C.-Q. Xia, X. Pan and H.-B. Shen, Bioinformatics, 2020, 36, 3018–3027 CrossRef CAS PubMed.
- X. Tang, Z. Wang, T. Lei, W. Zhou, S. Chang and D. Li, Phys. Chem. Chem. Phys., 2018, 20, 5591–5605 RSC.
- V. Salmaso and S. Moro, Front. Microbiol., 2018, 9, 923 CrossRef PubMed.
- E. M. Eichenberger and J. T. Thaden, Antibiotics, 2019, 8, 37 CrossRef CAS PubMed.
- B. Mehrad, N. M. Clark, G. G. Zhanel and L. J.-P. III, Crit. Care Med., 2015, 147, 1413–1421 Search PubMed.
- M. Costa, M. Cardo, M. C. d'Anjo and A. Leite, Zoonoses Public Health, 2022, 00, 1–13 Search PubMed.
- E. Avershina, V. Shapovalova and G. Shipulin, Front. Microbiol., 2021, 12, 707330 CrossRef PubMed.
- T. J. Laird, D. Jordan, Z. Z. Lee, M. O'Dea, M. Stegger, A. Truswell, S. Sahibzada, R. Abraham and S. Abraham, J. Antimicrob. Chemother., 2022, 77, 400–408 CrossRef CAS PubMed.
- S. Santajit and N. Indrawattana, BioMed Res. Int., 2016, 2016, 2475067 Search PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. Guzman Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. van der Meer, N. Van Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F.-L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- Z. Pang, R. Raudonis, B. R. Glick, T.-J. Lin and Z. Cheng, Biotechnol. Adv., 2019, 37, 177–192 CrossRef CAS PubMed.
- I. Alav, J. Kobylka, M. S. Kuth, K. M. Pos, M. Picard, J. M.-A. Blair and V. N. Bavro, Chem. Rev., 2021, 121, 5479–5596 CrossRef CAS PubMed.
- H. Nikaido, Res. Microbiol., 2018, 169, 363–371 CrossRef CAS PubMed.
- D. Du, X. Wang-Kan, A. Neuberger, H. W. van Veen, K. M. Pos, L. J.-V. Piddock and B. F. Luisi, Nat. Rev. Microbiol., 2018, 16, 523–539 CrossRef CAS PubMed.
- K. Tsutsumi, R. Yonehara, E. Ishizaka-Ikeda, N. Miyazaki, S. Maeda, K. Iwasaki, A. Nakagawa and E. Yamashita, Nat. Commun., 2019, 10, 1520 CrossRef PubMed.
- A. Zahedi Bialvaei, M. Rahbar, R. Hamidi-Farahani, A. Asgari, A. Esmailkhani, Y. Mardani Dashti and S. Soleiman-Meigooni, Microb. Pathog., 2021, 153, 104789 CrossRef CAS PubMed.
- H. I. Zgurskaya, G. Malloci, B. Chandar, A. V. Vargiu and P. Ruggerone, Curr. Opin. Microbiol., 2021, 61, 115–123 CrossRef CAS PubMed.
- H. A.-A. Ezelarab, S. H. Abbas, H. A. Hassan and G. E.-D. A. Abuo-Rahma, Arch. Pharm., 2018, 351, e1800141 CrossRef PubMed.
- G. Krishnamoorthy, I. V. Leus, J. W. Weeks, D. Wolloscheck, V. V. Rybenkov and H. I. Zgurskaya, mBio, 2017, 8, e01172–17 CrossRef CAS PubMed.
- S. J. Cooper, G. Krishnamoorthy, D. Wolloscheck, J. K. Walker, V. V. Rybenkov, J. M. Parks and H. I. Zgurskaya, ACS Infect. Dis., 2018, 4, 1223–1234 Search PubMed.
- H. I. Zgurskaya, J. K. Walker, J. M. Parks and V. V. Rybenkov, Acc. Chem. Res., 2021, 54, 930–939 CrossRef CAS PubMed.
- J. Kobylka, M. S. Kuth, R. T. Müller, E. R. Geertsma and K. M. Pos, Ann. N. Y. Acad. Sci., 2020, 1459, 38–68 CrossRef CAS PubMed.
- V. K. Ramaswamy, A. V. Vargiu, G. Malloci, J. Dreier and P. Ruggerone, Front. Microbiol., 2018, 9, 1144 CrossRef PubMed.
- M. Glavier, D. Puvanendran, D. Salvador, M. Decossas, G. Phan, C. Garnier, E. Frezza, Q. Cece, G. Schoehn, M. Picard, J.-C. Taveau, L. Daury, I. Broutin and O. Lambert, Nat. Commun., 2020, 11, 4948 CrossRef CAS PubMed.
- R. Nakashima, K. Sakurai, S. Yamasaki, K. Hayashi, C. Nagata, K. Hoshino, Y. Onodera, K. Nishino and A. Yamaguchi, Nature, 2013, 500, 102–106 CrossRef CAS PubMed.
- P. A. Klenotic, M. A. Moseng, C. E. Morgan and E. W. Yu, Chem. Rev., 2021, 121, 5378–5416 CrossRef CAS PubMed.
- M. Zwama, S. Yamasaki, R. Nakashima, K. Sakurai, K. Nishino and A. Yamaguchi, Nat. Commun., 2018, 9, 124 CrossRef PubMed.
- A. Ornik-Cha, J. Wilhelm, J. Kobylka, H. Sjuts, A. V. Vargiu, G. Malloci, J. Reitz, A. Seybert, A. S. Frangakis and K. M. Pos, Nat. Commun., 2021, 12, 6919 CrossRef CAS PubMed.
- R. Nakashima, K. Sakurai, S. Yamasaki, K. Nishino and A. Yamaguchi, Nature, 2011, 480, 565–569 CrossRef CAS PubMed.
- R. T. Müller, T. Travers, H. Jea Cha, J. L. Phillips, S. Gnanakaran and K. M. Pos, J. Mol. Biol., 2017, 429, 3863–3874 CrossRef PubMed.
- K. Sakurai, S. Yamasaki, K. Nakao, K. Nishino, A. Yamaguchi and R. Nakashima, Sci. Rep., 2019, 9, 4359 CrossRef PubMed.
- D. Dey, L. G. Kavanaugh and G. L. Conn, Antimicrob. Agents Chemother., 2020, 64, e00496 CrossRef CAS PubMed.
- R. K. Roy and N. Patra, ChemPhysChem, 2020, 21, 2516–2524 CrossRef PubMed.
- A. Yamaguchi, R. Nakashima and K. Sakurai, Front. Microbiol., 2015, 6, 327 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- A. T. McNutt, P. Francoeur, R. Aggarwal, T. Masuda, R. Meli, M. Ragoza, J. Sunseri and D. R. Koes, J. Cheminf., 2021, 13, 43 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- Y. Takatsuka, C. Chen and H. Nikaido, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6559–6565 CrossRef CAS PubMed.
- A. Atzori, G. Malloci, J. D. Prajapati, A. Basciu, A. Bosin, U. Kleinekathöer, J. Dreier, A. V. Vargiu and P. Ruggerone, J. Phys. Chem., 2019, 123, 4625–4635 CrossRef CAS PubMed.
- T. Eicher, H. Jea Cha, M. A. Seeger, L. Brandstätter, J. El-Delik, J. A. Bohnerta, W. V. Kern, F. Verrey, M. G. Grütter, K. Diederichs and K. M. Pos, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5687–5692 CrossRef CAS PubMed.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G.-W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci., 2018, 27, 112–128 CrossRef CAS PubMed.
- ChemAxon, Marvin version 21.14, https://www.chemaxon.com, 2021, https://www.chemaxon.com.
- S. Babić, A. J. Horvat, D. M. Pavlović and M. Kaśtelan-Macan, Trends Anal. Chem., 2007, 11, 1043–1061 CrossRef.
- H.-K. Tam, V. N. Malviya, W.-E. Foong, A. Herrmann, G. Malloci, P. Ruggerone, A. V. Vargiu and K. M. Pos, J. Mol. Biol., 2020, 432, 861–877 CrossRef CAS PubMed.
- J. Mehla, G. Malloci, R. Mansbach, C. A. López, R. Tsivkovski, K. Haynes, I. V. Leus, S. B. Grindstaff, R. H. Cascella, N. D'Cunha, L. Herndon, N. W. Hengartner, E. Margiotta, A. Atzori, A. V. Vargiu, P. D. Manrique, J. K. Walker, O. Lomovskaya, P. Ruggerone, S. Gnanakaran, V. V. Rybenkov, H. I. Zgurskaya and G. D. Wright, mBio, 2021, 12, e02785 CrossRef CAS PubMed.
- M. A. Seeger, A. Schiefner, T. Eicher, F. Verrey, K. Diederichs and K. M. Pos, Science, 2006, 313, 1295–1298 CrossRef CAS PubMed.
- G. Sennhauser, P. Amstutz, C. Briand, O. Storchenegger and M. G. Grütter, PLoS Biol., 2007, 5, e7 CrossRef PubMed.
- C. Ostwald, H.-K. Tam and K. M. Pos, Nat. Commun., 2016, 7, 13819 CrossRef PubMed.
- G. Sennhauser, M. A. Bukowska, C. Briand and M. G. Grütter, J. Mol. Biol., 2009, 389, 134–145 CrossRef CAS PubMed.
- W. Xue, T. Fu, S. Deng, F. Yang, J. Yang and F. Zhu, ACS Chem. Neurosci., 2022, 13, 340–351 CrossRef CAS PubMed.
- G. Tu, T. Fu, F. Yang, J. Yang, Z. Zhang, X. Yao, W. Xue and F. Zhu, ACS Chem. Neurosci., 2021, 12, 2013–2026 CrossRef CAS PubMed.
- Y. Zhang, G. Zheng, T. Fu, J. Hong, F. Li, X. Yao, W. Xue and F. Zhu, Phys. Chem. Chem. Phys., 2020, 7, 5132–5144 RSC.
- S. Gervasoni, G. Malloci, A. Bosin, A. V. Vargiu, H. I. Zgurskaya and P. Ruggerone, Sci. Data, 2022, 9, 148 CrossRef PubMed.
- J. Shao, S. W. Tanner, N. Thompson and T. E. Cheatham, J. Chem. Theory Comput., 2007, 3, 2312–2334 CrossRef CAS PubMed.
-
D. Case, V. Babin, J. Berryman, R. Betz, Q. Cai, D. Cerutti, T. Cheatham, III, T. Darden, R. Duke, H. Gohlke, A. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossviáry, A. Kovalenko, T. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. Merz, F. Paesani, D. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. Simmerling, W. Smith, J. Swails, R. Walker, J. Wang, R. Wolf, X. Wu and P. Kollman, AMBER 2018, University of California, San Francisco Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- T. V. Pyrkov, A. O. Chugunov, N. A. Krylov, D. E. Nolde and R. G. Efremov, Bioinformatics, 2009, 25, 1201–1202 CrossRef CAS PubMed.
- T. V. Pyrkov, Y. A. Kosinsky, A. S. Arseniev, J. P. Priestle, E. Jacoby and R. Efremov, Proteins, 2007, 66, 388–398 CrossRef CAS PubMed.
- R. G. Efremov, A. O. Chugunov, T. V. Pyrkov, J. P. Priestle, A. S. Arseniev and E. Jacoby, Curr. Med. Chem., 2007, 14, 393–415 CrossRef CAS PubMed.
- A. V. Vargiu and H. Nikaido, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20637–20642 CrossRef CAS PubMed.
- A. S. Rose, A. R. Bradley, Y. Valasatava, J. M. Duarte, A. Prlic’ and P. W. Rose, Bioinformatics, 2018, 34, 3755–3758 CrossRef CAS PubMed.
-
The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC Search PubMed.
- A. V. Vargiu, V. K. Ramaswamy, G. Malloci, I. Malvacio, A. Atzori and P. Ruggerone, Res. Microbiol., 2018, 169, 384–392 CrossRef CAS PubMed.
- A. V. Vargiu, V. K. Ramaswamy, I. Malvacio, G. Malloci, U. Kleinekathöfer and P. Ruggerone, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 836–845 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Giuliano
Malloci
a,
Giuliano
Malloci